Over the past decade, the availability of DNA methylation analyses has not only led to major advances in diagnostic tumor classification, but has also brought about significant improvements in oncological management through optimized stratification of tumor patients. The interpretation of results is largely based on the comparison of a tissue sample with thousands of reference cases. This comparison is typically performed using supervised or unsupervised machine learning, whereby the composition and quality of the available reference data sets determine the diagnostic granularity. In addition to the diagnosis of tumors of the nervous system, this approach mitigates further challenges, e.g., in organ mapping in CUP syndrome, low-invasive tumor detection in cerebrospinal fluid or other body fluids (so-called «liquid biopsy»), as well as grading of tumors and their prognostic stratification. Enabling broad access to clinicians, native tissue and liquid biopsies can easily be shipped at room temperature in cytological and liquid biopsy preservation media.
Die Verfügbarkeit von DNA-Methylomanalysen hat in den zurückliegenden 10 Jahren nicht nur zu entscheidenden Fortschritten in der diagnostischen Tumorklassifikation geführt, sondern durch optimierte Stratifizierung von Tumorpatienten auch signifikante Verbesserungen des onkologischen Managements mit sich gebracht. Die Befundauswertung beruht derzeit überwiegend auf dem Datenabgleich einer Gewebeprobe mit tausenden Referenzfällen. Dieser Abgleich erfolgt typischerweise mittels supervidiertem oder unsupervidiertem maschinellen Lernen, wobei die Zusammensetzung und Qualität der Kurierung der zur Verfügung stehenden Referenzdatensätze die diagnostische Granularität determinieren. Neben der Diagnostik von Tumoren des Nervensystems löst dieser Ansatz weitere Herausforderungen, beispielsweise bei der Organzuordnung beim CUP-Syndrom, der gering-invasiven Tumor-Detektion im Liquor oder anderen Körperflüssigkeiten (sog. «liquid biopsy»), wie auch der Dignitätseinstufung von Tumoren und deren prognostischer Stratifizierung. Die Methode ist für die Klinik breit verfügbar, da natives Gewebe und «liquid biopsies» in zytologischen bzw. für «liquid biopsy» vorgesehenen Medien bei Raumtemperatur mit der Post versandt werden können.
Keywords: DNA-methylation, Liquid Biopsy (CSF/cfDNA), tumor classification
Same-Day Tumor Classification
Even before the Heidelberg brain tumor classifier (1), utilizing DNA methylation-specific microarrays, was released in 2018, the underlying reference brain tumor database draft was made available for nanopore sequencing data, enabling same-day tumor classification (2). Subsequently, this approach was successfully adapted for routine use (3) and also expanded into the non-CNS tumor spectrum (4–6). A proof-of-concept study from Norway demonstrated feasibility even for intraoperative diagnostics during open brain surgery (7, 8), which has since then been recapitulated in larger cohorts by several centers (9). Despite its clinical significance, the approach remains hindered by rapid product cycles of the manufacturer (Oxford Nanopore Technologies) which can compromise the supply chain of sequencing hardware and reagents. Hence, routine use currently remains so far limited to academic institutions with required technological expertise at hand to continuously adapt their diagnostic workflows.
