
Erwachsene mit angeborenen Herzfehlern sind eine rasch wachsende, neue und komplexe Patientengruppe in der Erwachsenenkardiologie. Auch nach erfolgreicher chirurgischer Reparatur der Herzfehler im Kindesalter sind diese Patienten nicht geheilt. Zu den häufigsten Komplikationen im Erwachsenenalter gehören die Herzrhythmusstörungen (vor allem das atypische Vorhofflattern) und die infektiöse Endokarditis. Eine gute Patientenedukation trägt zur frühen Diagnose und Behandlung dieser Komplikationen bei und vermindert damit Morbidität und Mortalität. Gelegentlich werden Herzfehler erst im Erwachsenenalter entdeckt. Zu solchen Herzfehlern gehören die Vorhofseptumdefekte, die Aortenisthmusstenose und die kongenital korrigierte Transposition der grossen Arterien.
Hintergrund und Übersicht
Angeborene Herzfehler sind die häufigsten Geburtsdefekte. Etwa 1/100 Lebendgeborenen ist betroffen und etwa 6/1000 Babys haben einen komplexen angeborenen Herzfehler (1). Bis zur Entwicklung der modernen Herzchirurgie, insbesondere der Herz-Lungen-Maschine in den 1950er Jahren starben die meisten betroffenen Patienten im frühen Kindesalter. Dies hat sich in den letzten Jahrzehnten dramatisch verändert. Heute gehen wir davon aus, dass über 90% der Kinder mit einem angeborenen Herzfehler – auch jene mit komplexen Defekten – das Erwachsenenalter erreicht (2). Diese Entwicklung führt dazu, dass die Kohorten der Erwachsenen mit angeborenen Herzfehlern rasch wachsen und die Zahl der Erwachsenen mit angeborenen Herzfehlern jene der betroffenen Kinder bereits bei Weitem übersteigt (3, 4).
Bezeichnung und Begriffe
Erwachsene mit angeborenen Herzfehlern sind eine neue Disziplin in der Erwachsenen-Kardiologie. Über die letzten Jahrzehnte sind verschiedene Bezeichnungen und Abkürzungen für diese Patientengruppe entstanden. In Europa wurden die Patienten lange als ‘GUCH-Patienten’ (grown up congenital heart disease patients) bezeichnet, während sich im Rest der Welt die Abkürzung ACHD (adult congenital heart disease) etablierte, die sich nun auch in Europa durchsetzt. In Deutschland wird gelegentlich die Abkürzung EMAH (Erwachsene mit angeborenen Herzfehlern) verwendet. Das Spektrum einzelner angeborener Herzfehler und der verschiedenen Reparaturtechniken ist riesig und eine detaillierte Beschreibung der einzelnen Entitäten sprengt den Rahmen einer solchen Übersichtsarbeit. Oft hilft eine schematische Darstellung von Herzanatomie und Operationstechnik zum besseren Verständnis der modifizierten Anatomie des individuellen Patienten. Eine ausgezeichnete Ressource ist die Bildsammlung des New Media Center der Universität Basel (www.congenital-heart-disease.ch), die allen Interessierten gratis zur Verfügung steht.
Herausforderungen im Erwachsenenalter
Geflickt ist nicht geheilt! Obwohl das Überleben ins Erwachsenenalter heute die Regel ist, haben betroffene Erwachsene ein deutlich erhöhtes Risiko für kardiovaskuläre Komplikationen und viele auch ein erhöhtes Sterberisiko als junge Erwachsene (4-7). Mehrere Studien konnten zeigen, dass eine spezialisierte Betreuung von Erwachsenen mit angeborenen Herzfehlern durch multidisziplinäre Teams das Risiko für Komplikationen vermindert und das Überleben verbessert (8, 9).
