Einleitung
Seit 1998 existiert in der Schweiz ein Weiterbildungsprogramm in Hausarztmedizin, welches in den Hausarztpraxen vermittelt wird (1). Zuerst als Pilotprojekt vom Kollegium für Hausarztmedizin (KHM) und den Fachgesellschaften der Schweizerischen Gesellschaft für Allgemeinmedizin (SGAM), der Schweizerischen Gesellschaft für Innere Medizin (SGIM), der Schweizerischen Gesellschaft für Pädiatrie (SGP) und dem Verband Schweizerischer Assistenz- und Oberärztinnen und -ärzte (VSAO) initiiert, entstand umgehend ein Praxisassistenzprogramm, welches anfänglich beim KHM angesiedelt wurde und 2009 in die Stiftung zur Förderung der Weiterbildung in Hausarztmedizin überführt wurde (2). Anlässlich des 15-jährigen Bestehens der Stiftung WHM FMF wird in dieser Publikation die Funktion der Stiftung WHM in der haus- und kinderärztlichen Weiterbildung vorgestellt. Es folgt eine Übersicht über die Entstehung der Praxisassistenz und die aktuellen kantonalen Weiterbildungsprogramme sowie eine Diskussion ausgewählter Aspekte der haus- und kinderärztlichen Weiterbildung. Ein abschliessender Ausblick nennt potenzielle Anpassungen und deren Auswirkungen für die haus- und kinderärztliche Weiterbildung, die im Zuge der Veränderungen im schweizerischen Gesundheitswesen notwendig werden können.
Methodik
Zur Erhebung der aktuellen Daten über das Angebot an mitfinanzierten Praxisassistenzarztstellen sowie zu den Hausarzt-Curricula wurden von Frühjahr bis Herbst 2024 die kantonalen Programmkoordinatorinnen und -koordinatoren via E-Mail oder Telefon durch die Geschäftsstelle der Stiftung WHM befragt. Die Daten zu den Lehrärztekursen stammen aus der Befragung von insg. 184 Kursteilnehmenden von insg. acht Lehrarztkursen zwischen März und Dezember 2023.
1. Die Stiftung WHM FMF
Die Geschichte der Stiftung WHM FMF beginnt 1998, als die FMH, die SGAM, die SGP, das KHM sowie der VSAO ein Pilotprojekt zur Weiterbildung in Hausarztpraxen entwickelten, um die genuinen Lerninhalte und Kompetenzen der Hausarztmedizin im Setting der Hausarztpraxis zu vermitteln (1). Mit dem Format der Praxisassistenz wurde ein strukturiertes Weiterbildungsprogramm aufgebaut und umgesetzt, welches auf den damals gültigen medizindidaktischen Erkenntnissen basierte. Die steigende Nachfrage nach Praxisassistenzarztstellen und der damit verbundene zunehmende administrative Aufwand machten die Überführung des Programms «Praxisassistenz KHM» in eine eigenständige Stiftung erforderlich. Dies führte 2008 zur Gründung der Stiftung WHM FMF, welche 2009 ihre Tätigkeit aufnahm (2). Zu den Gründungsmitgliedern der Stiftung WHM FMF gehören die FMH, die SGAM – nachfolgend die SGAIM, die SGP (heute Pädiatrie Schweiz), das KHM und der VSAO. Seit Dezember 2024 sind sowohl die FMH als auch das SIWF jeweils mit einem eigenen Sitz im Stiftungsrat vertreten. Bis 2007 war das Kollegium für Hausarztmedizin (KHM) die einzige Institution, welche eine strukturierte Weiterbildung in Haus- und Kinderarztpraxen im Rahmen der Praxisassistenz anbot.
Positionierung der Stiftung WHM FMF
Die Stiftung bietet folgende Dienstleistungen an (3):
– Aus- und Fortbildung für Lehrärztinnen und Lehrärzte AIM und KJM sowie für Ärztinnen und Ärzte anderer Fachrichtungen, die eine strukturierte Weiterbildung in der Praxis anbieten möchten
– Subsidiäre Mitfinanzierung von Assistenzarztstellen in der Praxis
– Angebot von HR-Dienstleistungen für Assistenzarztstellen in der Praxis (Dienstleistung für Kantone und selbst zahlende Praxen)
– Kursangebot spezifisch für Bedürfnisse von Praxisassistenzärztinnen und Praxisassistenzärzten
– Qualitätssicherung (Evaluation) der Praxisweiterbildungsstätten
– Stellenplattform für Weiterbildungsstellen in der Praxis
– Wissenschaftliche Tätigkeit zur Weiterbildung in der Praxis
Die Stiftung WHM und ihr gesundheitspolitisches Umfeld
Ohne Kenntnis der Veränderungen im gesundheitspolitischen Umfeld der medizinischen Grundversorgung, welche mit der Hausärztedemonstration 2006 in Bern ihren Anfang nahmen und 2014 mit der Verankerung der medizinischen Grundversorgung in der Bundesverfassung endeten, ist die Entwicklung der Weiterbildung in der Praxisassistenz nicht zu verstehen (4). Der drohende Mangel an Hausärztinnen und Hausärzten, welcher auf die zunehmende Unzufriedenheit der Ärzteschaft, den fehlenden Nachwuchs in der Hausarztmedizin sowie auf die fehlende akademische Verankerung der Hausarztmedizin an den medizinischen Fakultäten der Universitäten zurückgeführt wurde, setzte politische Massnahmen in Gang. Im Jahr 2009 gründeten die Fachgesellschaften der Allgemeinmediziner, Internisten und Pädiater den Berufsverband «Hausärzte Schweiz» und lancierten im selben Jahr die Volksinitiative «Ja zur Hausarztmedizin» (5). An den Universitäten kam es zur Einrichtung von Hausarztprofessuren und Hausarztinstituten (6). Der Bund reagierte 2012 auf die Forderungen der Hausärzteschaft mit dem Einsetzen des Masterplans, welcher die kantonalen Weiterbildungsprogramme entscheidend förderte (7). Verschiedene Kantone entwickelten ihre eigenen Praxisassistenzprogramme, wobei die Programmstruktur der Stiftung WHM vielen als Vorlage diente. Am Prozess der Entwicklung und der Koordinierung der kantonalen Programme war die Stiftung zusammen mit der Konferenz der kantonalen Gesundheitsdirektorinnen und -direktoren (GDK) und dem Bundesamt für Gesundheit (BAG) wesentlich mitbeteiligt und hat über die Entwicklung der hausärztlichen Weiterbildung in der Schweiz 2014 und 2019 einen Bericht verfasst (8, 9). Seit 2013 dient die Stiftung als Koordinations- und Austauschplattform und nimmt eine Drehscheibenrolle im schweizerischen Netzwerk «Praxisassistenz» ein. Sie unterstützt zudem die Kantone bei der Weiterentwicklung ihrer Praxisassistenzprogramme.