Robust Tumor Typing for Everyone
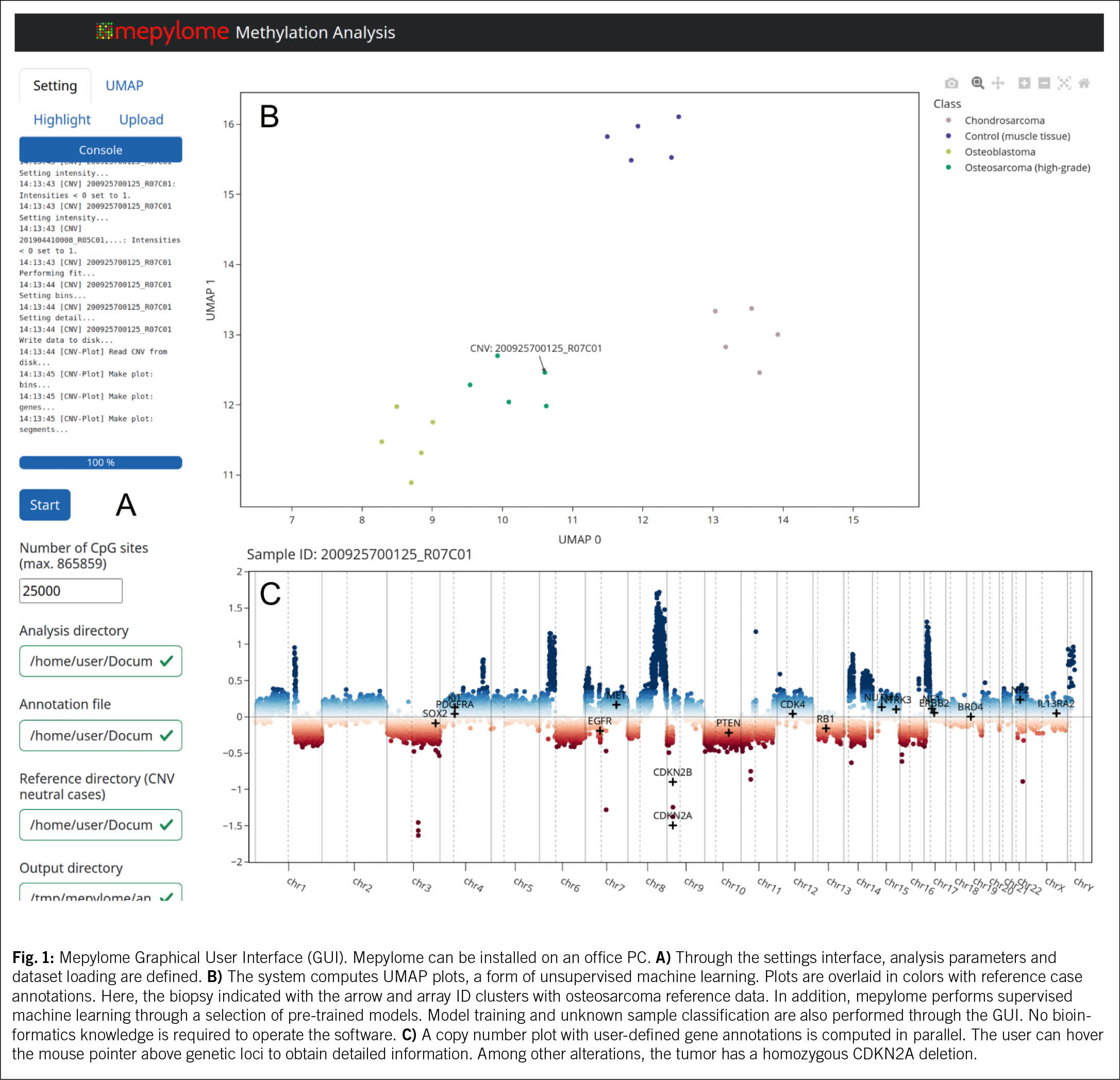
As anticipated during the early years of brain tumor methylation classification, the 2021 edition of the WHO CNS tumor classification (10) has dramatically gained in granularity. Beyond the Heidelberg classifier, a plethora of additional classifiers has emerged, with the shortcoming that these systems often behave like «black boxes». This lack of transparency in decision-making processes impedes clinical implementation of gained information. Recent work from our lab (11) now democratizes patient-centered methylome and copy number profiling (Fig. 1). In addition to static supervised (1, 2, 12) and unsupervised (5) classification algorithms, tumor methylome array data can now be examined on a standard office computer. This very recent development now enables relevant insights into individual tumors, such as information on potentially targetable chromosomal gains and losses, available to clinical oncologists in real-time.
From a practical perspective, native tissue and body fluids can be submitted by regular mail at room temperature using preservation media (tissue in ThinPrep® or SurePath® / liquid biopsies in Streck BCT® tubes), enabling universal clinical access (5). Of note, formalin is unsuitable for nanopore sequencing, as it fragments the DNA. According to previous experience, cost approval is not required.
Why Expand Methylome Analysis into Liquids?
