

Hämophilie A und B sind angeborene, X-chromosomal vererbte Erkrankungen, bei denen es zu einer Verminderung der Gerinnungsfaktoren VIII (Hämophilie A) bzw. IX (Hämophilie B) kommt. In diesem Artikel werden nach einem kurzen Überblick über die konventionelle Behandlung neue Therapieoptionen präsentiert, welche sowohl Verbesserungen in Bezug auf Faktor-Substitution wie auch Faktor-freie Behandlungsmöglichkeiten umfassen.
Hämophilien werden abhängig von der restlichen Faktoren-Aktivität in 3 Schweregrade unterteilt: schwer < 1%; mittelschwer 1-5%; mild 6-40% (1). Epidemiologisch treten sie weltweit auf. Die Inzidenz der Hämophilie A beträgt 1/5000 männliche Geburten (Prävalenz 1/12 000) und die der Hämophilie B 1/25 000 männliche Geburten (Prävalenz 1/19 000). Durch die verminderte Faktoren-Aktivität kommt es zu einer Blutungsneigung – neben akuten Blutungen sind hier auch die Folgen rezidivierender Gelenkblutungen, die zu schwerer Arthrose führen, relevant (1).
Erwähnenswert und abzugrenzen ist die Gruppe der sogenannten «erworbenen» Hämophilien (Autoimmunhämophilie), bei denen es zu einer Antikörper-Entwicklung und infolge dessen zu einer Verminderung der Gerinnungsfaktoren (am häufigsten Faktor VIII) kommt (Inzidenz 1:1 Mio. in der allgemeinen Bevölkerung).
«Konventionelle» Therapie der Hämophilie
Der Grundpfeiler der Hämophilie-Therapie besteht seit langem in der Substitution des jeweiligen Gerinnungsfaktors durch i.v. Injektion. Hierfür standen bisher plasmatische (aus menschlichem Plasma gewonnene) und rekombinante (synthetisierte) Präparate zur Verfügung. Bei der Auswahl müssen das Risiko einer möglichen Infektions-Übertragung (heute durch effiziente Pathogeninaktivierungsverfahren praktisch eliminiert) und das Risiko einer Immunisierung gegen das Präparat abgewogen werden. Bei der Therapie werden zwei Prinzipien unterschieden:
- die bedarfsweise Faktoren-Gabe («on-demand») bei Blutungen oder geplanten Eingriffen (alle Hämophilie-Formen) und
- die prophylaktische Faktoren-Gabe (regelmässig 2 x-3 x/Woche, schwere und z.T. mittelschwere Hämophilie).
Eine prophylaktische Therapie wird üblicherweise bereits in der frühen Kindheit nach der ersten relevanten Gelenkblutung (grosses Gelenk) initiiert mit dem Ziel, die basale Faktoren-Aktivität anzuheben (2). Mehrfach konnte gezeigt werden, dass eine konsequente Prophylaxe das Auftreten von Gelenkschädigungen verhindern kann (3,4). Die Halbwertzeit der in der Vergangenheit verfügbaren Faktoren-Präparate lag bei ca. 12h (Faktor VIII) bzw. ca. 20h (Faktor IX). Dadurch waren Injektionen 3 x /Woche (Faktor VIII) bzw. 2 x /Woche (Faktor IX) notwendig. Viele Hämophilie-Patienten lernen, die intravenöse Substitution selbstständig durchzuführen. Die häufigen Injektionen stellen jedoch für die Betroffenen und Ihre Familien eine z.T. erhebliche Belastung dar (2).
Neue Therapieoptionen
Nachdem die beschriebenen Therapie-Optionen seit Ende der 70er Jahre lange Zeit unverändert waren, haben letzte Entwicklungen grosse Bewegung in die Therapie-Möglichkeiten der Hämophilie gebracht. Hier sind insbesondere relevant:
- Präparate mit längerer Wirkdauer
- Neue Wirkmechanismen (Alternativen zu der Faktoren-Gabe)
- Neue Applikationsarten (subkutane Gabe)
- Gentherapie
Faktoren-Präparate mit längerer Halbwertzeit
Um die Injektions-Häufigkeit zu reduzieren und höhere basale Faktoren-Spiegel zu erreichen, haben verschiedene Hersteller Präparate mit einer verlängerten Halbwertzeit entwickelt. Dies wurde durch verschiedenartige Modifikationen des Faktor VIII-Moleküls erreicht (5):
- Pegylierung: Konjugation mit Polyethylenglycol (PEG) als Schutz vor Abbau und Verminderung der renalen Ausscheidung
- Fusionsmoleküle: Kopplung des Gerinnungsfaktors an ein Molekül mit längerer Halbwertzeit (Albumin oder IgG-Fc-Protein).