Die Stiftung WHM FMF und die hausärztliche Weiterbildung
Weiterbildungsprogramm AIM
Im Hinblick auf den Zusammenschluss der Schweizerischen Gesellschaft für Allgemeinmedizin (SGAM) und der Schweizerischen Gesellschaft für Innere Medizin (SGIM) zur Schweizerischen Gesellschaft für Allgemeine Innere Medizin SGAIM (2015) musste ein neues Weiterbildungsprogramm entwickelt werden, welches zum neu geschaffenen Facharzttitel Fachärztin/Facharzt für Allgemeine Innere Medizin führte. Dieses Weiterbildungsprogramm berücksichtigt die Laufbahn zur Hausärztin/zum Hausarzt sowie diejenige zur Spitalinternistin/zum Spitalinternist. Die Praxisassistenz wurde neu als mögliche ambulante Weiterbildung in die Basisweiterbildung und in die curriculare Aufbauweiterbildung implementiert. Dabei wurde die Struktur des Praxisassistenzprogramms der Stiftung WHM in das Weiterbildungsprogramm AIM übernommen (10). Mit dieser Gewichtung der Praxisassistenz innerhalb des Weiterbildungsprogramms AIM gewann die hausärztliche Weiterbildung an Attraktivität, was die Nachfrage nach hausärztlichen Weiterbildungsstellen weiter steigerte.
Aus- und Fortbildung von Lehrärztinnen und Lehrärzten – Lehrarztkurse
Seit 1998 bildet die Stiftung Lehrärztinnen und Lehrärzte in medizindidaktischen Kursen für die Lehre in den Praxen aus. Die Lehrinhalte und Lehrformate werden kontinuierlich den geltenden Standards angepasst und weiterentwickelt. Die medizinische Aus- und Weiterbildung hat sich mit der Einführung der kompetenzbasierten medizinischen Lehre (Competency-based Medical Education, CBME) wesentlich verändert. Die Stiftung WHM FMF integrierte neue, den Prinzipien der CBME entsprechende Lerninhalte in die Lehre, um eine optimale und praxisnahe Vorbereitung auf die Lehrtätigkeit in der Praxis sicherzustellen (11, 12).
Für die Anerkennung einer Arztpraxis als Weiterbildungsstätte wird vom SIWF die Absolvierung des Lehrarztkurses der Stiftung WHM FMF vorausgesetzt. Die Lehrarztkurse werden in Deutsch, Französisch und seit 2024 auch in Italienisch angeboten. Seit 2019 bietet die Stiftung Refresher-Kurse an, welche als «Teach the Teacher»-Kurse an den Kongressen der Schweizerischen Gesellschaft für Allgemeine Innere Medizin (SGAIM) und des Kollegiums für Hausarztmedizin (KHM) durchgeführt werden. In diesen Refresher-Kursen werden die aktuellen didaktischen Erneuerungen den «bestandenen» Lehrärztinnen und Lehrärzten vorgestellt. Sie bieten zudem die Gelegenheit zum gegenseitigen kollegialen Erfahrungsaustausch. Die Aus- und Fortbildung von Lehrärztinnen und Lehrärzten ist eine zentrale Dienstleistung der Stiftung, welche in den letzten Jahren in qualitativer wie auch quantitativer Hinsicht an Bedeutung zugenommen hat.
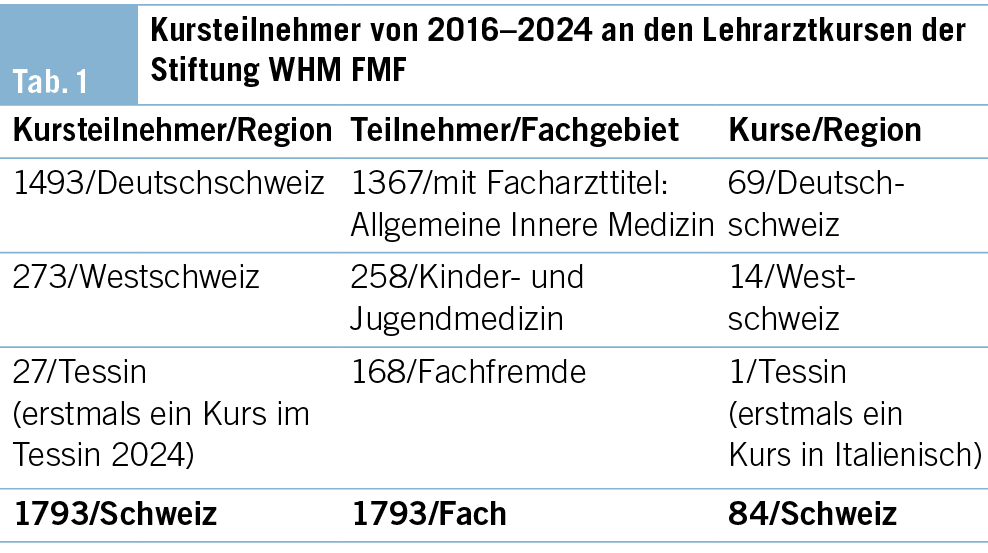
Kurswesen Lehrärztinnen und Lehrärzte 2016–2024 (Tab. 1)
Von 2016–2021 (in 5 Jahren) wurden 46 Kurse, in den Jahren 2022–2024 (in nur 3 Jahren) 38 Kurse durchgeführt, was doch eine bemerkenswerte Dynamik im Kurswesen erkennen lässt und das grosse Interesse und die Bedeutung des Kursangebots unterstreicht.
