Anamnese und Befunde
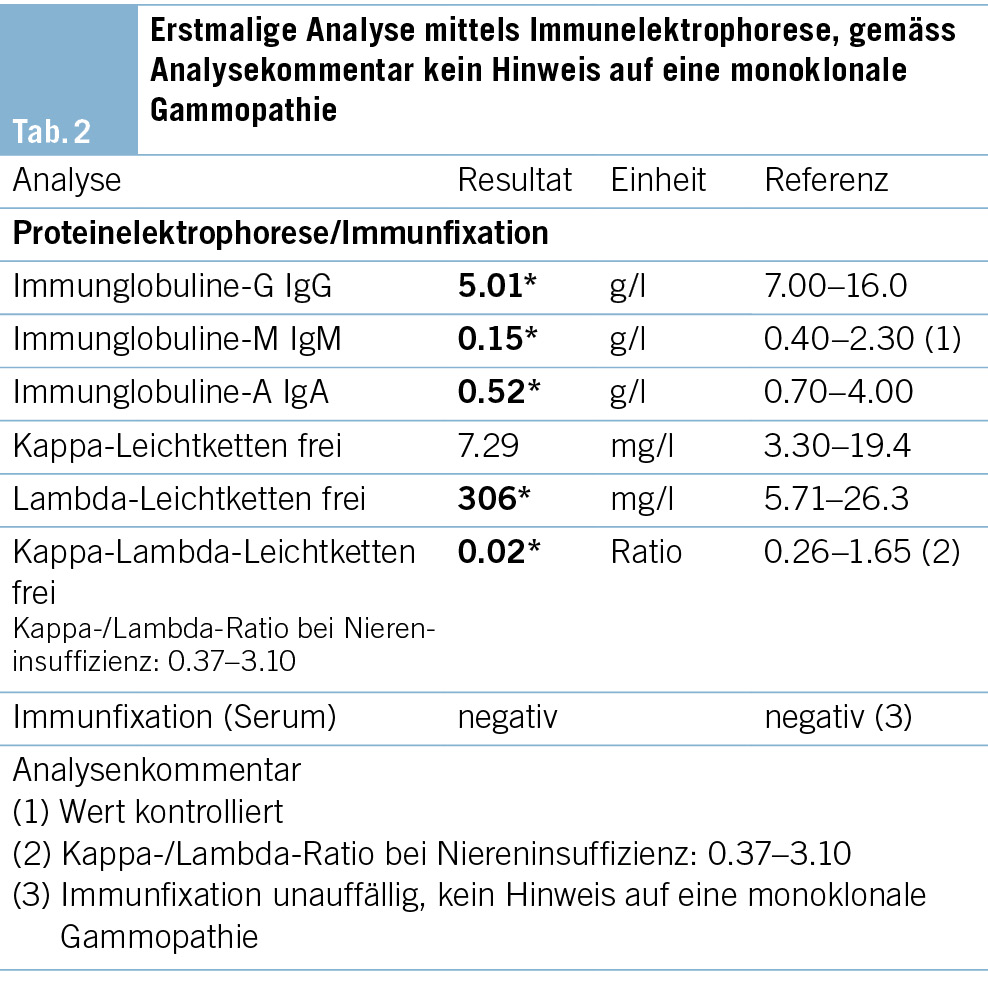
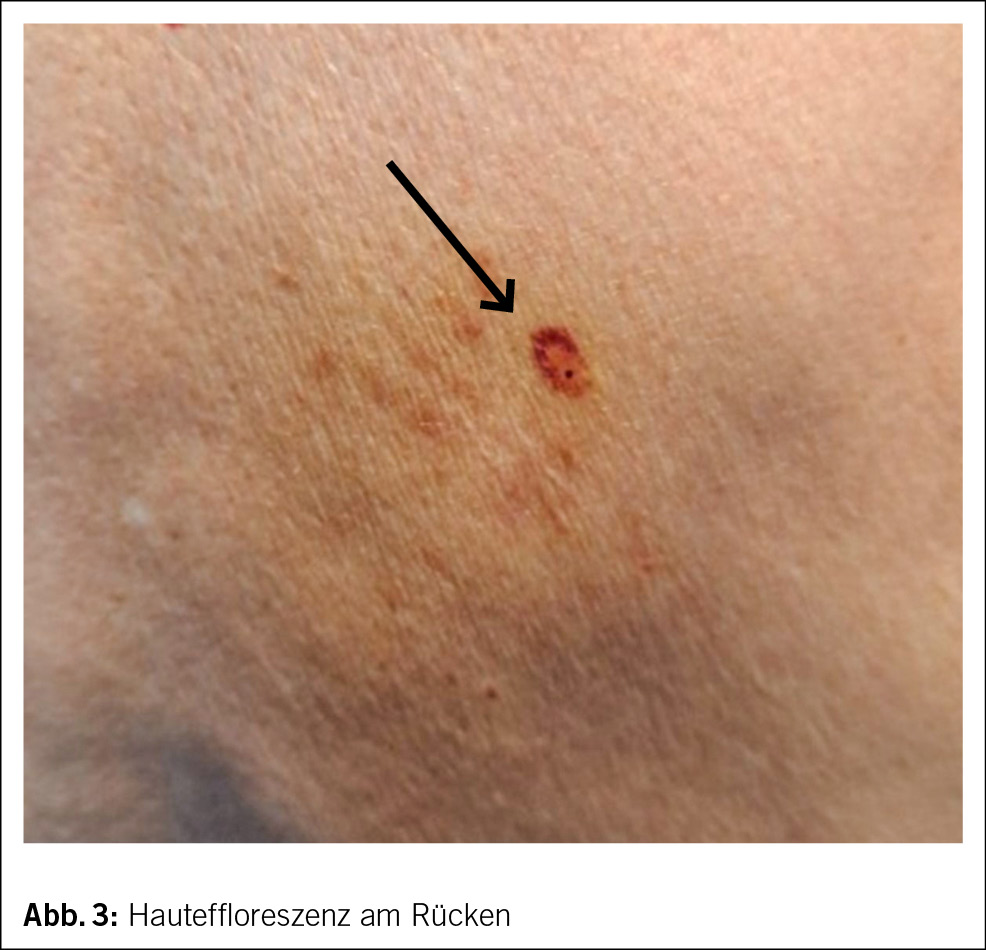
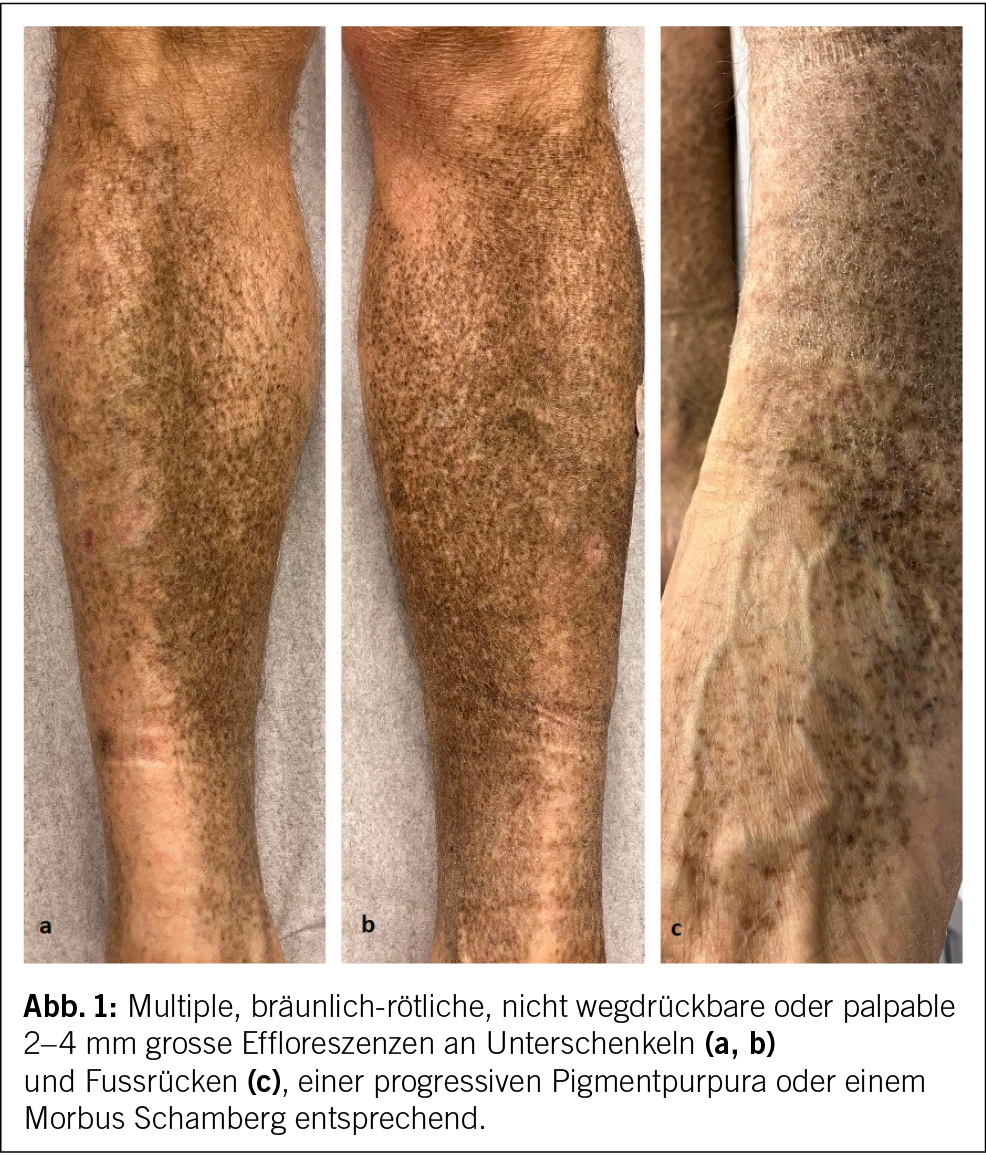
Ein 62-jähriger Patient stellt sich mit seit 2 Jahren bestehendem trockenen Reizhusten, Gewichtsverlust von 10 kg und vermehrter Müdigkeit, ohne wesentliche Einschränkung in seinen Aktivitäten, beim Hausarzt vor. Weitere B-Symptome werden verneint. Der Patient ist therapeutisch antikoaguliert und in regelmässiger kardiologischer Kontrolle, da 13 Jahre zuvor aufgrund eines