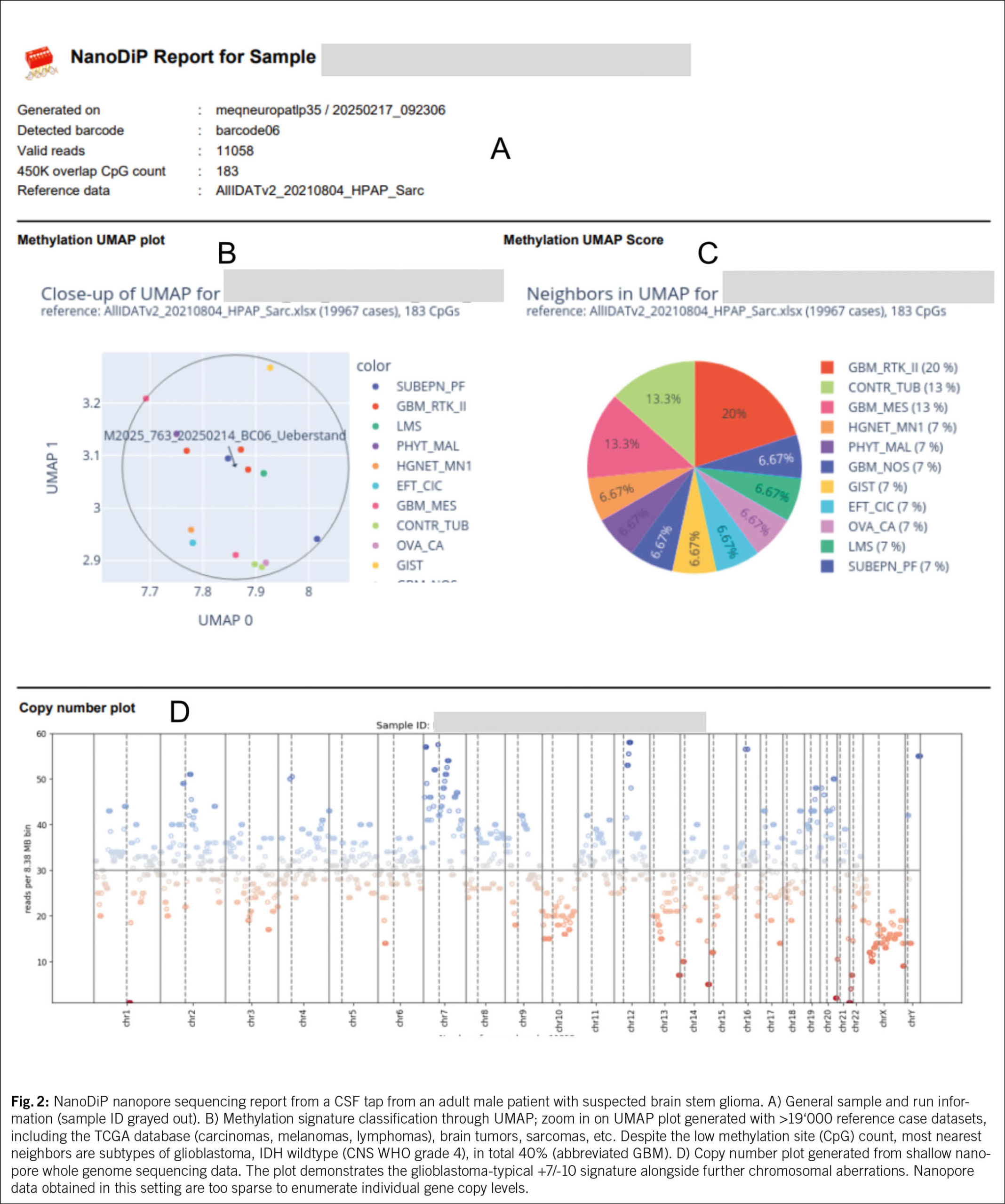
The «liquid biopsy» concept was practically introduced with PSA screening for early prostate cancer detection, at that time under the umbrella term «biomarker screening». The analysis target is a protein and the method stems from an era when nucleic acid characterization was too complex for routine implementation on low analyte quantities. Decades later, the term «liquid biopsy» became mostly restricted to circulating tumor DNA (ctDNA) and circulating tumor cell analyses in blood and other body fluids (13). Despite its vast potential for cancer progression and therapy resistance detection, tumor diagnostic utilization of somatic variants present in fluids remained technically unresolved. In contrast to blood which physiologically contains substantial DNA amounts, cerebrospinal fluid (CSF) from healthy individuals mostly consists of water. In case of a rapidly growing (i.e. malignant) neoplasm that has direct contact to the CSF, quantitatively sufficient ctDNA can be shed into CSF through necrosis and apoptosis. The same happens in inflammatory processes where leukocyte DNA shedding increases. Apart from CSF, this diagnostic approach is also applicable to pleural effusions, where ctDNA levels are usually much higher compared to CSF. Mutation analysis typically targets a single stretch of each genome in the analyte to detect mostly heterozygous oncogenic variants and is blind for non-neoplastic phenomena. By contrast epigenetic DNA methylation marks are found on many of the ~500 bp DNA fragments released from necrotic cells. This vastly increases the pool of analytes from one mutant nucleotide in ~12*106 DNA fragments per decayed cell to ~5 methylation sites per fragment, representing a 60*106-fold increase. Additionally, classification of DNA methylation patterns is mostly based on supervised (1) or unsupervised (5) machine learning and is highly