Die Praxisassistenz
Das Praxisassistenzprogramm der Stiftung WHM FMF
Das Praxisassistenzprogramm der Stiftung, welches pro Jahr rund 15 Praxisassistenzarztstellen umfasst, finanziert seine Weiterbildungsstellen im Sinne einer Mitfinanzierung subsidiär; insbesondere dann, wenn die Kapazität des jeweiligen kantonalen Programms ausgeschöpft ist oder formale Gründe die Nutzung eines kantonalen Programms ausschliessen. Es steht auch den Ärztinnen und Ärzten der Kinder- und Jugendmedizin zur Verfügung, welche nicht in allen kantonalen Programmen berücksichtigt werden (vgl. unten.) Das Programm der Stiftung wird erfreulicherweise jährlich ausgeschöpft. Zudem bietet die Stiftung ihre HR-Dienstleistungen für die Praxisassistenz Kantonen und Praxen an, welche keine Mitfinanzierung erhalten (3).
2. Praxisassistenz und Curriculaweiterbildung (Rotationsstellen) in der Schweiz
Kantonale Programme – Praxisassistenzprogramme
PA-Stellen für Allgemeine Innere Medizin
Die Praxisassistenz als hausärztliches Weiterbildungsformat in den Praxen ist schweizweit erfolgreich etabliert. Alle Kantone verfügen über ein kantonales Programm (13).
Anfang 2025 stehen in der Schweiz 303 kantonale Praxisassistenzarztstellen zur Verfügung. Mit den Praxisassistenzstellen der Stiftung WHM FMF können insgesamt 318 mitfinanzierte Stellen angeboten werden. Dabei handelt es sich um Stellen zu 100 % Arbeitszeit bei einer Anstellung von 6 Monaten. Teilzeitarbeit mit längerer Praxisassistenzdauer wird in allen Programmen vergeben. Die Finanzierung der Programme ist kantonal unterschiedlich. Grösstenteils werden die Kosten zwischen Kanton und der Lehrpraxis nach kantonal verschiedenen Vorgaben aufgeteilt; selten beteiligen sich auch Spitäler an der Finanzierung (61 % der Kosten entfallen auf die Kantone, 31 % auf die Lehrarztpraxen und 8 % auf die Spitäler) (13). In 24 von 26 Kantonen wird ein Bruttolohn entsprechend der Weiterbildungszeit, also analog der Entlöhnung im Spital, ausbezahlt. 2 Kantone kennen einen fixen Lohn (GE, LU) (13).
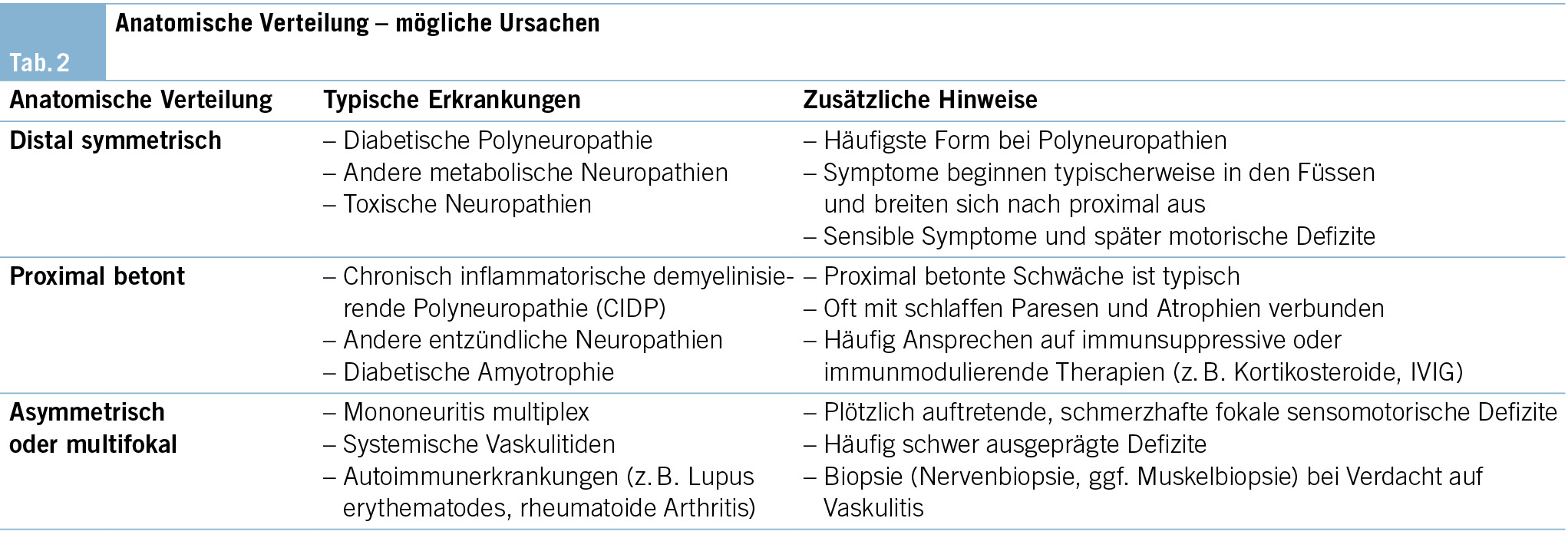
Die kantonalen Programme werden von kantonalen Programmkoordinatorinnen und -koordinatoren geleitet. Die Organisation sowie die Verantwortlichkeiten und Aufgaben der Koordinatorinnen und Koordinatoren sind kantonal unterschiedlich geregelt (Tab. 2).