Für Patientengruppen mit sehr hoher Komplexität, zum Beispiel Patienten mit univentrikulären Herzen, nicht operierten zyanotischen Herzfehlern oder Patienten mit schwerer Herzinsuffizienz ist eine exklusive Betreuung an einem spezialisierten Zentrum sinnvoll (www.sgk-watch.ch). Da in der Schweiz ein Netz an gut ausgebildeten niedergelassenen Kardiologen besteht, hat sich für Patienten mit weniger komplexen Herzfehlern die gemeinsame Betreuung in enger Zusammenarbeit von spezialisierten Zentren mit niedergelassenen Kardiologen sehr bewährt (10).
Ziel dieses Artikels:
- Diskussion wichtiger Langzeitkomplikationen (die sich bei Herzfehlerpatienten zuweilen atypisch präsentieren!)
- Neudiagnose von Herzfehlern im Erwachsenenalter – ein Überblick
- Behandlungsstrategien zur Vermeidung von Komplikationen
Langzeitkomplikationen
Herzrhythmusstörungen
Arrhythmien sind die mit Abstand häufigste Langzeitkomplikation bei Patienten mit angeborenen Herzfehlern. Bis zum 50. Altersjahr sind mehr als 50% aller Patienten davon betroffen (11). Hauptursache der Arrhythmien sind myokardiale Narben, oft entstanden bei chirurgischen Reparatureingriffen. Hämodynamische Residuen, wie Klappeninsuffizienzen oder Stenosen, sowie Myokardinsuffienz / Herzinsuffizienz können Arrhythmien zusätzlich begünstigen.
Aufgrund der modifizierten Herzanatomie präsentiert sich das atypische Vorhofflattern (auch intra-atriale Reentry-Tachykardie oder kurz IART genannt) oft ungewöhnlich und wird deshalb nicht selten anfangs verpasst. Insbesondere bei Patienten mit deutlich vergrösserten Herzvorhöfen präsentiert sich das Vorhofflattern mit 2:1-Überleitung oft mit ungewöhnlich tiefer Ventrikelfrequenz (Abb. 1). Gerade der raschen Erkennung des Vorhofflatterns