Sinus-Valsalvae-Aneurysmas mit schwerer Aortenklappeninsuffizienz ein Aortenwurzelersatz mit mechanischer Doppelflügelprothese erfolgte. Der Patient hat eine feste Lebenspartnerin und geht einer Bürotätigkeit nach. Als Freizeitbeschäftigung (Mountainbiking) hält er sich häufig im Wald auf und ist als Hobbyimker tätig. Als Haustier hält er eine Katze. Ein Zeckenstich ist nicht erinnerlich. Der Patient wurde bei oben beschriebenen Symptomen und den in Tab. 1 gezeigten Blutbild-/Laborveränderungen mit leichter Thrombopenie, stark erhöhter Blutsenkungsreaktion bei fast normalem CRP, leichter Hepatopathie und Paraproteinämie in die hämatologische Sprechstunde zugewiesen. Er präsentierte sich in gutem Allgemeinzustand mit normalen Vitalparametern. Klinisch fanden sich an den distalen unteren Extremitäten Effloreszenzen, passend zu einer Purpura pigmentosa progressiva (Abb. 1), anamnestisch seit 2 Jahren bestehend, welche vorgehend durch einen Dermatologen klinisch und bioptisch abgeklärt wurde. Ansonsten keine weiteren pathologischen wegweisenden Befunde.
