

Die Diagnose eines Systemischen Lupus Erythematodes (SLE) stellt bei klassischer Präsentation keine allzu hohen Anforderungen. Allerdings gehört die Erkrankung mit all ihren Facetten und Eigenarten zu den komplexesten und heterogensten, die wir kennen. Die korrekte und vor allem auch zeitnahe Diagnose stellt damit bei vielen Patient/-innen noch immer eine diagnostische Herausforderung dar. Entsprechend gibt es auf die Frage, wann man an einen Lupus denken soll, keine einfache oder pauschale Antwort. Bestimmte anamnestische und klinische Hinweise erlauben jedoch, die Wahrscheinlichkeit für das Vorliegen oder die Abwesenheit eines SLE zu erhöhen. Bei klinischem Verdacht auf das Vorliegen eines SLE kann die Bestimmung von Antinukleären Antikörpern (ANA) für die weitere Diagnostik wegweisend sein.
Diagnosing systemic lupus erythematosus (SLE) is not overly difficult when the classic presentation is present. However, with all its facets and peculiarities, the disease is among the most complex and heterogeneous conditions we know. Consequently, making a correct and, above all, timely diagnosis remains a diagnostic challenge for many patients. Accordingly, there is no simple or blanket answer to the question of when to consider lupus. However, certain anamnestic and clinical clues allow us to increase the probability of the presence or absence of SLE. If SLE is clinically suspected, the detection of antinuclear antibodies (ANA) can be decisive for further diagnosis.
Keywords: Systemischer Lupus Erythematodes, ANA, Klassifikationskriterien
Einführung
Trotz jahrzehntelanger Erforschung des Systemischen Lupus Erythematodes (SLE) ist dieser Prototyp einer Autoimmunerkrankung bis heute nur teilweise verstanden. Erfreulicherweise gab es in den letzten Jahren dennoch erhebliche Fortschritte und – für betroffene Patient/-innen am wichtigsten – wesentliche Entwicklungen bei den therapeutischen Möglichkeiten. Diese lenken die Aufmerksamkeit zunehmend auch auf die Langzeitauswirkungen der Erkrankung und ihre Therapie. Während dieser Artikel sich auf die initiale Diagnosestellung fokussiert, sind die Entwicklungen in Krankheitsverständnis und Therapiemöglichkeiten Gegenstand regelmässiger und umfassender Übersichtsarbeiten (1, 2).
Was ist ein Systemischer Lupus Erythematodes?
Der SLE ist eine Autoimmunerkrankung, also eine Erkrankung, bei der sich das Immunsystem gegen den eigenen Körper richtet. Die Erkrankung betrifft v.a. jüngere Frauen (ca. 20. bis 40. Lebensjahr), mit zunehmendem Alter der Patienten jedoch auch Männer. Die Ursachen und krankheitsbildenden Vorgänge sind nur teilweise verstanden. Das aktuelle Konzept geht davon aus, dass der SLE bei den meisten Patient/-innen auf dem Boden einer genetischen Prädisposition entsteht, jedoch zusätzliche (externe) Faktoren für die Krankheitsentwicklung nötig sind. Bezüglich Prädisposition steht die sogenannte Abfall-Entsorgungs-Hypothese im Vordergrund (3). Nach dieser Hypothese ist bei Patienten mit SLE die Beseitigung von physiologisch sterbenden Zellen (Apoptose) gestört, so dass es zu einer Fehlleitung des Immunsystems mit der konsekutiven Bildung von Autoantikörpern, die gegen Bestandteile der apoptotischen Zellen gerichtet sind, und der Aktivierung der Komplementkaskade kommt. Auch wenn diese Hypothese durch zahlreiche Studien gestützt wird, genügt sie nicht, um das Spektrum der klinischen und experimentellen Beobachtungen umfassend zu erklären. Neben genetisch determinierten Mechanismen scheinen auch extrinsische Faktoren Einfluss auf die Entstehung und den Verlauf der