mit 2:1-Überleitung kommt grosse Bedeutung zu! Bei starrer Herzfrequenz über Stunden oder Tage kommt es bei den betroffenen Patienten oft rasch zur Ausbildung einer ausgeprägten Tachy-Myopathie mit schwer eingeschränkter Ventrikelfunktion. So erleben wir im Alltag immer wieder Situationen, bei denen sich wegen der verzögerten Diagnose eines Vorhofflatterns, aus einer eigentlich banalen, gut beherrschbaren Herzrhythmusstörung, ein lebensbedrohlicher Zustand entwickelt. Diesbezüglich kommen einer guten Patientenedukation und einer raschen Abklärung bei Auftreten von Symptomen eine hohe Bedeutung zu.
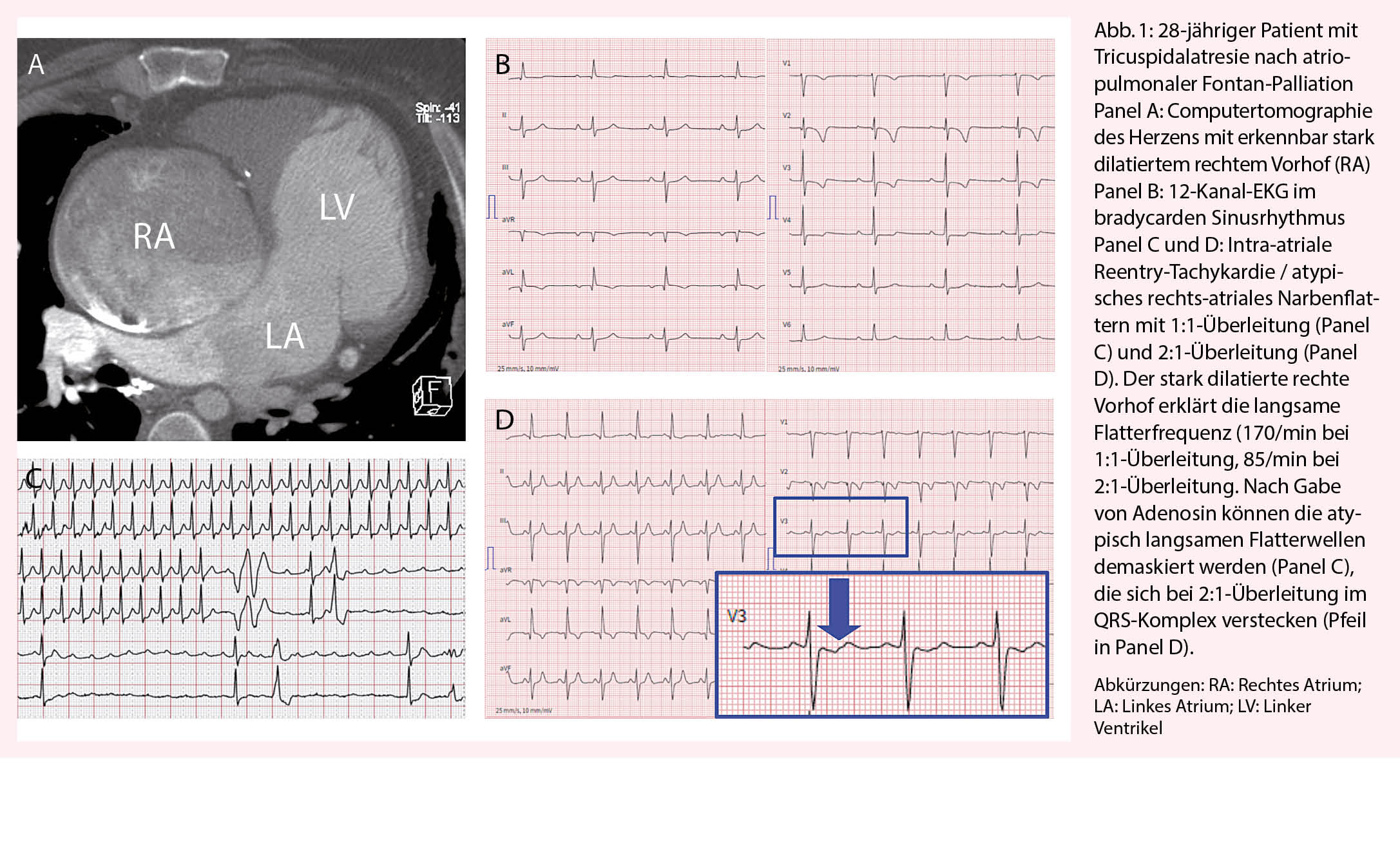
Auch ventrikuläre Arrhythmien und der plötzliche Herztod treten bei Patienten mit angeborenen Herzfehlern gehäuft auf. Aufgrund der Heterogenität der einzelnen Herzfehler (selbst innerhalb einzelner Entitäten!), der unterschiedlichen Operationstechniken und der spezifischen hämodynamischen Residuen des individuellen Patienten ist die Abschätzung des Risikos im Einzelfall schwierig und bedarf der Integration aller Befunde.
Zur Behandlung von Arrhythmien kommt das gesamte Spektrum der modernen Rhythmologie zum Einsatz. Insbesondere interventionelle Ablationsbehandlung kommen bei rezidivierenden Herzrhythmusstörungen immer häufiger zum Einsatz. Die Fortschritte der interventionellen Techniken, insbesondere durch verbesserte Bildgebung und damit besserer Navigation der Ablationskatheter bei komplexer Herzanatomie haben die Erfolgsraten stetig verbessert. Aufgrund des oft komplexen arrhythmischen Substrates mit multiplen Myokardnarben sind die Erfolgsraten aber weiterhin deutlich tiefer als bei Patienten mit vergleichbaren Rhythmusstörungen und strukturell normalem Herzen.
