Prävalenz und Epidemiologie: welche Erkrankungen gehören zur «progressiven pulmonalen Fibrose» ?
Für die Schweiz gibt es keine genauen Prävalenzdaten für das Vorkommen von ILDs (Interstitial lung disaease), aufgrund internationaler vergleichbarer Daten darf von einer Prävalenz von ca. 76/100 000 in Europa ausgegangen werden.
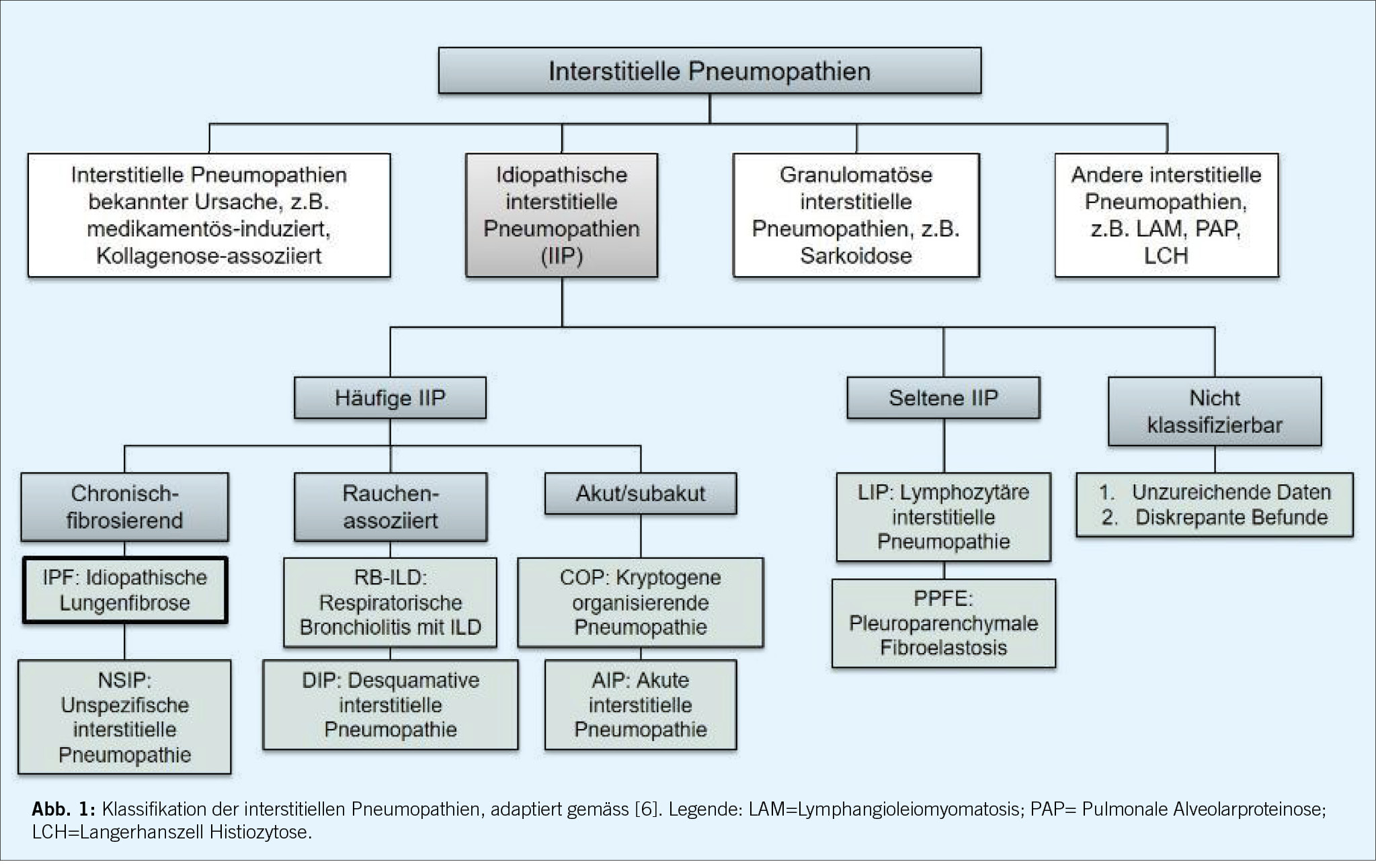
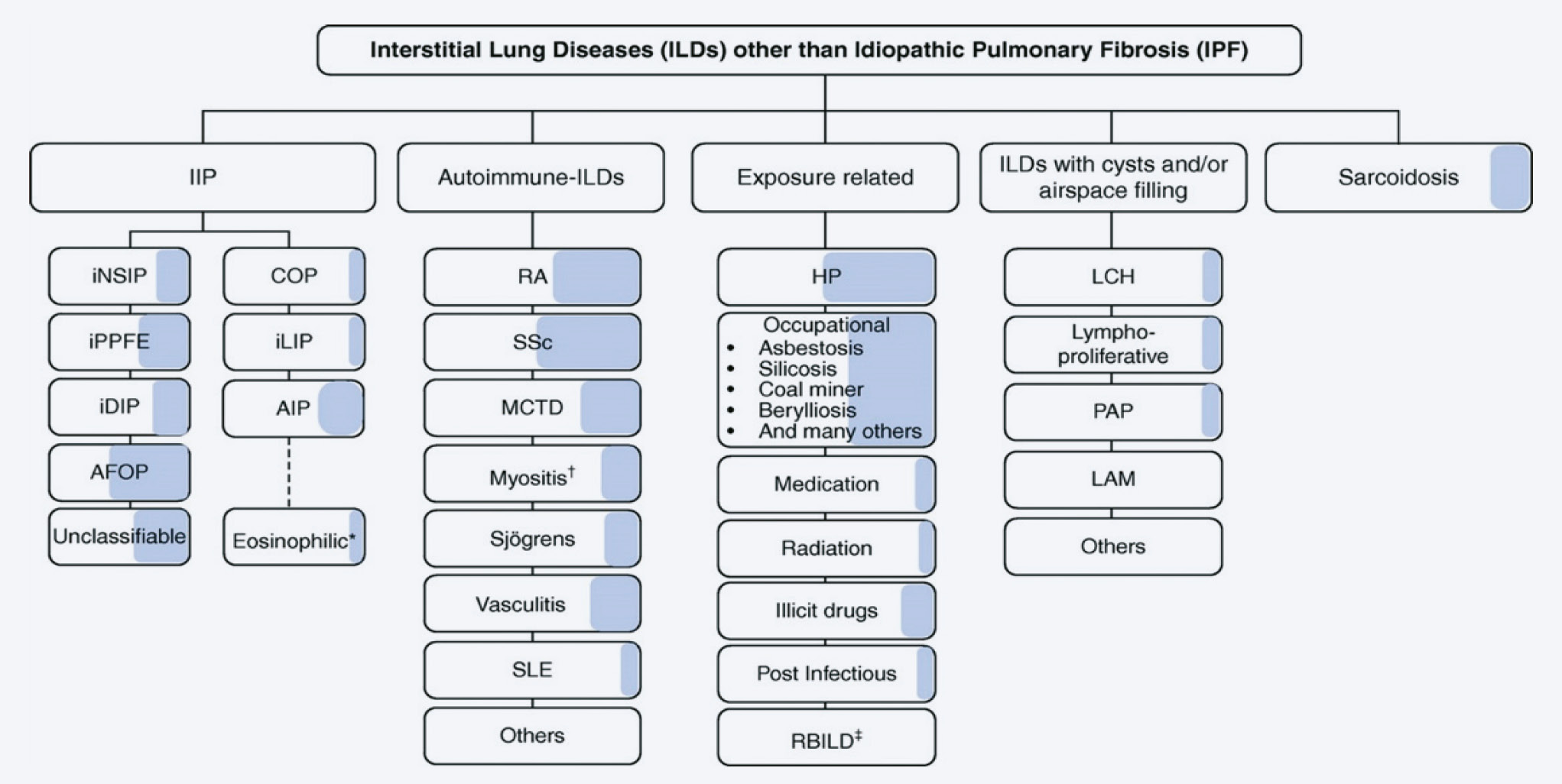
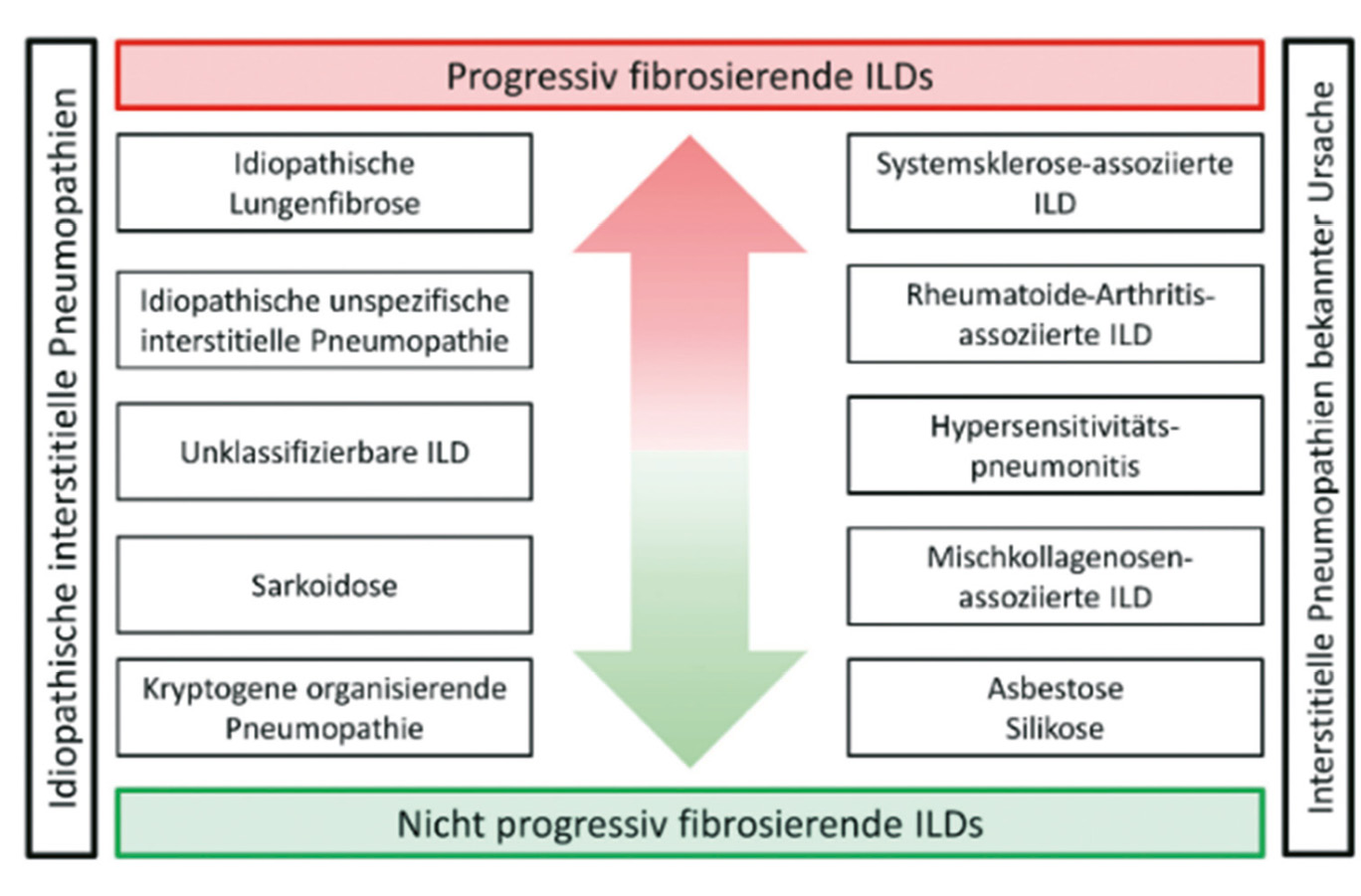
Die interstitiellen Lungenerkrankungen sind eine heterogene Gruppe von Lungenparenchymerkrankungen, wobei sich die klinische Präsentation und die radiologischen Muster der verschiedenen Ätiologien zum Teil überlappen, zum Teil aber auch deutlich unterscheiden. Diese verschiedenen Lungenparenchymerkrankungen werden gemäss geltenden Richtlinien eingeteilt. Eine Übersicht gibt die Abbildung 1a. Es werden die idiopathischen Interstitiellen Pneumonien (IIP) von Autoimmunvermittelten Interstitiellen Lungenerkrankungen, sowie der Hypersensitivitätspneumonitis, der Sarkoidose und einigen weiteren gut beschriebenen seltenen Lungenerkrankungen (z.B. LAM Lymphyngioleiomyomatose) abgegrenzt.