Differenzialdiagnostische Überlegungen und Weg zur Diagnose
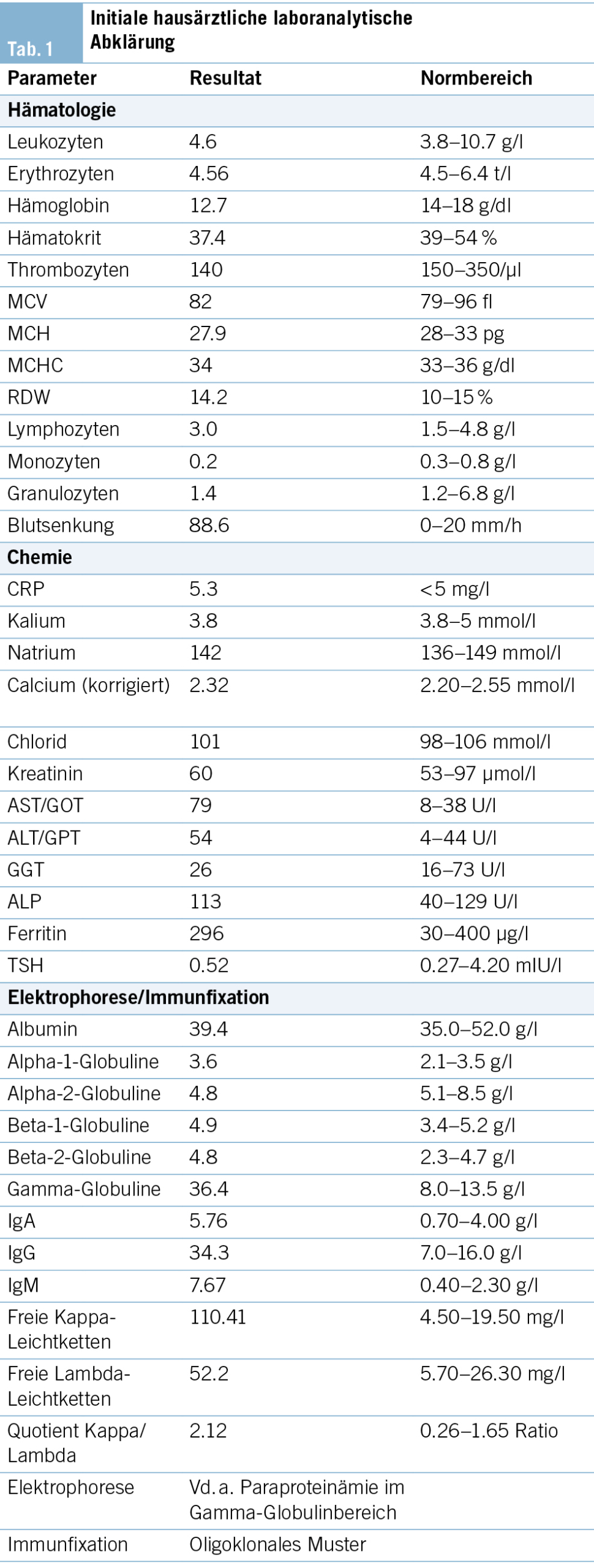
Symptome wie Müdigkeit, Gewichtsverlust, Leistungseinschränkung sind unspezifisch. Neoplasien, chronische Infektionen, rheumatologische Erkrankungen und Depressionen u. a. können alle mit solchen Symptomen einhergehen. Bei chronischer Symptomatik und fehlendem Fieber wäre eine akute Infektion hier sehr unwahrscheinlich. Eine Hepatitis-B-, -C- oder HIV-Infektion konnte serologisch ausgeschlossen werden. Die Blutkulturen zeigten kein Erregerwachstum. Die Paraproteinämie stellte sich als oligoklonal heraus, was auf einen chronischen Infekt hinweisend sein kann. Die ANA und ANCA waren negativ, die Komplementfaktoren C3 und C4 waren normal. Bei negativen ANA in der Immunfluoreszenz wurde der Nachweis von dsDNA-Antikörper von 23 IU/ml (cut-off 15 IU/ml) als unspezifisch gewertet.