Erkrankung zu nehmen. Besonders hervorzuheben ist die Beobachtung, dass eine vorangegangene Infektion mit dem Epstein-Barr-Virus (EBV) ein notwendiger Schritt bei der Entstehung des SLE zu sein scheint. Bei fast allen (ca. 99 %) erwachsenen Patienten mit SLE lässt sich eine solche zurückliegende EBV-Infektion serologisch nachweisen und insofern häufiger als bei an das Alter angepassten Kontrollen (4). Es gibt verschiedene Möglichkeiten, wie eine EBV-Infektion die Entstehung von Autoimmunität ermöglicht, aber auch diese Mechanismen genügen nicht, um die Krankheit vollständig zu erklären (5). Viel seltener lassen sich bei SLE-Patient/-innen rein monogenetische Ursachen nachweisen, und sind hier am besten beschrieben für Patient/-innen mit hereditärer Defizienz des klassischen Komplementweges (Defizienz auf Niveau Komplement C1q, C1r/s, C4, C2) (6). Da die Gründe, die zu einem SLE führen, nicht nur komplex, sondern die Folge ganz unterschiedlicher genetischer Risikofaktoren, gepaart mit mehr oder weniger vielen Aussenfaktoren, sind, handelt es sich sehr wahrscheinlich nicht um eine umschriebene Krankheit, sondern um ein Syndrom. Bei SLE-Patient/-innen handelt es sich also um Patient/-innen mit individuellen und heterogenen Ursachen, die sich in ihrer Auswirkung aber klinisch mit gewissen Ähnlichkeiten präsentieren. Die Eigenschaft des SLE, eher ein Syndrom als eine Krankheit zu sein, erklärt dabei einen Teil der Schwierigkeiten, Patient/-innen korrekt und zeitnah zu diagnostizieren.
Klinische Präsentation
In der Regel präsentiert sich ein SLE als Triade aus konstitutionellen bzw. unspezifischen Symptomen (Gewichtsverlust, vor allem nachmittägliche Müdigkeit, Fieber, Lymphknotenschwellungen), entzündlichen Organmanifestationen und typischen Laborbefunden, konkret dem Auftreten bestimmter Autoantikörper (Anti-Nukleäre Antikörper (ANA), Autoantikörper gegen native DNA (Anti-dsDNA)) und Zeichen der Aktivierung der Komplementkaskade (tiefe Werte für C4, C3 und/oder Bestimmung der Gesamtkomplementaktivität, früher als «CH50» bezeichnet). Insgesamt hat die Erkrankung die Tendenz, sich über längere Zeiträume zu entwickeln. Das bedeutet einerseits, dass sich die Hauptmanifestation der Erkrankung im Verlauf verändern kann. Andererseits können initial unspezifische Symptome und eine unklare Laborkonstellation vorliegen, so dass eine definitive Diagnose oft erst Monate oder Jahre nach Symptombeginn gestellt wird. Am anderen Ende des Spektrums gibt es Patient/-innen, die sich mit einem relativ akuten Vollbild eines schweren Schubes eines SLE als Erstmanifestation ihrer Erkrankung präsentieren. Aufgrund des systemischen Charakters der Erkrankung kann sie theoretisch alle Organe und Organsysteme involvieren. Manifestationen an Haut, Gelenken und Blutbildveränderungen treten dabei häufiger auf als beispielsweise am zentralen Nervensystem (ZNS), an der Lunge oder am Herz (7). Dagegen ist die Beteiligung anderer Organe zwar seltener, aber als schwerwiegender einzustufen (v.a. Niere und ZNS). Eine ZNS-Beteiligung ist in diesem Kontext trotz MRI und/oder Lumbalpunktion oft schwer zu beweisen und kann sich sehr heterogen präsentieren. Dadurch wird sie bisher möglicherweise in ihrer Häufigkeit unterschätzt. Damit wird klar, dass das klinische Bild des SLE nicht nur insgesamt, sondern auch innerhalb der betroffenen Organe heterogen ist. Darüber hinaus zeigt jede Krankengeschichte einen sehr individuellen Verlauf. Als Konsequenz daraus fällt der SLE in die Differentialdiagnose vieler anderer Krankheiten.
Wann sollte man an SLE denken?