Die in der Abbildung 1b hervorgehobenen ILDs gehen mit einem progressiv fibrosierenden Phänotyp einher (1). Die Schraffierung in der Abbildung 1a visualisiert zudem gemäss Schätzung eines internationalen Expertenkommittees der prozentuale Anteil der Erkrankungen, welche progredient fibrosieren (2).
Studien früher und heute – Von der IPF zum Konzept der «Progressiven pulmonalen Fibrose»
In der jüngeren Vergangenheit wurde die idiopathische Lungenfibrose (engl.: idiopathic pulmonary fibrosis, IPF) besonders hervorgehoben, da diese Erkrankung verglichen mit anderen ILDs mit einer erheblichen Morbidität und Mortalität einhergeht. Die IPF wurde als überwiegend inflammatorische Erkrankung verstanden, die aufgrund gestörter Reparaturprozesse in einer irreversiblen Fibrose endet. Mit diesem Krankheitskonzept war der langjährige Einsatz von Kortikosteroiden und antiinflammatorischer Medikation (überwiegend Azathioprin) begründet. Die Bewertung der Wirksamkeit wurde intensiv und kontrovers diskutiert und insbesondere die potentiellen relevanten Nebenwirkungen erwähnt (3).
Im Verlauf wurde diskutiert, dass Sauerstoffradikale zum epithelialen Schaden beitragen, es wurde N-Acetylcystein als Antioxidans eingesetzt. Im Jahr 2005 wurde der IFIGENIA (Idiopathic Pulmonary Fibrosis International Group Exploring N-Acetylcystein I Annual)-Trial durchgeführt, die Studie untersuchte den möglichen Benefit von hochdosiertem N-Acetylcystein zusätzlich zur damaligen Standard-Therapie (Kortikosteroide und Azathioprin). Die Patienten unter Triple-Therapie zeigten nach einem Jahr einen Benefit bezüglich Erhalt der Lungenfunktion, nicht aber bezüglich der Mortalität.
Das zwischenzeitlich weiter entwickelte pathophysiologische Verständnis und das fehlende histopathologische Merkmal der aktiven Entzündung bei der IPF führten zum PANTHER-IPF (Prednisone, Azathioprine , and N-Acetlycystein: A Study That Evaluates Respones in Idiopathic Pulmonary Fibrosis)-Trial, der die antiinflammatorische und antioxidative Therapie-Strategie versus Placebo untersuchte (4). 2011 wurde eine geplante Interims-Analyse durchgeführt und die Studie aufgrund schwerer Nebenwirkungen (u.a. Tod) gestoppt.
Diese Studie führte nun also dazu, dass Kortikosteroide und antiinflammatorische Medikamente bei IPF nicht mehr eingesetzt wurden und werden (5), wohingegen diese bei anderen ILDs wie z.B. der Hypersensitivitätspneumonitis oder Autoimmunassoziierten-ILDs zumindest in einem frühen Stadium weiterhin erfolgreich verabreicht werden können.
Die klinische Forschung fokussierte sich im weiteren Verlauf auf den Einsatz der antifibrotischen Substanzen Pirfenidon (6,7) und Nintedanib (8) bei der idiopathischen Lungenfibrose.
Beide Therapiestrategien führen bei der IPF zu einer Verzögerung der lungenfunktionellen Verschlechterung, insbesondere der weiteren Reduktion der FVC (forcierte Vitalkapazität). Somit wurden Pirfenidon und Nintedanib für die Behandlung der idiopathischen Lungenfibrose zugelassen und entsprechende internationale Guidelines zur Diagnose und Therapie der IPF publiziert (9).
Es blieb jedoch zunächst unklar, ob andere primär entzündliche ILDs, welche im Verlauf eine Fibrosierung aufwiesen, ebenso auf die antifibrotische Therapie ansprechen würden.
Im Sinne der Präzisionsmedizin muss das pathophysiologische Verständnis für die ILDs mittels Anwendung von Biomarkern, genetischen Profilen und Umgebungsfaktoren weiter konkretisiert werden und entsprechende Therapieverfahren entwickelt werden. Dies wird vermutlich zu einer weiteren Aufsplittung der IPF und anderen ILDs, zu noch nicht definierten, aber präziseren Subtypen führen (10).
Im Kontrast dazu werden im Konzept der «Progressiven Pulmonalen Fibrose» mehrere ILDs, die sich trotz unterschiedlicher pathophysiologischer Mechanismen im Verlauf mit einer zunehmenden Fibrosierung ähnlich verhalten, zusammengefasst.
Wie wird der «Phänotyp» der progressiven pulmonalen Fibrose definiert ?