Endokarditis
Durch die zunehmende Verwendung prothetischer Klappensubstitute und transvenöser Schrittmacher- und Defibrillator-Elektroden stieg in den vergangenen Jahren die Zahl der Herzfehler-Patienten, die eine infektiöse Endokarditis erlitten stetig an. Insbesondere die häufige Implantation prothetischer Pulmonalklappen trägt zu diesem Anstieg bei. Das Risiko einer Pulmonalklappen-Prothesen-Endokarditis beträgt – je nach Klappenprothese – bis zu 3% pro Patientenjahr und manifestiert sich zudem häufig atypisch. Auch bei der Endokarditis gilt, dass eine gute Patientenedukation und eine rasche Abklärung bei Auftreten von Symptomen die Morbidität und Mortalität dieser schwerwiegenden Komplikation vermindert.
Aneurysmen / Pseudoaneurysmen
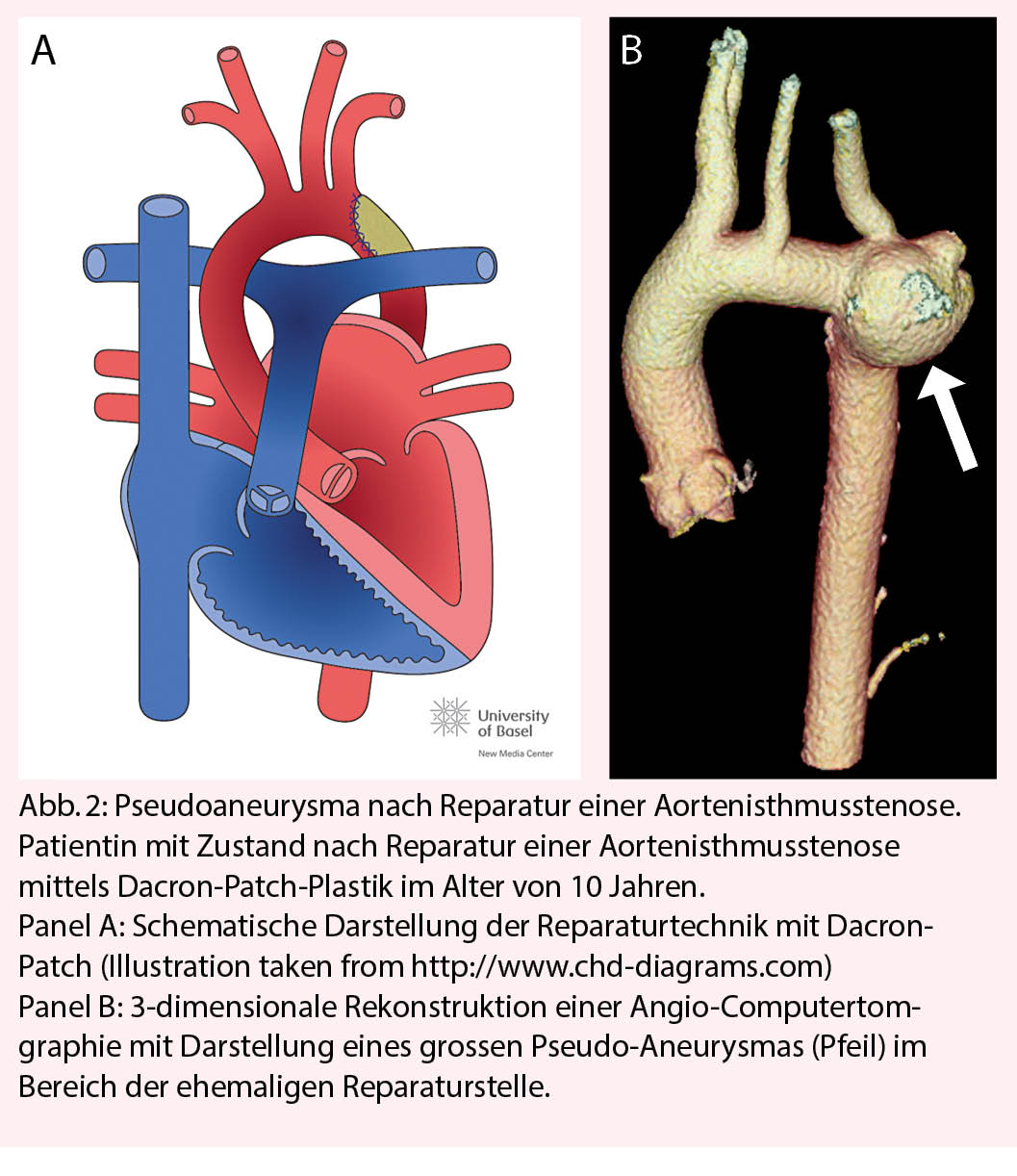
Patienten mit vorgängigen Operationen der Aorta haben ein erhöhtes Risiko für die Ausbildung von Aneurysmen und Pseudoaneurysmen. Dies gilt insbesondere für Patienten mit Bindegewebeerkrankungen (Marfan-Syndrom, Loeys-Dietz-Syndrom, etc.), aber auch Patienten mit reparierter Aortenisthmusstenose (Abb. 2). Bei diesen Patienten sind regelmässige Schnittbilduntersuchungen zur frühzeitigen Erkennung dieser Komplikation wichtig. Die Frequenz der Schnittbilduntersuchung richtet sich nach dem individuellen Risiko, insbesondere der verwendeten Operationstechnik der Aortenisthmusstenose und sollte je nach Situation mindestens alle 5-10 Jahre erfolgen.
Schwangerschaft
Je nach Art des Herzfehlers und residueller Läsionen kann das Risiko für kardiovaskuläre Komplikationen während einer Schwangerschaft erhöht sein. Wenn immer möglich sollte vor einer Schwangerschaft eine umfassende Abklärung und Beratung erfolgen. Das Management während Schwangerschaft und peripartal muss individuell festgelegt werden. Eine besondere Herausforderung bezüglich des Schwangerschaftsmanagements stellen Frauen mit mechanischen Herzklappen dar. Diese Frauen und andere Frauen mit erhöhtem und hohem Schwangerschaftsrisiko profitieren von einer Betreuung durch spezialisierte, multidisziplinäre Teams.