Wie ausgeführt sind die vielfältigen Symptome des SLE Teil der Differentialdiagnose bei einem breiten Spektrum initial unklarer Erkrankungen. Dabei gilt zu beachten, dass die Erkrankung trotz steigender Inzidenz insgesamt noch immer eher im Bereich der seltenen Erkrankungen anzusiedeln ist und damit per se eine niedrige Ausgangswahrscheinlichkeit bei den meisten differentialdiagnostischen Überlegungen hat. Die Wahrscheinlichkeit der Diagnose eines SLE steigt vor allem, wenn die betroffenen Patienten weiblich und im Altersbereich von 20–40 Jahren sind. Auch haben Patient/-innen mit genetisch asiatischem, afrikanischem oder hispanischem Ursprung ein erhöhtes Risiko für einen SLE (8). Bezüglich der Beschwerden lassen v.a. Allgemeinbeschwerden (z. B. ausgeprägte Müdigkeit, Fieber, Gewichtsverlust) gepaart mit Symptomen, welche die Haut (inkl. Haarausfall, Photosensitivität, Raynaud und andere) und/oder die Gelenke (v. a. Finger, Hände, Schultern und Knie) betreffen, aber auch primär myalgieforme Beschwerden und Sehnenentzündungen, eher einmal an einen SLE denken. Bezüglich der durchaus häufigen Hautbeteiligung bleibt jedoch anzumerken, dass nur etwa die Hälfte aller Patient/-innen jemals ein klassisches Schmetterlingserythem entwickelt, das damit deutlich weniger krankheitstypisch ist, als häufig der Eindruck vermittelt wird (Tab. 1).
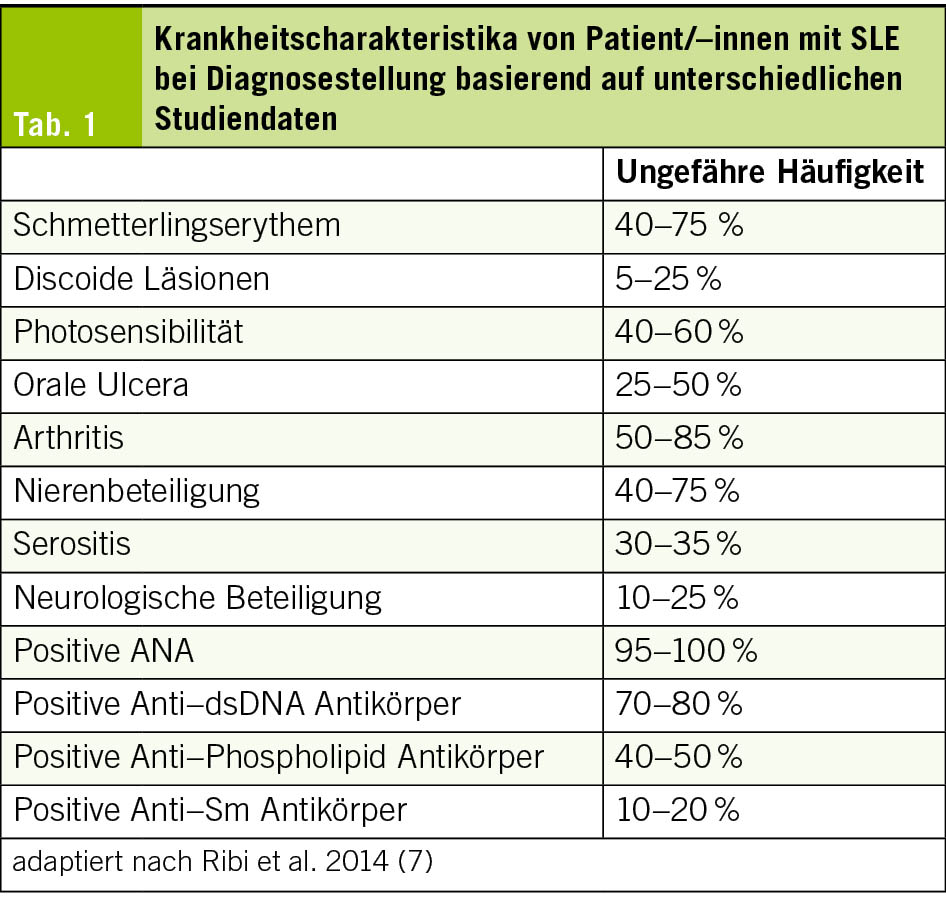
Auch eine positive Familienanamnese für entzündlich-rheumatische Erkrankungen kann ein wichtiger Hinweis sein. Verschiebt man den Blickwinkel zurück auf die Sicht der Patient/-innen, dann kann ein bereits 1980 vorgestellter Fragenkatalog von Liang et al. (9) bei der Frage weiterhelfen, ob man die Diagnostik in Richtung eines SLE weiterverfolgen soll. Bejaht eine Patientin oder ein Patient mindestens 3 der in Tab. 2 genannten Fragen, dann ist die Diagnose eines SLE ernsthaft zu erwägen und weitere Diagnostik angebracht. Im Labor können insbesondere unerwartete Blutbildveränderungen im Sinne von einer oder mehreren Zytopenien (Lympho-/Leukopenie, Anämie, Thrombozytopenie) den Verdacht aufkommen lassen. Ein