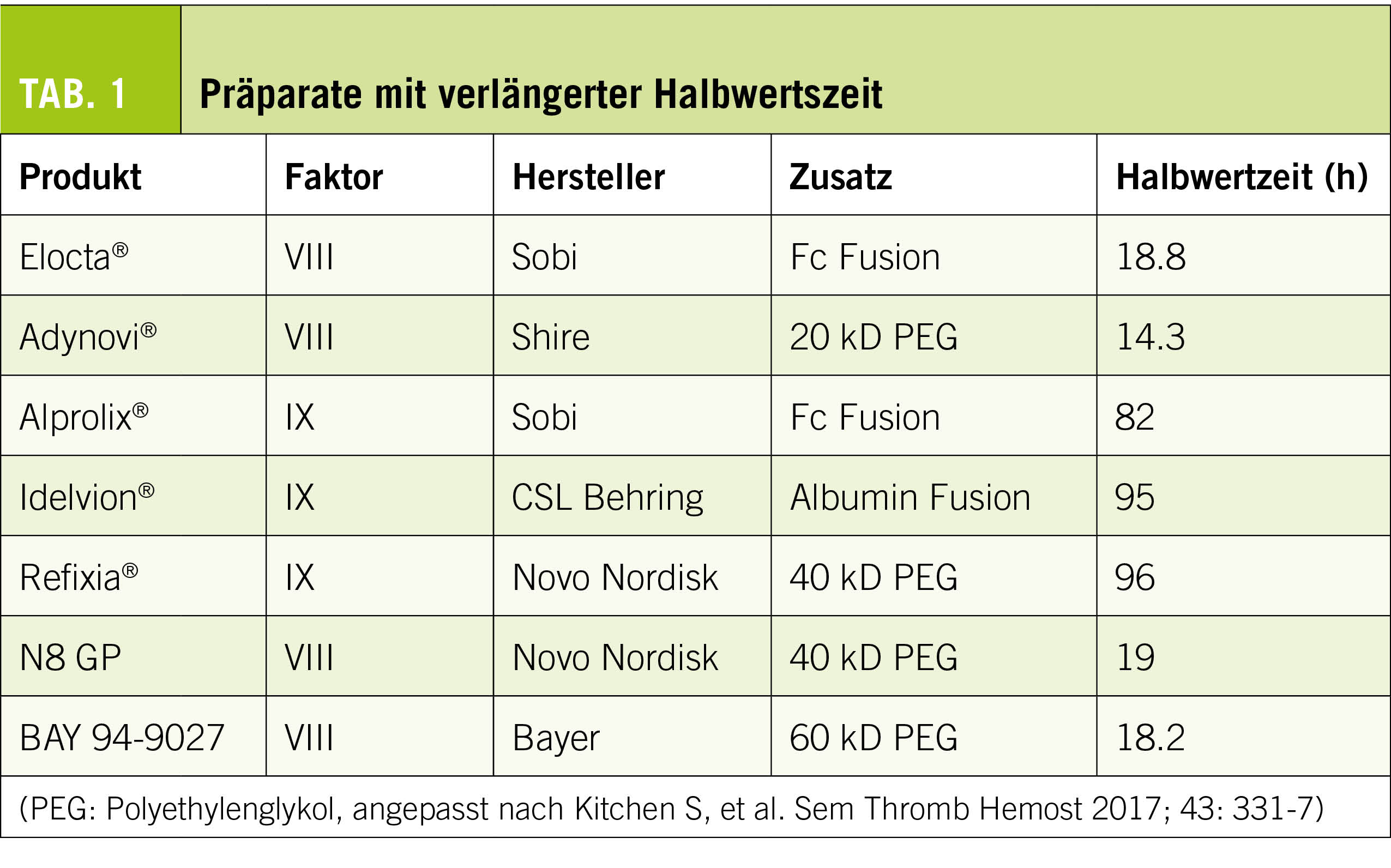
Derzeit sind in der Schweiz fünf Faktoren-Präparate mit verlängerter Halbwertzeit verfügbar, zwei weitere befinden sich im Zulassungsverfahren (Tab. 1).
Diese Modifikationen haben für Faktor VIII eine Verlängerung der Halbwertzeit (HWZ) auf das ca. 1,5 fache (d.h. ca. 18h) erreichen können. Die Injektionsfrequenz in der prophylaktischen Therapie konnte dadurch je nach Situation auf 2 x /Woche reduziert werden. Faktor VIII wird physiologisch an das Von Willebrand-Molekül gebunden transportiert und ist damit von dessen HWZ abhängig. Dieser Umstand erschwert eine weitere Verlängerung der Wirkdauer (6).