Neudiagnose eines Herzfehlers im Erwachsenenalter
Zuweilen werden Herzfehler erst im Erwachsenenalter diagnostiziert. Am häufigsten sind dies Vorhofseptumdefekte und bicuspide Aortenklappen. Seltener werden auch Aortenisthmusstenosen oder eine kongenital korrigierte Transposition der grossen Arterien erst im Erwachsenenalter diagnostiziert.
Vorhofseptumdefekte
Ein Vorhofseptumdefekt wird gelegentlich erst im Erwachsenenalter diagnostiziert. Grund für die Abklärung sind oft abnormale klinische und paraklinische Untersuchungen (Herzgeräusch, abnormales EKG) oder das Auftreten von Komplikationen (Vorhofflattern, Vorhofflimmern, paradoxe Embolien, pulmonale Hypertonie, progrediente Tricuspdialinsuffizienz).
Bei der kardiologischen Abklärung fällt häufig primär die Vergrösserung der rechtsseitigen Herzhöhlen bei der Echokardiographie auf. Der eigentliche Defekt ist bei Erwachsenen manchmal schwierig zu erkennen, insbesondere bei Sinus venosus-Defekten. Bei Verdacht auf eine intra-kardiale Shuntverbindung ist die Bubble-Kontrast-Echokardiographie ein einfacher, günstiger und sensitiver Test und sollte grosszügig eingesetzt werden. Zur genaueren Charakterisierung des Defektes ist oft eine transösophageale Echokardiographie nötig, insbesondere zur Beurteilung der Möglichkeit eines interventionellen Device-Verschlusses. Dabei ist es entscheidend, dass die Gewebesäume (rims) über die ganze Zirkumferenz des Defektes sorgfältig dargestellt werden. Bei Zweifeln bezüglich hämodynamischer Relevanz von Defekten kann eine kardiale Magnetresonanz-Untersuchung mit Messung der rechtsventrikulären Volumina und Bestimmung der pulmonalen Flusswerte hilfreich sein. Die Schnittbilduntersuchung erlaubt auch die Darstellung und Quantifizierung allfälliger falsch mündender Lungenvenen.