deutlich erhöhtes CRP ist bei Patient/-innen mit SLE dagegen eher ungewöhnlich. Als charakteristisch beim Vorliegen eines floriden SLE wird eine erhöhte Blutsenkung (BSG) angesehen, die sich in ihrem Ausmass nicht bereits aus einem erhöhten CRP oder einer schweren Anämie ableiten lässt. Jedoch nimmt die BSG als Screeningtool nur einen begrenzten Stellenwert ein. Eine klinisch-anamnestische Sonderstellung haben Schwangerschaftskomplikationen, insbesondere Aborte, sowie thromboembolische Ereignisse. Letztere sind besonders relevant bei ungewöhnlicher Charakteristik (z. B. ungewöhnlicher Ort und/oder erhöhter Häufigkeit oder kombinierte venöse und arterielle Manifestationen) oder bei fehlendem Risikoprofil, etwa Herzinfarkt oder Schlaganfall bei jüngeren Patient/-innen ohne typisches kardiovaskuläres Risikoprofil. Solche Präsentationen lassen an das Vorliegen eines häufig mit einem SLE vergesellschafteten Anti-Phospholipidantikörpersyndromes denken.

Diagnosestellung
Vorweg ist zu betonen, dass die häufig verwendeten Kriterien der internationalen Fachgesellschaften nicht primär dafür gedacht waren und auch weiterhin nicht sind, die Diagnose eines SLE zu stellen. Sie haben das Ziel, einen SLE von anderen entzündlich-rheumatischen Erkrankungen abzugrenzen und auch für Studienzwecke zu «homogenisieren». Trotzdem kann man die beschriebenen Klassifikationskriterien natürlich für die Diagnosestellung heranziehen, muss aber beachten, dass es eine Reihe von Erkrankungen gibt, welche die Kriterien eines SLE erfüllen, tatsächlich aber eine völlig andere Pathogenese haben. Das gilt z. B. für gewisse maligne Erkrankungen, v.a. solche aus dem hämatologischen Formenkreis, aber auch z. B. für bestimmte chronische Infektionskrankheiten. Insofern kann man Klassifikationskriterien eigentlich nur für die Diagnose eines SLE benutzen, wenn Infekte und/oder Malignome ausgeschlossen sind. Seit 2019 sind die gemeinsamen Klassifikationskriterien der European Alliance of Associations for Rheumatology/ American College of Rheumatology (EULAR/ACR) in Gebrauch und haben sich durchgesetzt (Abb. 1) (10).