Generell handelt es sich um eine Gruppe der interstitiellen Lungenerkrankung mit heterogener Ätiologie, welche per definitionem nicht den Kriterien der IPF entspricht und unter etablierter initialer Standardtherapie eine Verschlechterung, im Sinne einer progredienten Fibrosierung aufweist.
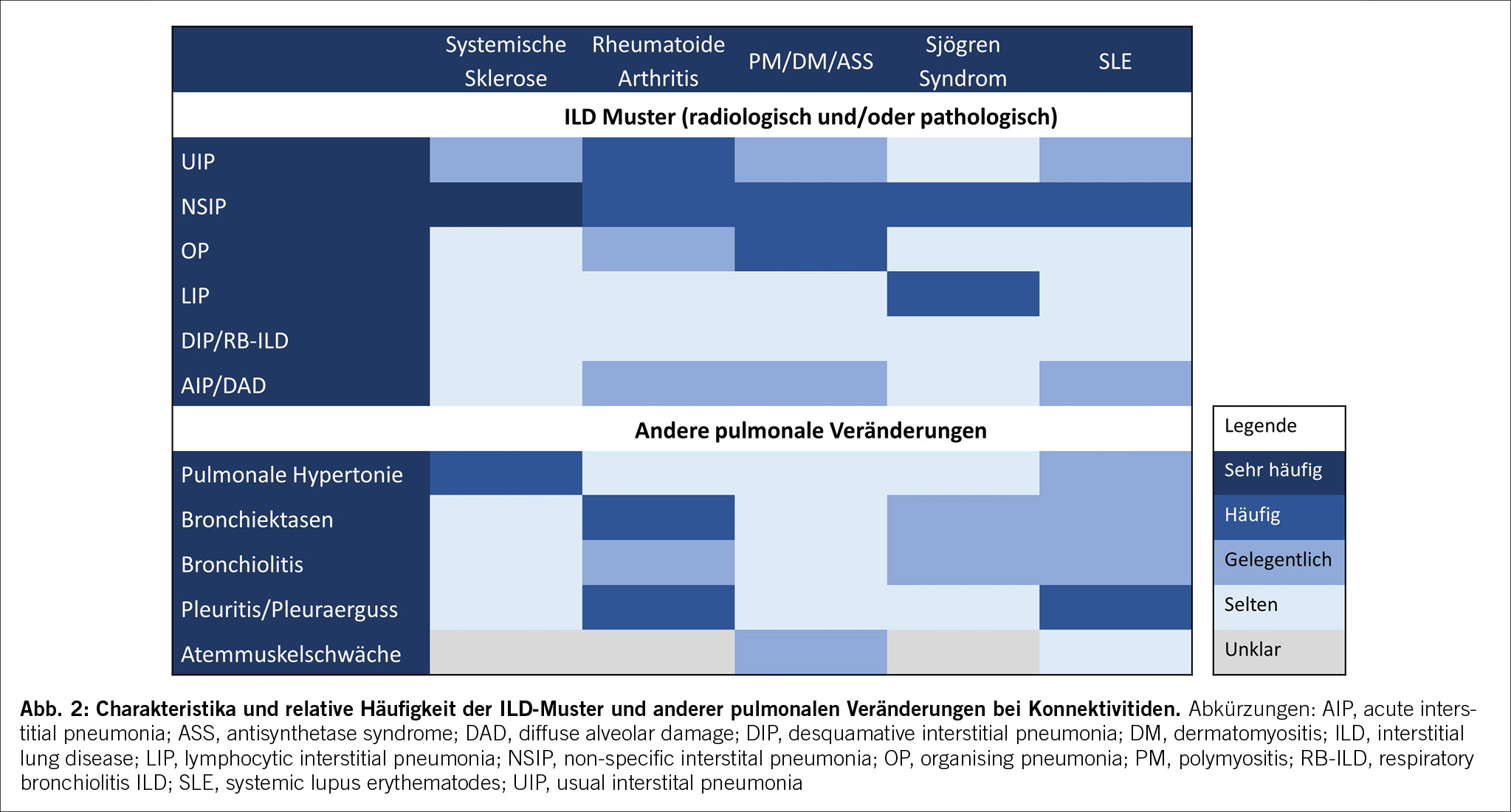
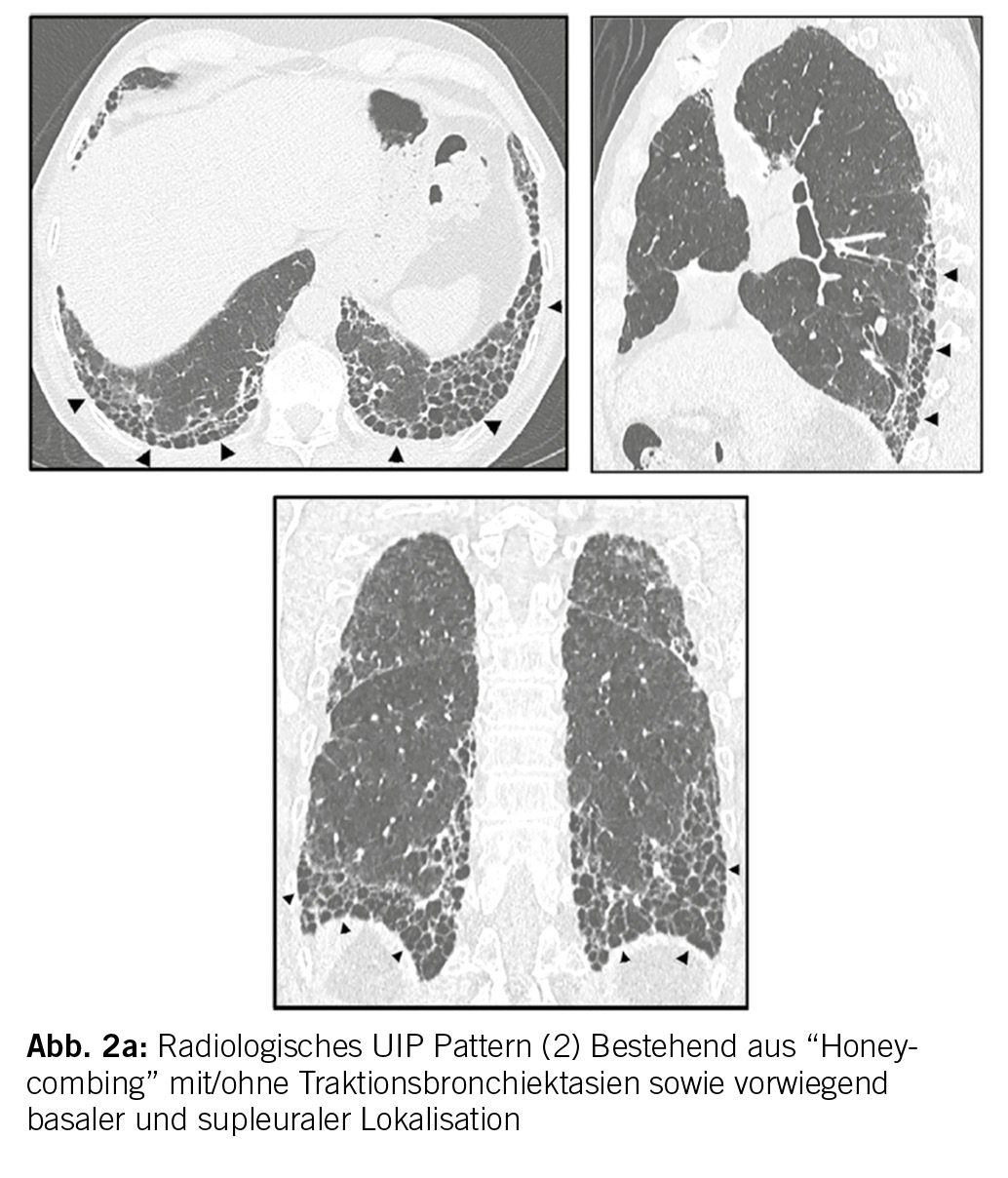