Aortenisthmusstenose
Die Diagnosestellung einer Aortenisthmusstenose erst im Erwachsenenalter ist selten. 50-80% der betroffenen Patienten haben zusätzlich eine bicuspide Aortenklappe. Die Diagnose einer Aortenisthmusstenose als sekundäre Ursache einer systemischen arteriellen Hypertonie darf aber nicht verpasst werden! Echokardiographisch findet sich oft eine Flussbeschleunigung über den Aortenisthmus und ein langsamer, kaum pulsatiler Fluss in der Aorta abdominalis mit typischem diastolischem Vorwärtsfluss. Insbesondere bei schwerer / subatretischer Aortenisthmusstenose fehlt aber häufig das typische Doppler-Signal von suprasternal! Die Diagnose lässt sich klinisch ganz einfach stellen mit Messung des Blutdrucks an Armen und Beinen. Dabei findet sich die typische Arm-Bein-Blutdruckdifferenz. Die Diagnose wird mit Magnet-Resonanz-Angiographie oder Computertomographie bestätigt. Die Schnittbilduntersuchungen sind auch wichtig zur Therapieplanung.
Kongenital korrigierte Transposition der grossen Arterien
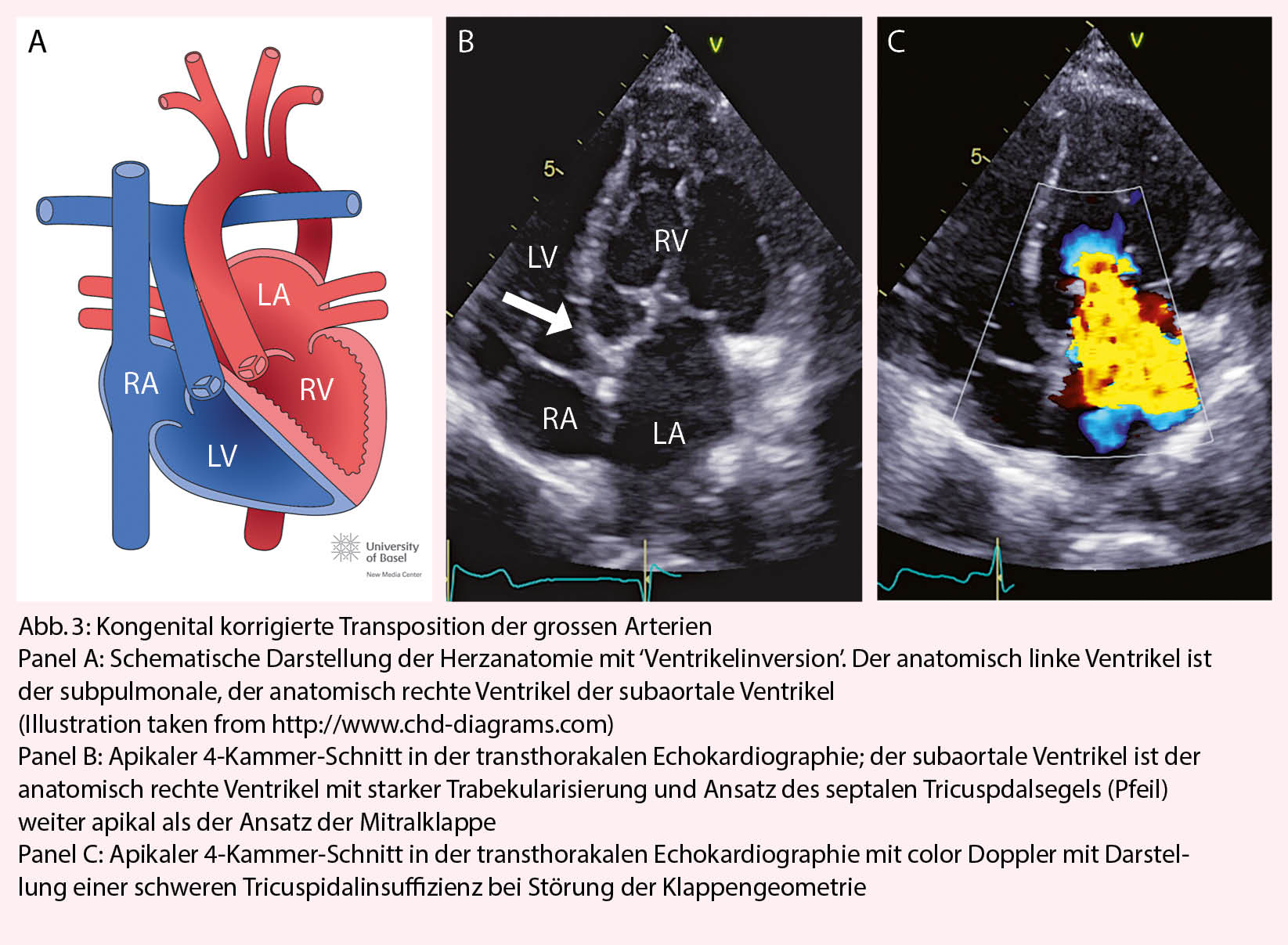
Die Ursache der kongenital korrigierten Transposition der grossen Arterien ist eine fehlerhafte ‘Faltung’ des Herzschlauches während der Embryonalentwicklung. Durch den L-loop kommt es physiologisch zur ‘Ventrikelinversion’. Das heisst, der rechte Vorhof konnektiert zum morphologisch linken Ventrikel, der linke Vorhof zum morphologisch rechten Ventrikel. Die Pulmonalarterie entspringt dem subpulmonalen linken, die Aorta dem subaortalen rechten Ventrikel (Abb. 3). Wenn zusätzliche Defekte vorliegen (am häufigsten sind Ventrikelseptumdefekte und die Pulmonalstenose) erfolgt die Diagnose meist im