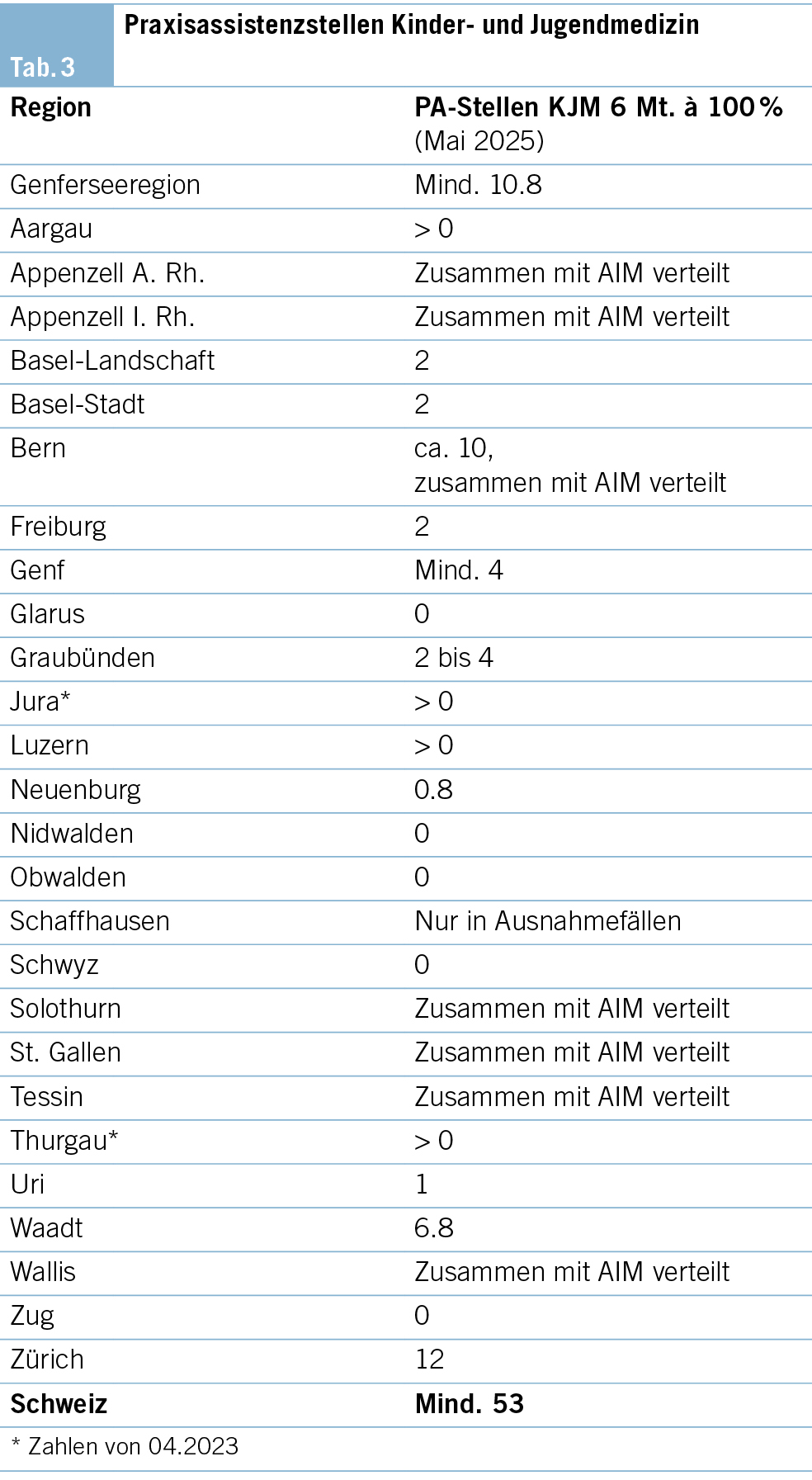
PA-Stellen für Kinder- und Jugendmedizin
Das Angebot an Praxisassistenzarztstellen für KJM ist von Kanton zu Kanton verschieden:
– 12 Kantone bieten keine Praxisassistenzstellen für Assistenzärztinnen und Assistenzärzte mit dem Ziel Kinder- und Jugendmedizin an.
– 8 Kantone stellen aus ihrem Praxisassistenzangebot eine definierte Anzahl Praxisassistenzstellen für die Kinder- und Jugendmedizin (KJM) zur Verfügung.
– 6 Kantone offerieren ihr Praxisassistenzangebot für die Allgemeine Innere Medizin(AIM)- wie für KJM-Assistenzärztinnen und -ärzte gleichermassen.