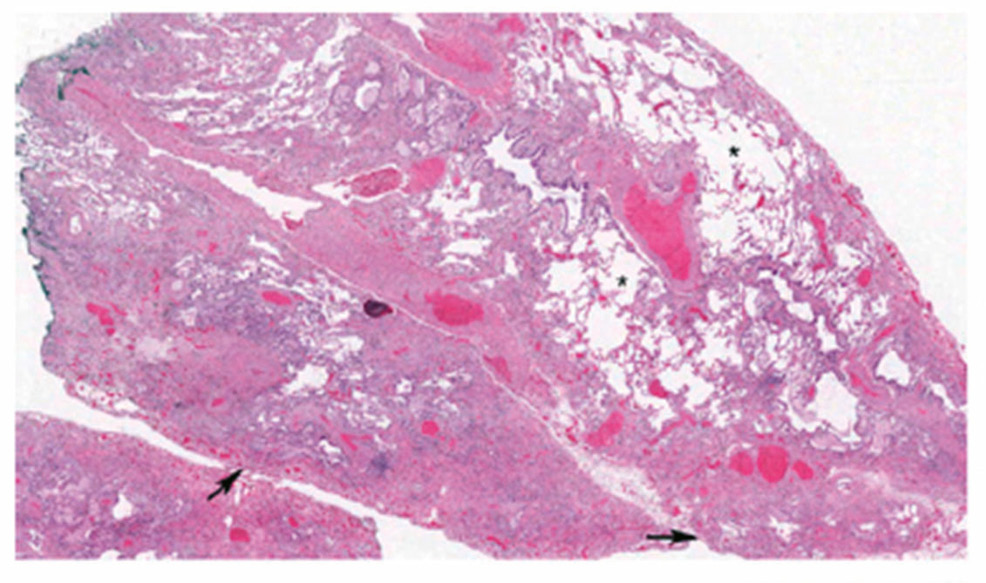
In der Bildgebung (HRCT (High resolution CT)) und/oder in den Biopsien finden wir dabei progrediente fibrotische Veränderungen. Insbesondere das Auftreten eines UIP-Pattern (usual interstitial pneumonia), siehe Abbildung 2a und 2b. geht mit einer Verschlechterung der Lungenfunktion und einer Zunahme der Morbidität und Mortalität einher.