gap-tolerant. As a rule of thumb, epigenetic tumor classification requires solely ~1000 methylation sites for rough and ~5000 for robust diagnostic differentiation (5, 14). It is hence possible to detect, e. g., diffuse large B-cell lymphoma (15) or inoperable high-grade glioma (16) from a CSF tap. Overall costs are in the range of 2nd generation parallel sequencing (so-called NGS panels). For CSF, the current primary choice is 3rd generation direct sequencing (through nanopores) which delivers both epigenomic and copy number variant information (Fig. 2). It is usually possible to have a nanopore sequencing-based «first glance» at the methylation signatures present on CSF cell-free DNA (cfDNA) and, depending on the findings, proceed with select confirmatory mutation-based tests on the leftover DNA, e. g., sequencing of MYD88 in case of a B-cell lymphoma methylome profile, or TERT promoter for suspected IDH-wildtype glioblastoma. In an internal retrospective study at the Institute for Medical Genetics and Pathology in Basel from 90 routinely examined CSF tap liquid biopsies, 5 yielded a comprehensive tumor diagnosis, eliminating the need for tissue biopsy. 35 taps had too low overall DNA content precluding further analysis. Of note, a negative finding due to low CSF DNA concentration suggests that the respective lesion does not release significant amounts of DNA.
Outlook – Future of Methylome Analysis
While early studies (17, 18) suggested DNA methylation analysis from blood for early cancer screening, current use remains limited to start-up companies and academic institutions (19). Nevertheless, already today, methylome profiling of CSF taps (15, 16) represents a viable approach to the diagnosis of malignant CNS tumors located adjacent to the ventricular
system, which minimizes inherent surgical risks in cases of presumably inoperable brain tumors such as CNS lymphoma and certain gliomas.