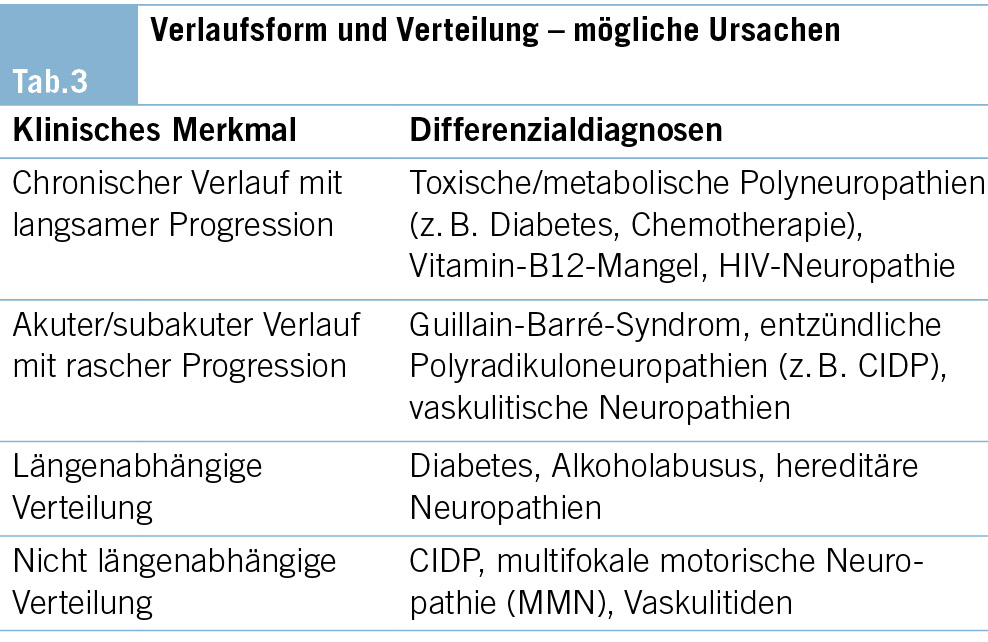
– 1 Kanton vergibt die Stelle der KJM, wenn keine AIM- Teilnehmenden gefunden werden (Tab. 3).
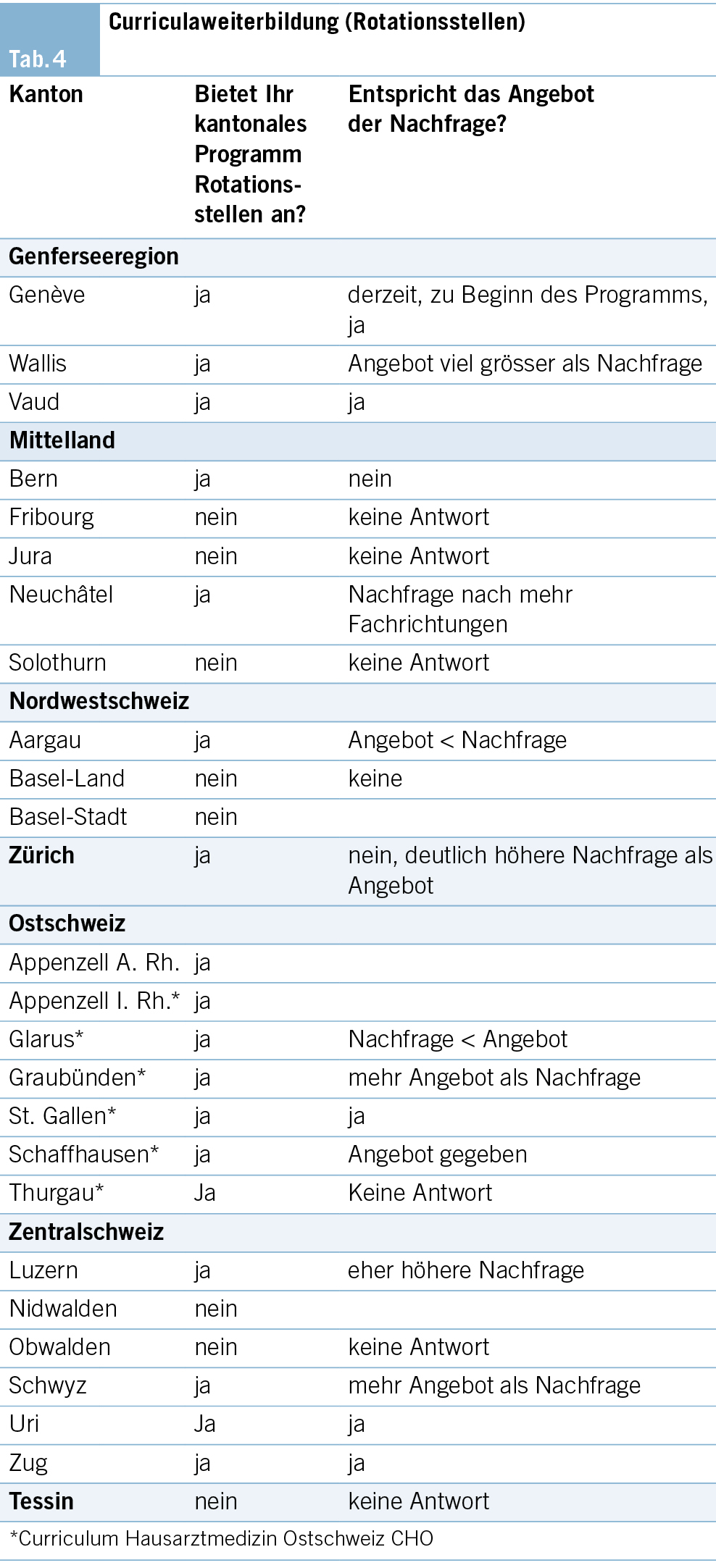
Kantonale Programme: Curriculaweiterbildung (Rotationsstellen)
Diese Weiterbildungsstellen bieten eine, auf die spätere Tätigkeit in der Hausarztpraxis ausgerichtete Weiterbildung in den Fachgebieten ausserhalb der Allgemeinen Inneren Medizin an (z. B. Chirurgie/Orthopädie, Psychiatrie, Pädiatrie, Dermatologie und andere). Sie fokussieren ihre praktischen Lerninhalte auf das Vermitteln der Kompetenzen im jeweiligen Fachgebiet, welche für eine eigenverantwortliche Arbeit in der Grundversorgung notwendig sind. Die curriculare Weiterbildung ist für den Erhalt der Breitenkompetenz in der Grundversorgung zentral (14, 15).
18 Kantone bieten Rotationsstellen mit unterschiedlichen Konzepten an (AG, AI, AR, BE, GE, GL, GR, LU, NE, SG, SH, SZ, TG, UR, VD, VS, ZG, ZH). 8 Kantone verfügen über kein Angebot.