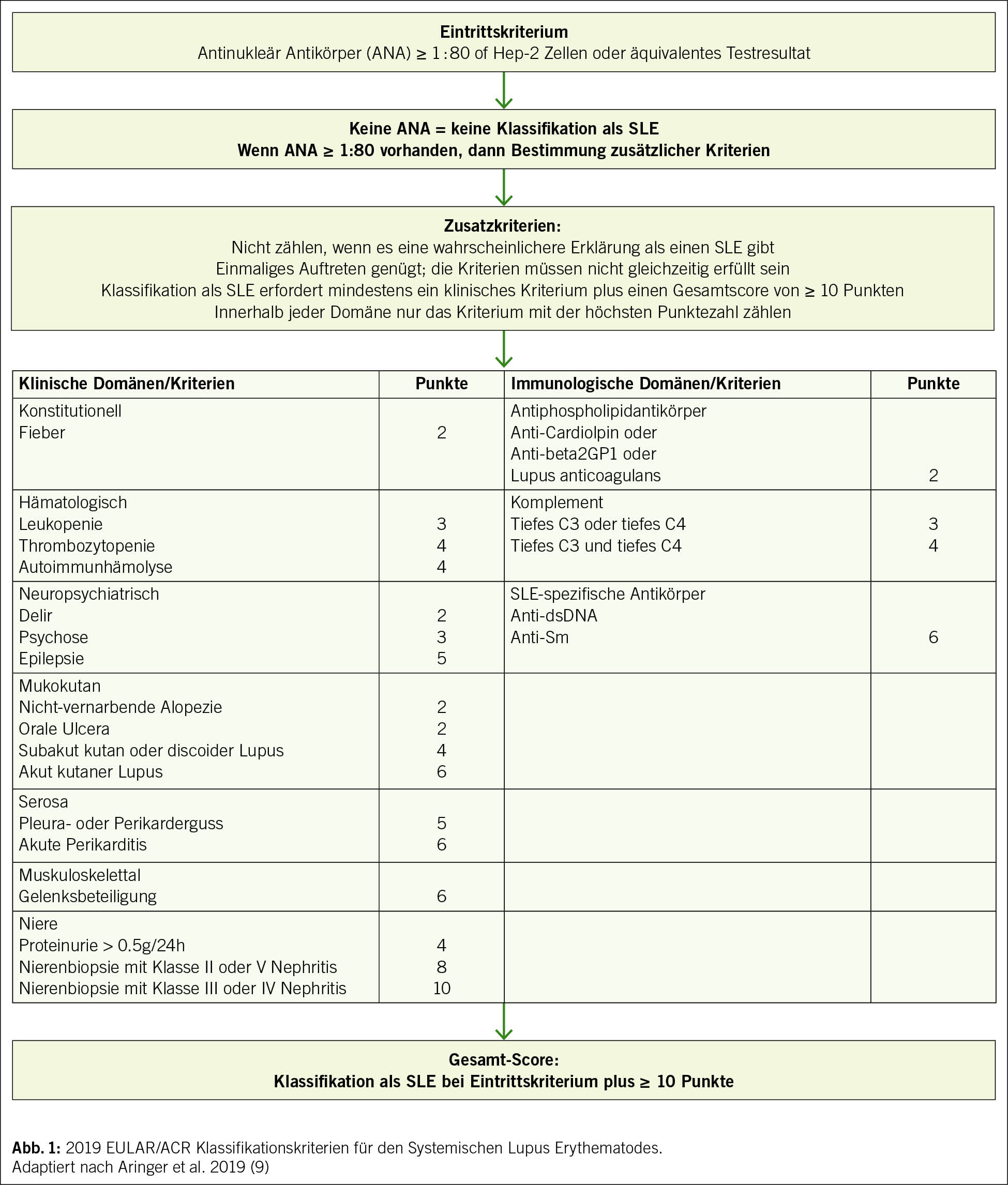
Gegenüber den älteren ACR-Kriterien von 1997 (11), die ihre Gültigkeit prinzipiell behalten haben, sind sie etwas sensitiver und damit in der Lage, eine SLE-Diagnose früher zu stellen. Ausserdem wurde ein ANA-Titer von zumindest 1 : 80 als Eintrittskriterium eingeführt, womit man den SLE neu zu einer Art «ANA-Erkrankung» macht. Da es sich um Klassifikations- und nicht um Diagnosekriterien handelt, kann man bei eindeutiger sonstiger Konstellation aus Klinik und Labor trotz negativer ANA jedoch immer noch die Diagnose eines SLE stellen, auch wenn das in der Praxis sicher eine Ausnahme bleiben wird. Auf der Basis der 2019 EULAR/ACR-Kriterien drängt sich aber die relativ einfache und breit verfügbare Bestimmung von ANA als Screening-Tool zum Ausschluss eines SLE auf. Sind die ANA negativ, wird eine Diagnose eines SLE sehr unwahrscheinlich, andere Differentialdiagnosen treten dagegen weiter in den Vordergrund. Umgekehrt kann man jedoch beim alleinigen Nachweis von ANA, insbesondere wenn nur niedrig titrig positiv, keinesfalls bereits die Diagnose eines SLE stellen, denn ANA finden sich auch bei anderen rheumatischen Erkrankungen und insbesondere auch bei Menschen ganz ohne Autoimmunerkrankung (12). Dem positiven ANA Nachweis muss zwingend eine gute klinische Gesamtbeurteilung folgen oder – viel besser – bereits vorangehen: Passen die Beschwerden insgesamt zu einem SLE? Gibt es andere Diagnosen, die die Beschwerden besser oder mindestens genauso gut erklären können? Wenn die zweite Frage mit Nein beantwortet wird, dann kommen zur Eingrenzung sekundär zusätzliche Laborbestimmungen ins Spiel: Bestimmung von Antikörpern gegen ENA (z. B. Anti-Ro, Anti-Sm), gegen dsDNA (Anti-dsDNA) und gegen Cardiolipin bzw. Beta-2-Glykoprotein 1, aber auch Untersuchungen auf einen Komplementverbrauch (C3 und C4) sowie die Bestimmung der Gesamtkomplementaktivität. Das Vorliegen von Autoantikörpern gegen dsDNA oder gar Anti-Smith (Anti-Sm) und auch Zeichen für einen Komplementverbrauch lassen das Vorliegen eines SLE rasch wahrscheinlicher werden. Für den Fall einer nicht messbar tiefen