Kindesalter. Bei Fehlen zusätzlicher Defekte wird die Diagnose gelegentlich erst im Erwachsenenalter gestellt. Dann meist bei Auftreten von Komplikationen (progrediente Tricuspidalinsuffizienz, Herzinsuffizienz oder atrioventrikulärer Block). Die Betreuung solcher Patienten sollte in Zusammenarbeit mit einem spezialisierten Zentrum erfolgen.
Prävention
Wie oben erwähnt kommt der Patientenedukation grosse Bedeutung zu. Über den Dachverband www.herznetz.ch wurden über die letzten Jahre verschiedene Edukationstools entwickelt, die zur Unterstützung eingesetzt werden können. Im Alltag als sehr hilfreich und effektiv hat sich ein kleines Faltblatt erwiesen, das die wichtigsten Komplikationen und die einschlägigen Vorsichtsmassnahmen zusammenfasst. Wir verwenden dies als Unterstützung der mündlichen Patienten-Edukation (Abb. 4). Dieses Faltblatt steht in deutscher, englischer und italienischer Sprache zur Verfügung (www.herznetz.ch/service/mini-kg).

Auch die neuen schweizerischen Endokarditisrichtlinien, die 2020 erarbeitet und publiziert wurden, unterstreichen die Wichtigkeit der Patientenedukation zur frühzeitigen Erkennung einer Endokarditis. Gemeinsam mit der Schweizerischen Herzstiftung wurde auch für die Endokarditis ein Edukations-Flyer entwickelt (www.endocarditis.ch).
Copyright bei Aerzteverlag medinfo AG
Abteilung für Angeborene Herzfehler
Kardiologie
Universitäres Herzzentrum Zürich, Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
Der Autor hat keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Saw J, Mancini GBJ, Humphries KH. Contemporary Review on Spontaneous
Coronary Artery Dissection. J Am Coll Cardiol. 2016;68(3):297-312.