4 Kantone bieten die Stellen für 6 Monate zu 100 %, 2 Kantone für 6–12 Monate an. 3 Kantone kennen keine definierte Dauer der Anstellung. 9 Kantone stellen maximal 18 Monate zu 100 % zur Verfügung.
16 Kantone geben an, eine strukturierte Weiterbildung anzubieten. 10 Kantone erlauben eine modulare Belegung dieser Angebote.
Die Kantone AR, AI, Glarus, Graubünden, Schaffhausen, St. Gallen und Thurgau bieten unter dem Namen «Curriculum Hausarztmedizin Ostschweiz CHO» ihre Praxisassistenzprogramme sowie die Rotationsstellen gemeinsam an (Tab. 4).
Evaluation der Weiterbildungsstätten
Die Praxisassistenz, vormals des KHM und seit 2009 der Stiftung WHM FMF, wird seit 1998 evaluiert (1, 5). Die wissenschaftliche Auswertung und Begleitung geschehen durch das Institut für Medizinische Lehre (IML) der Universität Bern. Die Evaluation der Weiterbildungsstätten, also der Lehrpraxen, wird in den kantonalen Programmen unterschiedlich gehandhabt. 16 Kantone evaluieren ihre Weiterbildung; davon lassen 9 Kantone ihre Evaluation von der Stiftung WHM FMF durchführen. In 5 Kantonen evaluiert die Koordinationsstelle selbst, und in 2 Kantonen ist das jeweilige Hausarztinstitut dafür verantwortlich. 4 Kantone lassen gelegentlich evaluieren, 4 Kantone kennen keine Evaluation, und von 2 weiteren Kantonen liegen keine Informationen vor.
Diskussion
Lehrärztinnen- und Lehrärztekurse der Stiftung WHM FMF
Die Ausbildung einer ausreichenden Zahl qualifizierter Lehrärztinnen und Lehrärzte für die vom SIWF anerkannte Weiterbildung im Rahmen der Praxisassistenz ist eine zentrale Voraussetzung für die Nachhaltigkeit des Weiterbildungsprogramms. Erfreulicherweise hat die Zahl der Kursteilnehmenden in den letzten Jahren deutlich zugenommen: Während zwischen 2016 und 2021 insgesamt 981 Personen an den Kursen teilnahmen, wurden in den drei Folgejahren (2022–2024) bereits 812 Teilnehmende registriert. Parallel dazu stieg auch das Kursangebot: Von 2016 bis 2021 wurden 46 Kurse durchgeführt, von 2022 bis 2024 waren es bereits 38 Kurse. Diese dynamische Entwicklung ist sehr positiv und verdeutlicht das starke, freiwillige Engagement der Hausärztinnen und Hausärzte für die Lehre. Zwar stellt die finanzielle Förderung der Praxisassistenz einen wichtigen Anreiz dar und erleichtert potenzielle Praxisnachfolgeregelungen, doch besonders hervorzuheben ist, dass zunehmend Ärztinnen und Ärzte an den Kursen teilnehmen, die selbst eine Praxisassistenz absolviert haben. So zeigte eine Befragung von Kursteilnehmenden der Lehrarztkurse im Zeitraum März bis Dezember 2023, dass mehr als die Hälfte der Teilnehmenden selbst eine Praxisassistenz während ihrer Weiterbildung absolviert hatte. Dies deutet darauf hin, dass die eigene positive Erfahrung mit der Praxisassistenz und die Freude an der Lehre selbst zur Lehrtätigkeit motivieren.
Praxisassistenz
Insgesamt stehen zurzeit 318 mitfinanzierte Praxisassistenzstellen zur Verfügung, was wiederum einen Anstieg der verfügbaren Weiterbildungsstellen bedeutet (13).
In der Kinder- und Jugendmedizin, welche auch zur Grundversorgung gehört, sind die verfügbaren Angebote an Praxisassistenzstellen deutlich geringer. Lediglich 14 Kantone beziehen die pädiatrische Weiterbildung in ihre Praxisassistenzprogramme mit ein. Und selbst in diesen Programmen wird die KJM nicht überall paritätisch berücksichtigt. Das ist bei dem aktuellen Mangel an Pädiaterinnen und Pädiatern in der medizinischen Grundversorgung nur schwer verständlich.
Die Beantwortung der Frage nach der Auslastung der Programme lässt sich für eine Jahresperiode meist nicht eindeutig beantworten: Sie unterliegt einer natürlichen Fluktuation, die schwerlich gesteuert werden kann. In kleinen Kantonen ist eine gleichmässige Auslastung ihrer Programme noch schwieriger zu erreichen, da die Anzahl der infrage kommenden Teilnehmenden oft klein ist. Zudem werden Programme immer wieder weiterentwickelt und verändert, sodass ein objektiver Vergleich der Programme schwierig ist. Zurzeit befinden sich 4 Kantone im Stadium der Weiterentwicklung.
Nach Angabe von 5 Kantonen ist das Angebot an Praxisassistenzstellen grösser als die Nachfrage, in 4 Kantonen übersteigt die Nachfrage das Angebot, und in 11 Kantonen sind das Angebot und die Nachfrage ausgeglichen. Von 2 Kantonen fehlen Daten.
Rotationsstellen – curriculare Weiterbildung
Die Notwendigkeit einer qualitativen Weiterbildung in Fachgebieten ausserhalb der Allgemeinen Inneren Medizin ist unbestritten, und eine solide hausärztliche Breitenkompetenz ist vor allem für eine ärztliche Tätigkeit in ländlichen Gebieten unabdingbar. Diese Breitenkompetenz muss daher mit einem adäquaten Weiterbildungsprogramm gesichert werden (14, 15). Die Angebote, die Finanzierungen, die Strukturen und allfällige Zulassungsbedingungen zur curricularen Weiterbildung sowie die Zuständigkeit für diese Programme unterscheiden sich häufig von Kanton zu Kanton.