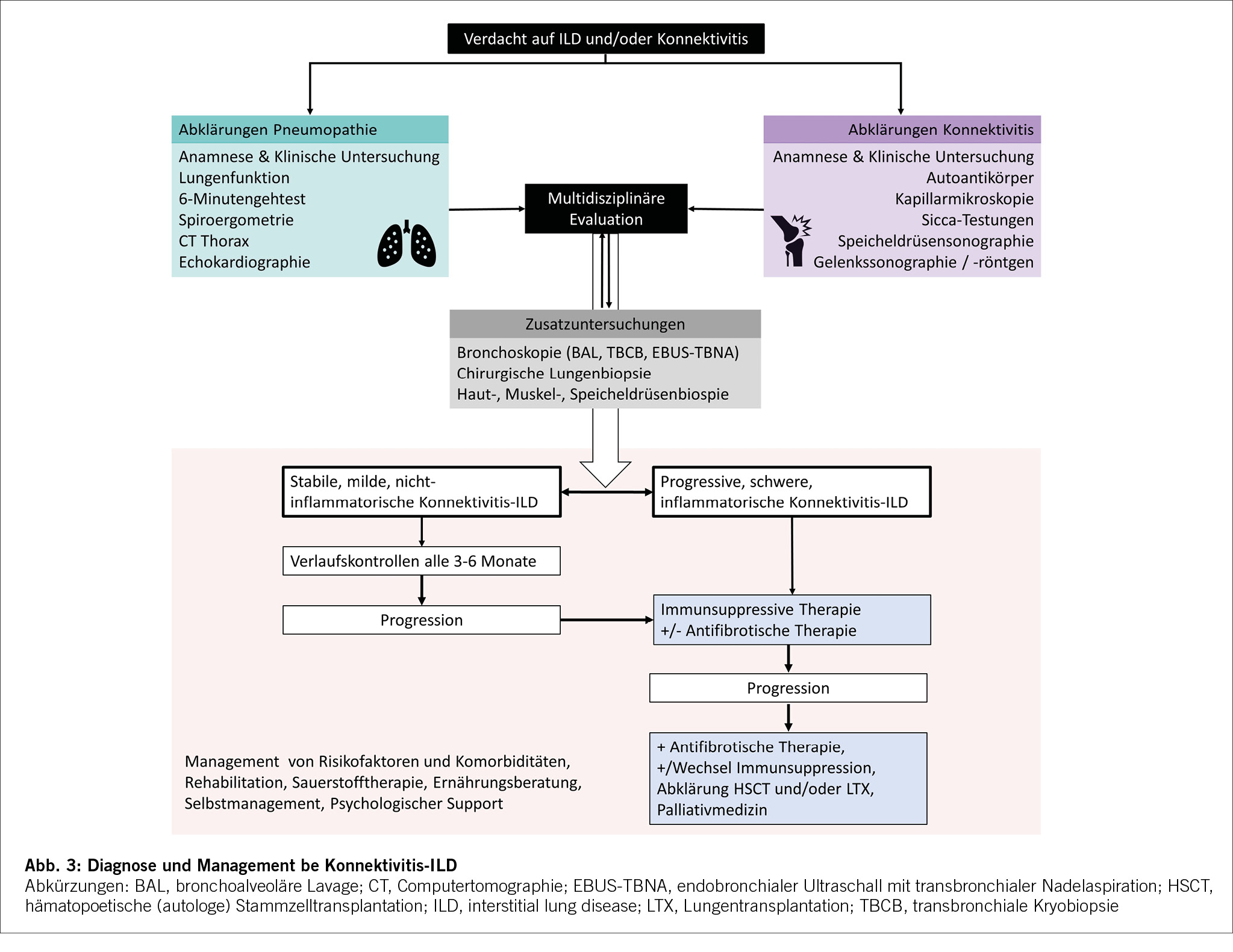
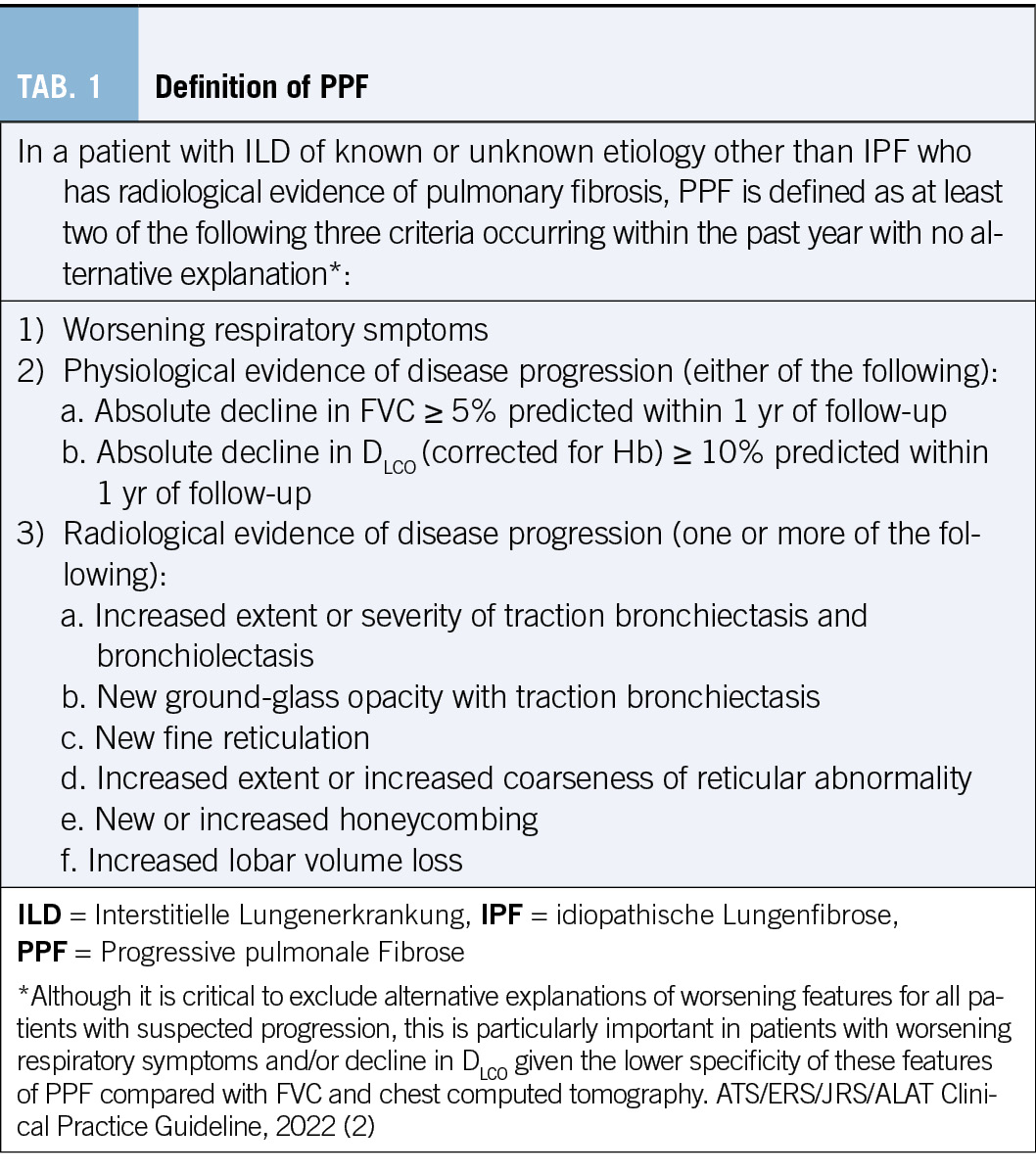
Der Phänotyp der progredienten pulmonalen Fibrosierung, definiert sich gemäss ATS Guidelines in 3 verschiedenen Kriterien (s. Tabelle 1) aus Klinik, Lungenfunktion und Radiologie. Zur Diagnosestellung einer progressiven pulmonalen Fibrose sind zwei von drei Kriterien innerhalb von 12 Monaten notwendig, eine alternative Ätiologie für die Progredienz der ILD muss ausgeschlossen sein.