2. Saw J, Mancini GB, Humphries K, Fung A, Boone R, Starovoytov A, et al. Angiographic appearance of spontaneous coronary artery dissection with intramural hematoma proven on intracoronary imaging. Catheter Cardiovasc Interv. 2016;87(2):E54-61.
3. Hayes SN, Kim ESH, Saw J, Adlam D, Arslanian-Engoren C, Economy KE, et al. Spontaneous Coronary Artery Dissection: Current State of the Science: A Scientific Statement From the American Heart Association. Circulation. 2018;137(19):e523-e57.
4. Hill SF, Sheppard MN. Non-atherosclerotic coronary artery disease associated with sudden cardiac death. Heart. 2010;96(14):1119-25.
5. Rogowski S, Maeder MT, Weilenmann D, Haager PK, Ammann P, Rohner F, et al. Spontaneous Coronary Artery Dissection: Angiographic Follow-Up and Long-Term Clinical Outcome in a Predominantly Medically Treated Population. Catheter Cardiovasc Interv. 2017;89(1):59-68.
6. Saw J, Humphries K, Aymong E, Sedlak T, Prakash R, Starovoytov A, et al. Spontaneous Coronary Artery Dissection: Clinical Outcomes and Risk of Recurrence.
J Am Coll Cardiol. 2017;70(9):1148-58.
7. Saw J, Aymong E, Sedlak T, Buller CE, Starovoytov A, Ricci D, et al. Spontaneous coronary artery dissection: association with predisposing arteriopathies and precipitating stressors and cardiovascular outcomes. Circ Cardiovasc Interv. 2014;7(5):645-55.
8. Faden MS, Bottega N, Benjamin A, Brown RN. A nationwide evaluation of spontaneous coronary artery dissection in pregnancy and the puerperium. Heart. 2016;102(24):1974-9.
9. Vaccarino V, Parsons L, Every NR, Barron HV, Krumholz HM. Sex-based differences in early mortality after myocardial infarction. National Registry of Myocardial Infarction 2 Participants. N Engl J Med. 1999;341(4):217-25.
10. Gilhofer TS, Saw J. Spontaneous coronary artery dissection: update 2019. Curr Opin Cardiol. 2019;34(6):594-602.
11. Lettieri C, Zavalloni D, Rossini R, Morici N, Ettori F, Leonzi O, et al. Management and Long-Term Prognosis of Spontaneous Coronary Artery Dissection. Am J Cardiol. 2015;116(1):66-73.
12. Gilhofer TS, Saw J. Spontaneous coronary artery dissection: a review of complications and management strategies. Expert Rev Cardiovasc Ther. 2019;17(4):
275-91.
13. Henkin S, Negrotto SM, Tweet MS, Kirmani S, Deyle DR, Gulati R, et al. Spontaneous coronary artery dissection and its association with heritable connective
tissue disorders. Heart. 2016;102(11):876-81.
14. Grover P, Fitzgibbons TP. Spontaneous coronary artery dissection in a patient with autosomal dominant polycystic kidney disease: a case report. J Med Case Rep. 2016;10:62.
15. Prakash R, Starovoytov A, Heydari M, Mancini GB, Saw J. Catheter-Induced
Iatrogenic Coronary Artery Dissection in Patients With Spontaneous Coronary Artery Dissection. JACC Cardiovasc Interv. 2016;9(17):1851-3.
16. Tweet MS, Hayes SN, Gulati R, Rose CH, Best PJ. Pregnancy after spontaneous coronary artery dissection: a case series. Ann Intern Med. 2015;162(8):598-600.
17. Olin JW, Gornik HL, Bacharach JM, Biller J, Fine LJ, Gray BH, et al. Fibromuscular dysplasia: state of the science and critical unanswered questions: a scientific statement from the American Heart Association. Circulation. 2014;129(9):1048-78.
18. Saw J. Coronary angiogram classification of spontaneous coronary artery dissection. Catheter Cardiovasc Interv. 2014;84(7):1115-22.

info@herz+gefäss
- Vol. 11
- Ausgabe 6
- Dezember 2021