Gut strukturierte Angebote weisen die Kantone AG, BE, GE, LU mit NW, OW, UR, ZH und die Ostschweizer Kantone AI, AR, GL, GR, SH, SG, TG mit dem Curriculum Hausarztmedizin Ostschweiz CHO auf.
Im Gegensatz zu den Praxisassistenzstellen, bei welchen die Zuständigkeit und Verantwortung für das Programm die kantonalen Koordinatoren innehaben, sind es bei den Rotationsstellen oft auch Kliniken, welche eigene Rotationsstellen in ihrem Fachbereich anbieten und selbst verwalten. Aus Rückmeldungen von gewissen Kantonen erfährt man, dass ihre Curricula-Angebote aus diversen Gründen (organisatorischer und finanzieller Art) noch nicht verbindlich implementiert sind und diese daher auch noch nicht ihre volle Wirksamkeit entfalten können. Vor allem in kleineren Kantonen sind curriculare Angebote, wenn überhaupt, nur mit beträchtlichem Aufwand möglich und können nur eine begrenzte Wirkung entfalten.
Kantone wie Luzern mit Obwalden und Nidwalden sowie das Curriculum Ostschweiz CHO (19) mit den oben genannten Ostschweizer Kantonen zeigen mit ihrem Vorgehen Wege zu einer sinnvollen Zusammenarbeit auf. Schon wiederholt wurde darauf hingewiesen, dass die ausgeprägte Heterogenität, welche der föderalen Struktur des Schweizer Gesundheitssystems geschuldet ist, der optimalen Nutzung der kantonalen Programme nicht sehr förderlich ist. Eine regionale Zusammenarbeit mit kleineren Kantonen verbessert das Angebot der hausärztlichen Weiterbildung auch in ländlichen Gebieten und Randregionen und kann so mithelfen, den Einstieg in eine hausärztliche Tätigkeit in diesen Gegenden zu fördern.
Die Evaluation der Praxisassistenz und der Rotationsstellen
Die Evaluation stellt ein elementares Instrument der Qualitätssicherung dar. Das Prozedere der Evaluation von Praxisassistenzstellen wird vom Schweizerischen Institut für Weiter- und Fortbildung (SIWF) festgelegt.
Während die Evaluation der Weiterbildungsstätten in der stationären Weiterbildung eine Selbstverständlichkeit ist, ist sie bei einer Weiterbildung im ambulanten Bereich einer Arztpraxis bis heute nicht verpflichtend. Die freiwillig durchgeführten Evaluationen von Praxisassistenzstellen unterscheiden sich oft erheblich in ihrer Qualität und genügen den Ansprüchen an eine korrekt durchgeführte und wissenschaftlich ausgewertete Evaluation nicht immer. Eine Evaluation der Praxisassistenzstellen soll grundsätzlich analog der stationären Weiterbildung, aber praktikabel und am speziellen Setting der Praxis angepasst, eingesetzt werden. Es sollten aber nicht nur die mitfinanzierten Praxisassistenzstellen der kantonalen Programme evaluiert werden – einige von ihnen lassen die Evaluation bei der Stiftung WHM durchführen oder aber sie evaluieren selbst –, sondern auch alle jene, welche an keinem Programm angeschlossen sind. Die Befragung von 184 Kursteilnehmenden an den Lehrarztkursen von März bis Dezember 2024 hat ergeben, dass 46 % der Lehrärztinnen und Lehrärzte ihre Praxisassistenz ohne Hilfe eines Praxisassistenzprogramms selbst finanzieren. Wenn man das Praxissetting kennt, in dem die befragten Ärztinnen und Ärzte arbeiten, und weiss, dass 84 % in einer Gruppen- oder Gemeinschaftspraxis tätig sind, versteht man, dass in diesen Praxisformen eine direkte Praxisassistenzanstellung häufig ist. Diese Praxisassistenzstellen, deren Anzahl nicht bekannt ist, werden bis jetzt nicht evaluiert.
Die kantonalen Koordinatorinnen und Koordinatoren
Je nach Organisation und Umfang der kantonalen Programme ist deren Einsatz unterschiedlich. So organisieren beispielsweise 10 Kantone freiwillig einen regelmässigen Austausch mit ihren Lehrärztinnen und Lehrärzten und/oder führen Visitationen bei ihren Lehrpraxen durch. Dies zeigt, dass schon jetzt einige Programme um eine gewisse Qualitätssicherung besorgt sind. Die Stiftung WHM lädt jährlich zu einem Treffen der kantonalen Koordinatorinnen und Koordinatoren ein, welches dem Informationsaustausch sowie der Diskussion von relevanten Themen der hausärztlichen Weiterbildung dient.