Die S2K-Leitlinie der deutschen Gesellschaft für Pneumologie hat in einer kürzlichen Überarbeitung eine leicht abgewandelte Definition erarbeitet. Hier muss eine Fibrosierung des Lungengewebes von mindestens 10% im Computertomogramm vorliegen sowie eine klinische, lungenfunktionelle oder radiologische Verschlechterung innerhalb von 24 Monaten (11).
Diese Empfehlungen verdeutlichen, dass ein regelmässiges Follow-Up der betroffenen ILD-Patienten (üblicherweise ca. 3-4 monatlich klinisch und lungenfunktionell, bei Verschlechterung auch mittels Bildgebung) angezeigt ist, um die allfällige Progression der Erkrankung festzustellen.
Es ist empfohlen, diese Patienten interdisziplinär an einem Board für interstitielle Lungenerkrankungen zu besprechen, um die Diagnose und das Therapiekonzept festzulegen.
Therapeutische und prognostische Aspekte – Was für Therapieoptionen stehen zur Verfügung?
Der Begriff PPF Progressive pulmonale Fibrose subsumiert auch Interstitielle Lungenerkrankungen (vgl. Abbildung 1), die u.a. mit Inflammation einhergehen, wie bereits erwähnt, ist die möglichst präzise Diagnosestellung wichtig. Die initiale, insbesondere antiinflammatorische Therapiestrategie wird bei Nutzen (allenfalls auch in Bezug auf weitere Manifestationen der Grunderkrankung) auch bei zunehmender pulmonaler Fibrosierung in der Regel weitergeführt.
Nintedanib und Pirfenidon haben antifibrotische Eigenschaften unabhängig vom pathophysiologischen Auslöser der Fibrosierung. Aufgrund der wissenschaftlichen Erkenntnisse in Anwendung zur antifibrotischen Therapie bei der idiopathischen Lungenfibrose (IPF) wurde diese Therapiestrategie bei anderen chronisch fibrosierenden ILDs evaluiert (12–14). Aufgrund der positiven Studienergebnisse bezüglich Erhalt der lungenfunktionellen Parameter (insbesondere der FVC (forcierte Vitalkapazität) wurden nun neue Empfehlungen publiziert (2,13,15).
Ist der «Phänotyp» einer progressiven pulmonalen Fibrose definiert worden, sollte daher eine antifibrotische Therapie in Betracht gezogen werden.
Ausnahmen stellen palliative Situationen, sehr hohes Alter oder Komorbiditäten dar, welche für den Medikamenteneinsatz kontraindiziert sind.
Auch sind Nebenwirkungen (Nintedanib: Diarrhoe, Gewichtsverlust, erhöhte Leberwerte. Pirfenidon: Nausea, Anorexie, Gewichtsverlust, Photosensitivität, erhöhte Leberwerte) und Interaktionen dieser Medikamente zu beachten.
Vor Einsatz muss ein entsprechendes Kostengutsprachegesuch eingereicht werden.
Für Nintedanib ist die Studienevidenz höher, weshalb primär dieses Präparat zur Anwendung kommt. Head-to-head Studien, die die Wirksamkeit dieser beiden Therapieoptionen vergleichen, gibt es allerdings nicht.
In Studien konnte analog zur IPF ein geringerer Abfall der Lungenvolumina (gemessen an der forcierten Vitalkapazität innerhalb von 12 Monaten) nachgewiesen werden und somit der fibrotische Prozess zumindest verlangsamt werden. Eine Verbesserung klinischer Parameter konnte sich bisher allerdings nicht nachweisen lassen.
Neben der antifibrotischen Therapie sind auch weitere therapeutische Massnahmen wichtig.
Patienten, die eine respiratorische Partialinsuffizienz mit einem pO2 von <7.3 (resp. <8kPa bei pulmonaler Hypertonie) in der arteriellen Blutgasanalyse aufweisen, sollten mit dauerhafter Heimsauerstofftherapie versorgt werden. Auch bei signifikanter Entsättigung unter Belastung kann eine Sauerstofftherapie erwogen werden(16).
Patienten mit interstitieller Lungenerkrankung und pulmonaler Hypertonie haben unter einer inhalativen Therapie mit Treprostinil, einem synthetischen Prostazyklin, eine deutliche Verbesserung der 6-Minuten-Gehstrecke aufgewiesen(17). Diese Therapie ist jedoch in der Schweiz noch nicht regulär erhältlich.
Rehabilitationsmassnahmen haben in Studien bei Patienten mit interstitiellen Lungenerkrankungen einen Benefit gezeigt und sollten den Patienten je nach Allgemeinzustand und Komorbiditäten angeboten werden(18).
Nicht zuletzt werden geeignete Patienten mit schwerem Befall frühzeitig in einem Zentrum für Lungentransplantationen vorgestellt.
Auch ist je nach Leidensdruck die Behandlung der Symptome durch Mukolytika oder auch Antitussiva und bei zunehmender Atemnot der Einsatz von Opiaten zu erwägen (19), ggf. auch mit Unterstützung durch einen Palliativdienst.