Für Faktor IX konnte eine wesentlich deutlichere HWZ-Verlängerung auf 80 – 100h (d.h. das 5 fache) erreicht werden. Die Injektionsfrequenz liess sich auf 1 x alle 1-2 Woche reduzieren – mit Faktor IX-Talspiegeln von > 3%. Die Verbesserung der Hämophilie B-Behandlung durch diese therapeutische Option ist immens.
Durch die umfangreichere Modifikation der Faktorenmoleküle besteht die Sorge, dass die Immunogenität und damit das Risiko einer Antikörper-Entstehung («Inhibitor») zunimmt. Die bisherigen – limitierten – Erfahrungen konnten dies erfreulicherweise nicht bestätigen (5).
Neue Wirkmechanismen / «non-factor-replacement»
Neben der Modifikation der Gerinnungsfaktoren VIII und IX wurden alternative Ansätze zur Beeinflussung und Verbesserung der Hämostase verfolgt. Diese Ansätze werden als «non-factor-replacement» bezeichnet. Sie stellen eine grosse therapeutische Hoffnung dar bei Patienten, die einen Antikörper gegen Faktor VIII oder IX entwickelt haben (im Rahmen der Faktoren-Substitution bei Hämophilie A oder B oder einer «erworbenen» Hämophilie). Zudem besteht die Möglichkeit einer subkutanen Applikation. Die derzeit entwickelten Medikamente sind:
- Bispezifische Antikörper: Emicizumab (Hemlibra®) ist ein bispezifischer, rekombinant hergestellter IgG-Antikörper, der – vereinfacht – in der plasmatischen Gerinnung die Funktion von Faktor VIII ersetzt, indem er aktivierten Faktor IX und Faktor X zusammenführt und dadurch Faktor X aktivieren kann. Emicizumab kann daher auch bei Faktor VIII-Inhibitoren eingesetzt werden. Der Antikörper wird s.c. verabreicht und hat eine Halbwertzeit von 4-5 Wochen, die Gabe erfolgt 1 x / Woche oder 1 x alle zwei Wochen. Eine «Normalisierung» der Gerinnung bei der schweren Hämophilie A wird formal nicht erreicht, jedoch war der Rückgang der Blutungsereignisse bei Patienten mit oder ohne Inhibitor in Phase III-Studien relevant gross (79-87% resp. 95-96%). Vorsicht ist bei dem gleichzeitigen Verabreichen von anderen plasmatischen Gerinnungsprodukten geboten, welche zum Teil aktivierte Faktorenmoleküle beinhalten (FEIBA factor eight bypassing agents). Unter deren kombinierter Wirkung traten Thromboembolien und thrombotische Mikroangiopathien auf. Emicizumab hat die Zulassung in der EU erhalten, in der Schweiz ist das Produkt von der Swissmedic zugelassen für die Hemmkörperhämophilie; das Zulassungsverfahren für die Hämophilie ohne Hemmkörper ist auch aktiviert (7, 8).
- Tissue Factor Pathway Inhibitor (TFPI)-Antikörper (z.B. Concizumab): In Entwicklung befinden sich verschiedene monoklonale Antikörper, die den TFPI blockieren und somit zu einer verbesserten Aktivierung des Tissue Factors (TF) führen (Blockade des negativen Feedback-Mechanismus). TF und Faktor VIIa aktivieren Faktor X zu Faktor Xa und gleichen so einen FVIII- oder FIX-Mangel teilweise aus. Concizumab ist in einer Phase I-Studie untersucht worden, eine Phase II-Studie ist derzeit aktiv (9); zwei weitere Anti-TFPI monoklonale Antikörper (Marstacimab und BAY-109) sind aktuell in Entwicklung.
- Antithrombin-Verminderung mittels siRNA (Fitusiran): Antithrombin hemmt Thrombin (FIIa), welches für die Thrombusbildung verantwortlich ist. Ein Antithrombinmangel führt daher zu einer potentiellen Gerinnungsaktivierung. Es besteht die Hoffnung, hierdurch den Faktoren Mangel der Hämophilie A und B «auszugleichen». «Antithrombin small interfering RNA» (siRNA) soll die Antithrombin-Bildung durch Blockierung der Antithrombin mRNA in den Hepatozyten reduzieren. Die Substanz Fitusiran wird s.c. einmal monatlich appliziert und konnte bei Hämophilie-Patienten den Antithrombin-Spiegel um bis zu 88% senken. Die Gerinnungsteste in dieser Konstellation zeigten einen deutlichen Anstieg der Thrombinbildung. Derzeit befindet sich das Produkt in Phase III-Studien (ATLAS Trial) (10).