Ausblick
Bis jetzt wird die haus- und ärztliche Weiterbildung in einem 1:1-Setting Lehrärztin/Lehrarzt – Praxisassistenzärztin/Praxisassistenzarzt vermittelt, dem höchsten didaktischen Lehrformat. Beim SIWF ist die Lehrärztin/der Lehrarzt in der Praxisassistenz ad personam akkreditiert; dies im Unterschied zu den anderen Weiterbildungsstätten, bei denen die Institution akkreditiert wird. Früher waren die Lehrarztpraxen in der Regel Einzelpraxen oder kleinere Gruppenpraxen. Das hat sich grundlegend geändert: Lehrpraxen sind heute überwiegend Gruppen- oder Gemeinschaftspraxen, Einzelpraxen sind die Ausnahme. Lehrärztinnen und Lehrärzte arbeiten mehrheitlich nicht mehr in einem 100 %-Arbeitspensum; die Praxisassistenzärztinnen- und -ärzte ebenfalls nicht. Grössere Praxiseinheiten können für die Praxisassistenz eine vermehrte Flexibilität der Arbeitszeit anbieten, was die Attraktivität solcher Stellen steigert. Angehende Hausärztinnen und Hausärzte bevorzugen für ihre Weiterbildung kleinere Spitäler und Praxen (17). Nicht wenige Landspitäler, welche diese Weiterbildungsmöglichkeit angeboten haben, sind im Zuge von kantonalen Sparprogrammen und Reorganisationen aufgehoben worden oder sind in ihrer Existenz gefährdet. Universitätskliniken und grosse Zentrumsspitäler bieten wohl eine breite Palette von Weiterbildungsstellen an. Diese sind aber aufgrund des selektionierten Patientenguts und der hochgradigen Spezialisierung vielfach für eine längerfristige Weiterbildung mit dem Ziel Hausärztin/Hausarzt weniger geeignet (15). Eine Ausnahme dürften die Kliniken und Abteilungen für Allgemeine Innere Medizin in den Spitälern darstellen, welche ein etwas weniger selektioniertes Patientengut aufweisen und für die stationäre Weiterbildung zur Hausarztmedizin geeignet sind. Eine weitere Verlagerung der hausärztlichen Weiterbildung in den ambulanten Bereich ist zu erwarten. Dies bedeutet, dass das Weiterbildungsprogramm AIM für den Track Hausarztmedizin sowie die Praxisweiterbildungsstätten den veränderten Gegebenheiten strukturell angepasst werden müssen. Zudem ist es für die Weiterbildung in der Hausarztpraxis existenziell, dass deren Finanzierung weiterhin gesichert bleibt. Nicht nur in der Weiterbildung zur Hausärztin/zum Hausarzt ist der Trend von stationär zu ambulant festzustellen; auch in anderen Fachdisziplinen wird diese Verlagerung zu beobachten sein. Somit wird klar, dass in Zukunft vermehrt Lehrärztinnen und Lehrärzte für die Lehre im ambulanten Bereich ausgebildet werden müssen.
Die verfügbaren Daten ermöglichen einen positiven Blick in die Zukunft: Die Praxisassistenz trägt nicht nur zur Sicherung der medizinischen Grundversorgung der Schweiz bei, sie sichert auch das Fortbestehen der haus- und kinderärztlichen Weiterbildung und ist zudem ein wirkungsvolles Instrument für eine nachhaltige Nachwuchsförderung (16). Dieses Erfolgsmodell mit seinem zukunftsweisenden Konzept ist weiterhin auf eine robuste finanzielle Unterstützung angewiesen und benötigt auch künftig ein klares politisches Commitment.
Christian Häuptle, Henrik Zimmermann, Sarina Keller, Réka Veress-Daugaard
Stiftung zur Förderung der Weiterbildung in Hausarztmedizin WHM FMF, Bern
Otmarweg 8, 9200 Gossau
haeuptle@hin.ch
Die Autorenschaft hat keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Schläppi P. Lerngewinn durch Praxisassistenz. Schweizerische Ärztezeitung 2000,81, N.36 1985-1993
2. Carobbio Guscetti M., von Erlach M. Stiftung zur Förderung der Weiterbildung in Hausarztmedizin Jahresbericht 2009. Primary Care 2011,11, N.1, 9-10
3. https://www.whm-fmf.ch Stiftung zur Förderung der Weiterbildung in Hausarztmedizin
4. https://www.hausärzteschweiz.ch Gesundheitspolitik
5. Stricker B., Die Geschichte der Volksinitiative «Ja zur Hausarztmedizin». Primary Care 2013, Edition 3-5
6. Gesundheit B.f. Bestandesaufnahme Institute für Hausarztmedizin. Bundesamt für Gesundheit 2013
7. Gesundheit B.f. Faktenblatt Masterplan und medizinische Grundversorgung 2013
8. Häuptle C., von Erlach M., Bauer W., Brinkley B. Koordination von Curricula (Rotationsstellen) und Praxisassistenzstellen. Praxis (Bern 1994) 2015;104(3):137-50
9. Häuptle C., von Erlach M., Weiterbildung in Hausarztmedizin: Praxisassistenz und Curricula- weiter-bildung (Rotationsstellen) in der Schweiz. Praxis (Bern1994).2019;108(1):63-72
10. https://www.siwf.ch/weiterbildung.cfm Schweizerisches Institut für ärztliche Weiter-und Fortbildung
11. Albermann K., Frick S., Grünig P., Meienberg A. “Bin ich eine gute Ärztin? Bin ich ein guter Arzt?“ Schweizerische Ärztezeitung 2022; 103 (08) 238-41
12. https://www.siwf.ch siwf-projekte cbme.cfm
13. Gerber T., Häuptle C., Denti F., Graf., Merlo C., Pasche O., Rodondi N., Rosemann T., Senn N., Som-mer J., Zeller A. Praxisassistenz in der Schweiz: eine Übersicht in den Kantonen. Prim Hosp Care2022; Allg Inn Medizin.22 (11) 331-334
14. Häuptle C. Weiterbildung zur Hausärztin und zum Hausarzt im Kanton St. Gallen. Prim Hosp Care 2019; Allg Inn Medizin.2022; 22(11) 331-334
15. Plate A., Di Gangi S., Pichierri G., Rosemann T., Senn O. Praxisassistenz und Curriculum: Bedeutung für den Nachwuchs in der Grundversorgung im Kanton Zürich. Praxis 2025; 114 (1) 11-15
16. Zimmerli L., Fluri M., Droste P., Cina C., Leupold F., Streit S., Erfolgreiche Nachwuchsförderung. Schweizerische Ärztezeitung 2020; 101; 948-9.
17. Rozsnayi Z., Tal.K., Bachofner M., et al. Swiss students and young physicians want a flexible goal-orientet PG training curriculum. Scan J Prim Health Care.2018 ; 36(3) 249-61
18. Felber S. Praxisassistenz:»» …die lehrreichste Zeit in meiner Ausbildung». Schweizerische Ärztezei-tung 2005,86, N19 1147-53
19. www.curriculum-ostschweiz.ch