Prognostische Einschätzungen sind aufgrund der Heterogenität der PPF schwierig. Klar ist, dass das rasche Voranschreiten der Fibrosierung die Mortalität der Patienten deutlich erhöht.
Eine verschlechterte Prognose weisen auch Patienten mit Exazerbationen auf. Dabei kommt es zu akuten Entzündungsschüben, welche mit einer klinischen und radiologischen Verschlechterung (CT-Thorax) einhergehen.
Hier ist die differentialdiagnostische Abgrenzung zu anderen Ätiologien für eine akute pulmonale Verschlechterung (z.B. infektiöse Aetiologie, kardialer Dekompensation) notwendig. Sollte es sich um eine Exazerbation einer interstitiellen Lungenerkrankung handeln, wird diese in der Regel mittels kurzfristiger Steroidtherapie behandelt, auch wenn für diese Behandlung keine hohe Studienevidenz vorliegt.
Insgesamt steht für die Behandlung der interstitiellen Lungenerkrankungen eine Strategie zur Verfügung, welche den Fibrosierungsprozess zumindest verlangsamen mag und einige weitere, neuere Wirkstoffe sind derzeit in Studien in Erprobung.
Dr. Rebekka Kleiner, rebekka.kleiner@kssg.ch
Leitende Ärztin
Dr. Susanne Pohle, Oberärztin mbF
Klinik für Pneumologie und Schlafmedizin
Kantonsspital St. Gallen, Rorschacher Strasse 95,
9007 St. Gallen
Interessenskonflikt: Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
Klinik für Pneumologie und Schlafmedizin
Kantonsspital St. Gallen, Rorschacher Strasse 95,
9007 St. Gallen
Literatur:
1. Cottin V, Hirani NA, Hotchkin DL, Nambiar AM, Ogura T, Otaola M, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018 Dec 31;27(150):180076.
2. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022 May 1;205(9):e18–47.
3. Idiopathic Pulmonary Fibrosis: Diagnosis and Treatment: International Consensus Statement. Am J Respir Crit Care Med. 2000 Feb 1;161(2):646–64.
4. The Idiopathic Pulmonary Fibrosis Clinical Research Network. Prednisone, Azathioprine, and N -Acetylcysteine for Pulmonary Fibrosis. N Engl J Med. 2012 May 24;366(21):1968–77.
5. Raghu G, Rochwerg B, Zhang Y, Garcia CAC, Azuma A, Behr J, et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015 Jul 15;192(2):e3–19.
6. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. The Lancet. 2011 May;377(9779):1760–9.
7. King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N Engl J Med. 2014 May 29;370(22):2083–92.
8. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N Engl J Med. 2014 May 29;370(22):2071–82.
9. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018 Sep 1;198(5):e44–68.
10. Wells AU, Brown KK, Flaherty KR, Kolb M, Thannickal VJ. What’s in a name? That which we call IPF, by any other name would act the same. Eur Respir J. 2018 May;51(5):1800692.
11. Behr, Jürgen et al. Pharmakotherapie der idiopathischen pulmonalen Fibrose (ein Update) und anderer progredienter pulmonaler Fibrosen, S2k-Leitlinie der Deutschen Gesellschaft für Pneumologie und Beatmungsmedizin e.V. [Internet]. AWMF online; 2022. Available from: https://register.awmf.org/assets/guidelines/020-025l_S2k_Idiopathische-Lungenfibrose-Update-medikamentoese-Therapie_2022-11.pdf
12. Flaherty KR, Wells AU, Cottin V, Devaraj A, Inoue Y, Richeldi L, et al. Nintedanib in progressive interstitial lung diseases: data from the whole INBUILD trial. Eur Respir J. 2022 Mar;59(3):2004538.
13. Ghazipura M, Mammen MJ, Herman DD, Hon SM, Bissell BD, Macrea M, et al. Nintedanib in Progressive Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. Ann Am Thorac Soc. 2022 Jun;19(6):1040–9.
14. Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. 2020 Feb;8(2):147–57.
15. Tzilas V, Tzouvelekis A, Ryu JH, Bouros D. 2022 update on clinical practice guidelines for idiopathic pulmonary fibrosis and progressive pulmonary fibrosis. Lancet Respir Med. 2022 Aug;10(8):729–31.
16. Sharp C, Adamali H, Millar AB. Ambulatory and short-burst oxygen for interstitial lung disease. Cochrane Airways Group, editor. Cochrane Database Syst Rev [Internet]. 2016 Jul 6 [cited 2023 Apr 3];2016(7). Available from: http://doi.wiley.com/10.1002/14651858.CD011716.pub2
17. Waxman A, Restrepo-Jaramillo R, Thenappan T, Ravichandran A, Engel P, Bajwa A, et al. Inhaled Treprostinil in Pulmonary Hypertension Due to Interstitial Lung Disease. N Engl J Med. 2021 Jan 28;384(4):325–34.
18. Dowman L, Hill CJ, May A, Holland AE. Pulmonary rehabilitation for interstitial lung disease. Cochrane Airways Group, editor. Cochrane Database Syst Rev [Internet]. 2021 Feb 1 [cited 2023 Apr 3];2021(2). Available from: http://doi.wiley.com/10.100 2/14651858.CD006322.pub4
19. Kreuter M, Bendstrup E, Russell AM, Bajwah S, Lindell K, Adir Y, et al. Palliative care in interstitial lung disease: living well. Lancet Respir Med. 2017 Dec;5(12):968–80.
20. Müller C, Mattig N, Geiser TK, Guler SA. Progressiv fibrosierende interstitielle Pneumopathien. Swiss Med Forum – Schweiz Med-Forum [Internet]. 2021 Mar 16 [cited 2023 Apr 3]; Available from: https://doi.emh.ch/smf.2021.08694