Gesamtkomplementaktivität rückt dabei nicht nur die mögliche Diagnose eines SLE weiter in den Vordergrund, sondern auch die Möglichkeit für das Vorliegen einer Komplementdefizienz. Ist die Diagnose eines SLE in den Bereich des Wahrscheinlichen gerückt, werden eine Reihe weiterer Untersuchungen erforderlich, die dem Charakter einer Systemerkrankung Rechnung tragen müssen und unter anderem auch das Ziel haben, schwerwiegende Organbeteiligungen nicht zu verpassen. Dies beinhaltet neben einer guten Anamnese und einem guten internistischen Status eine Reihe von Laboruntersuchungen inkl. einer Urinuntersuchung mit der Frage nach einer möglichen Nierenbeteiligung (Proteinurie, glomeruläre Erythrozyturie). Ausserdem könnten je nach Klinik zusätzliche Bilddiagnostik (z. B. Ultraschall der Pleura, Echokardiographie, MRI des Gehirns) und/oder histologische Untersuchungen (z. B. Haut-/Nierenbiopsie) nötig werden. Für diesen Zweck erscheint uns die zumindest initiale Beurteilung an einem Zentrum mit Erfahrung in der Betreuung von Patient/-innen mit SLE sinnvoll.
Zusammenfassend ist die Diagnosestellung eines SLE komplex und erfordert klinische Erfahrung. Frühzeitige anamnestische Hinweise, typische klinische Befunde und korrespondierende diagnostische Auffälligkeiten können jedoch richtungsweisend sein, um die Erkrankung auszuschliessen oder eine weiterführende Diagnostik zur Diagnosesicherung einzuleiten.
Copyright
Aerzteverlag medinfo AG
Klinik für Innere Medizin
Petersgraben 4
Universitätsspital Basel
4031 Basel
Klinik für Innere Medizin
Petersgraben 4
Universitätsspital Basel
4031 Basel
Die Autorenschaft hat keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Siegel CH, Sammaritano LR. Systemic Lupus Erythematosus: A Review. JAMA. 2024 May 7;331(17):1480-1491. doi: 10.1001/jama.2024.2315. Erratum in: JAMA. 2024 Jun 25;331(24):2136. doi: 10.1001/jama.2024.10468.
2. Hoi A, Igel T, Mok CC, Arnaud L. Systemic lupus erythematosus. Lancet. 2024 May 25;403(10441):2326-2338. doi: 10.1016/S0140-6736(24)00398-2. Epub 2024 Apr 17. Erratum in: Lancet. 2024 May 25;403(10441):2292. doi: 10.1016/S0140-6736(24)01044-4.