- Prokoagulatorische Moleküle: In experimenteller Entwicklung befinden sich weitere Konzepte, wie ein Protein C-Inhibitor oder prokoagulatorisch wirkende monoklonale Antikörper gegen Faktor IXa oder Faktor X.
Gentherapie
Mit den Fortschritten der Gentherapie der letzten Jahre besteht seit langem auch die Hoffnung, Hämophilie A und B heilen zu können (11).
Prinzip: Das Therapieprinzip beruht aktuell auf dem Transfer eines funktionierenden F8- oder F9-Gens oder Genteiles in die Hepatozyten der Person mit Hämophilie. Der Transfer erfolgt via virale Vektoren, am besten haben sich dafür aktuell die «gutted» Adenoviren bezüglich Effizienz und Nebenwirkungen bewährt. Das transferierte Genom der Vektoren (F8- oder F9-Gen) nutzt die Umgebung der Hepatozyten aus, um FVIII- oder FIX-Moleküle zu produzieren ohne sich ins Genom der Hostzelle zu integrieren. Die Herausforderung besteht im Erreichen eines adäquaten FVIII- oder FIX-Spiegels (z.B. > 2%) über längere Zeit. Die Transformation der Hämophilie von der schweren (FVIII < 1%) in die mittelschwere oder milde Form (FVIII 2-4% oder höher) würde die Lösung des klinischen Problems bedeuten.
Vektoren und Ergebnisse: Erfolgreiche Studien wurden mit dem Transfer des F9-Gens und mit einem an der Länge reduzierten F8-Gen durchgeführt. In beiden Ansätzen haben die Patienten Faktorenspiegel von 10-50% über längere Zeit (aktuell 2-4 Jahre nach Beginn der Studien) erreicht. Es ist noch unbekannt wie haltbar diese Intervention bleiben kann (11).
Nebenwirkungen: Als häufige Nebenwirkungen wurden eine – meist transiente – Hepatitis beobachtet, welche gut auf Steroid-Therapie anspricht. Unsicher ist derzeit, wie lange die Gen-Expression anhält, zudem sind langfristige, derzeit unbekannte, Folgen der Vektor-Integration in die Hepatozyten unklar (11).
Diagnostische Hämatologie
Universitätsspital Basel
Petersgraben 4
4031 Basel
SYNLAB Suisse SA
Alpenquai 14
6002 Luzern
dimitrios.tsakiris@usb.ch
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Beitrag deklariert.
Literatur:
1. Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A. Definitions in hemophilia: Communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935–9.
2. Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1): e1-47.
3. Manco-Johnson MJ, Soucie JM GJ. Joint Outcomes Committee of the Universal Data Collection USHTCN. Prophylaxis usage, bleeding rates, and joint outcomes of hemophilia, 1999 to 2010: a surveillance project. Blood. 2017;129:2368–74.
4. Manco-Johnson MJ, Kempton CL, Reding MT, Lissitchkov T, Goranov S, Gercheva L, et al. Randomized, controlled, parallel-group trial of routine prophylaxis vs. on-demand treatment with sucrose-formulated recombinant factor VIII in adults with severe hemophilia A (SPINART). J Thromb Haemost. 2013;11(6):1119–27.
5. Laffan M. New products for the treatment of haemophilia. Br J Haematol. 2016;172(1):23–31.
6. Tiede A. Half-life extended factor VIII for the treatment of hemophilia A. J Thromb Haemost. 2015;13(S1):S176–9.
7. Oldenburg J, Mahlangu JN, Kim B, Schmitt C, Callaghan MU, Young G, et al. Emicizumab Prophylaxis in Hemophilia A with Inhibitors. N Engl J Med 2017; 377: 809-18
8. Mahlangu JN, Oldenburg, J, Paz-Priel I, et al. Emicizumab prophylaxis in patients who have hemophiia A without inhibitors. N ENgl J Med 2018; 379: 811-22.
9. Chowdary P, Lethagen S, Friedrich U, Brand B, Hay C, Abdul Karim F, et al. Safety and pharmacokinetics of anti-TFPI antibody (concizumab) in healthy volunteers and patients with hemophilia: A randomized first human dose trial. J Thromb Haemost. 2015;13(5):743–54.
10. Pasi KJ, Rangarajan S, Georgiev P, Mant T, Creagh MD, Lissitchkov T, et al. Targeting of Antithrombin in Hemophilia A or B with RNAi Therapy. N Engl J Med 2017; 377: 819-28.
11. Pierce GF, Iorio A. Past, present and future of haemophilia gene therapy: From vectors and transgenes to known and unknown outcomes. Haemophilia 2018; 24 Suppl. 6: 60-7.


der informierte @rzt
- Vol. 9
- Ausgabe 1
- Januar 2019