PD. Dr. med. Jürgen Hench
Dr. rer. nat. Claus Hultschig
Dr. rer. nat. Benjamin Freyter
Dr. rer. nat. Ivana Bratic Hench
Prof. Dr. med. Stephan Frank
Universitätsspital Basel
Institut für Medizinische Genetik und Pathologie
Schönbeinstrasse 40
4031 Basel
Copyright
Aerzteverlag medinfo AG
Universitätsspital Basel
Institut für Medizinische Genetik und Pathologie
Schönbeinstrasse 40
4031 Basel
Universitätsspital Basel
Institut für Medizinische Genetik und Pathologie
Schönbeinstrasse 40
4031 Basel
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469-474. doi:10.1038/nature26000
2. Euskirchen P, Bielle F, Labreche K, et al. Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol (Berl). 2017;134(5):691-703. doi:10.1007/s00401-017-1743-5
3. Kuschel LP, Hench J, Frank S, et al. Robust methylation-based classification of brain tumours using nanopore sequencing. Neuropathol Appl Neurobiol. Published online November 9, 2022. doi:10.1111/nan.12856
4. Jurmeister P, Leitheiser M, Arnold A, et al. DNA Methylation Profiling of Salivary Gland Tumors Supports and Expands Conventional Classification. Mod Pathol. 2024;37(12):100625. doi:10.1016/j.modpat.2024.100625
5. Hench J, Hultschig C, Brugger J, et al. EpiDiP/NanoDiP: a versatile unsupervised machine learning edge computing platform for epigenomic tumour diagnostics. Acta Neuropathol Commun. 2024;12(1):51. doi:10.1186/s40478-024-01759-2
6. Yuan D, Jugas R, Pokorna P, et al. crossNN is an explainable framework for cross-platform DNA methylation-based classification of tumors. Nat Cancer. Published online June 6, 2025. doi:10.1038/s43018-025-00976-5
7. Djirackor L, Halldorsson S, Niehusmann P, et al. Intraoperative DNA methylation classification of brain tumors impacts neurosurgical strategy. Neuro-Oncol Adv. Published online October 10, 2021:vdab149. doi:10.1093/noajnl/vdab149
8. Brändl B, Steiger M, Kubelt C, et al. Rapid brain tumor classification from sparse epigenomic data. Nat Med. Published online February 28, 2025. doi:10.1038/s41591-024-03435-3
9. Vermeulen C, Pagès-Gallego M, Kester L, et al. Ultra-fast deep-learned CNS tumour classification during surgery. Nature. Published online October 11, 2023. doi:10.1038/s41586-023-06615-2
10. WHO Classification of Tumours Editorial Board, ed. Central Nervous System Tumours. 5th edition. International Agency for Research on Cancer; 2021.
11. Brugger J, Hultschig C, Baumhoer D, et al. Mepylome: A Point-of-Care Tumor Diagnostic Toolkit for Tumor DNA Methylation and Copy Number Analysis. Adv Intell Syst. Published online December 18, 2025:e202500778. doi:10.1002/aisy.202500778
12. Koelsche C, Schrimpf D, Stichel D, et al. Sarcoma classification by DNA methylation profiling. Nat Commun. 2021;12(1):498. doi:10.1038/s41467-020-20603-4
13. Hench IB, Hench J, Tolnay M. Liquid Biopsy in Clinical Management of Breast, Lung, and Colorectal Cancer. Front Med. 2018;5:9. doi:10.3389/fmed.2018.00009
14. Afflerbach AK, Rohrandt C, Brändl B, et al. Classification of Brain Tumors by Nanopore Sequencing of Cell-Free DNA from Cerebrospinal Fluid. Clin Chem. Published online August 25, 2023:hvad115. doi:10.1093/clinchem/hvad115
15. Hench J, Hultschig C, Bratic Hench I, et al. Rapid brain lymphoma diagnostics through nanopore sequencing of cytology-negative cerebrospinal fluid. Acta Neuropathol (Berl). 2024;148(1):36. doi:10.1007/s00401-024-02793-z
16. Sol N, Kooi EJ, Pagès-Gallego M, et al. Glioblastoma, IDH-wildtype with primarily leptomeningeal localization diagnosed by nanopore sequencing of cell-free DNA from cerebrospinal fluid. Acta Neuropathol (Berl). 2024;148(1):35. doi:10.1007/s00401-024-02792-0
17. Widschwendter M, Evans I, Jones A, et al. Methylation patterns in serum DNA for early identification of disseminated breast cancer. Genome Med. 2017;9(1). doi:10.1186/s13073-017-0499-9
18. Widschwendter M, Zikan M, Wahl B, et al. The potential of circulating tumor DNA methylation analysis for the early detection and management of ovarian cancer. Genome Med. 2017;9(1). doi:10.1186/s13073-017-0500-7
19. Li N, Song K, Chen H, Dai M. Advance and challenge of DNA methylation as cancer biomarkers for risk stratification, screening and early detection. J Natl Cancer Cent. 2025;5(2):108-112. doi:10.1016/j.jncc.2024.12.007


info@onco-suisse
- Vol. 16
- Ausgabe 2
- Mai 2026