3. Botto M, Walport MJ. C1q, autoimmunity and apoptosis. Immunobiology. 2002 Sep;205(4-5):395-406. doi: 10.1078/0171-2985-00141.
4. Hanlon P, Avenell A, Aucott L, Vickers MA. Systematic review and meta-analysis of the sero-epidemiological association between Epstein-Barr virus and systemic lupus erythematosus. Arthritis Res Ther. 2014 Jan 6;16(1):R3. doi: 10.1186/ar4429.
5. Jog NR, James JA. Epstein Barr Virus and Autoimmune Responses in Systemic Lupus Erythematosus. Front Immunol. 2021 Feb 3;11:623944. doi: 10.3389/fimmu.2020.623944.
6. Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol. 2000;76:227-324. doi: 10.1016/s0065-2776(01)76021-x.
7. Ribi C, Trendelenburg M, Gayet-Ageron A, Cohen C, Dayer E, Eisenberger U, Hauser T, Hunziker T, Leimgruber A, Lindner G, Koenig K, Otto P, Spertini F, Stoll T, Von Kempis J, Chizzolini C; Swiss Systemic Lupus Erythematosus Cohort Study Group. The Swiss Systemic lupus erythematosus Cohort Study (SSCS) – cross-sectional analysis of clinical characteristics and treatments across different medical disciplines in Switzerland. Swiss Med Wkly. 2014 Aug 7;144:w13990. doi: 10.4414/smw.2014.13990.
8. Izmirly PM, Parton H, Wang L, McCune WJ, Lim SS, Drenkard C, Ferucci ED, Dall‘Era M, Gordon C, Helmick CG, Somers EC. Prevalence of Systemic Lupus Erythematosus in the United States: Estimates From a Meta-Analysis of the Centers for Disease Control and Prevention National Lupus Registries. Arthritis Rheumatol. 2021 Jun;73(6):991-996. doi: 10.1002/art.41632.
9. Liang MH, Meenan RF, Cathcart ES, Schur PH. A screening strategy for population studies in systemic lupus erythematosus. Series design. Arthritis Rheum. 1980 Feb;23(2):153-7. doi: 10.1002/art.1780230204.
10. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, Smolen JS, Wofsy D, Boumpas DT, Kamen DL, Jayne D, Cervera R, Costedoat-Chalumeau N, Diamond B, Gladman DD, Hahn B, Hiepe F, Jacobsen S, Khanna D, Lerstrøm K, Massarotti E, McCune J, Ruiz-Irastorza G, Sanchez-Guerrero J, Schneider M, Urowitz M, Bertsias G, Hoyer BF, Leuchten N, Tani C, Tedeschi SK, Touma Z, Schmajuk G, Anic B, Assan F, Chan TM, Clarke AE, Crow MK, Czirják L, Doria A, Graninger W, Halda-Kiss B, Hasni S, Izmirly PM, Jung M, Kumánovics G, Mariette X, Padjen I, Pego-Reigosa JM, Romero-Diaz J, Rúa-Figueroa Fernández Í, Seror R, Stummvoll GH, Tanaka Y, Tektonidou MG, Vasconcelos C, Vital EM, Wallace DJ, Yavuz S, Meroni PL, Fritzler MJ, Naden R, Dörner T, Johnson SR. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019 Sep;71(9):1400-1412. doi: 10.1002/art.40930.
11. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997 Sep;40(9):1725. doi: 10.1002/art.1780400928.
12. Tan EM, Feltkamp TE, Smolen JS, Butcher B, Dawkins R, Fritzler MJ, Gordon T, Hardin JA, Kalden JR, Lahita RG, Maini RN, McDougal JS, Rothfield NF, Smeenk RJ, Takasaki Y, Wiik A, Wilson MR, Koziol JA. Range of antinuclear antibodies in „healthy“ individuals. Arthritis Rheum. 1997 Sep;40(9):1601-11. doi: 10.1002/art.1780400909.


der informierte @rzt
- Vol. 16
- Ausgabe 4
- April 2026