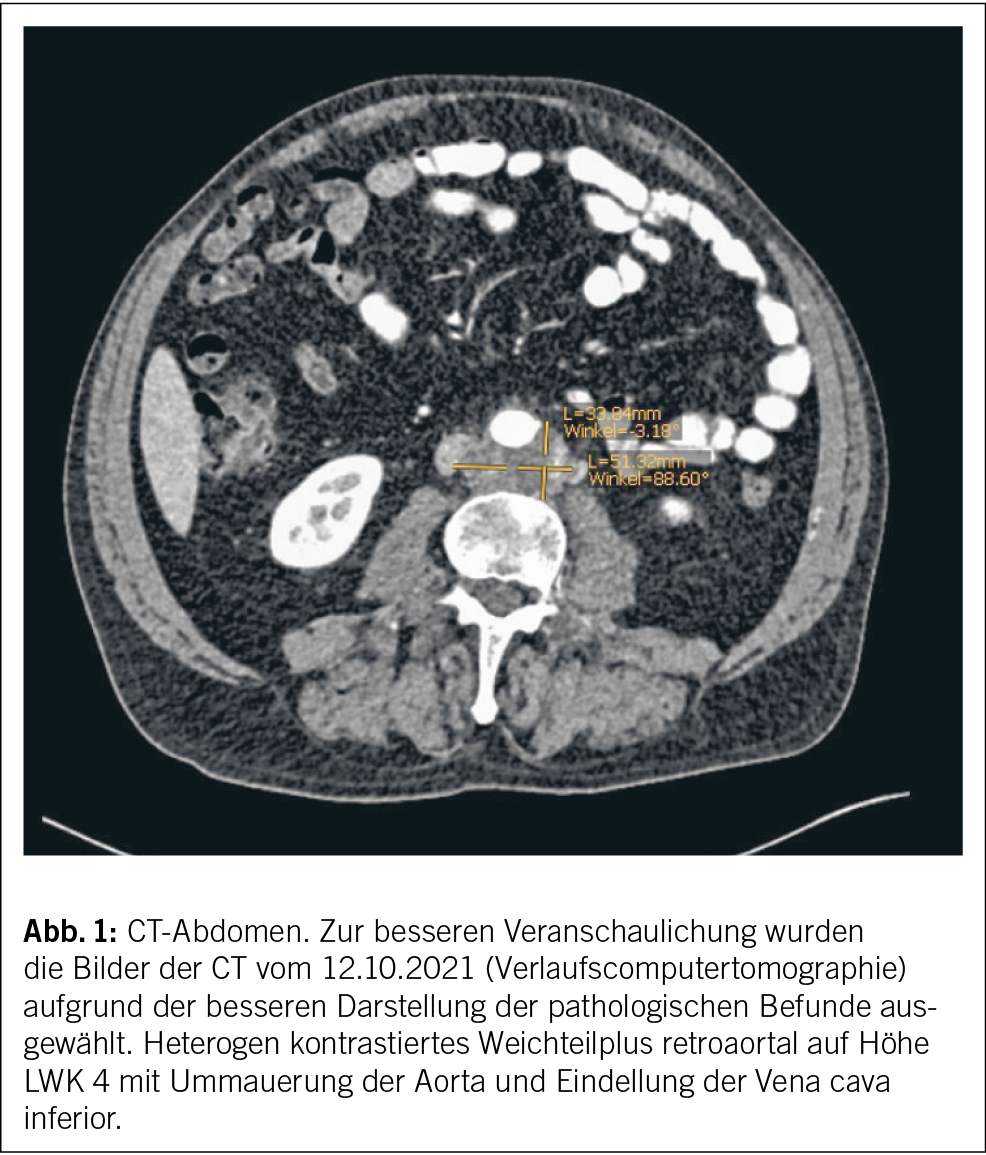
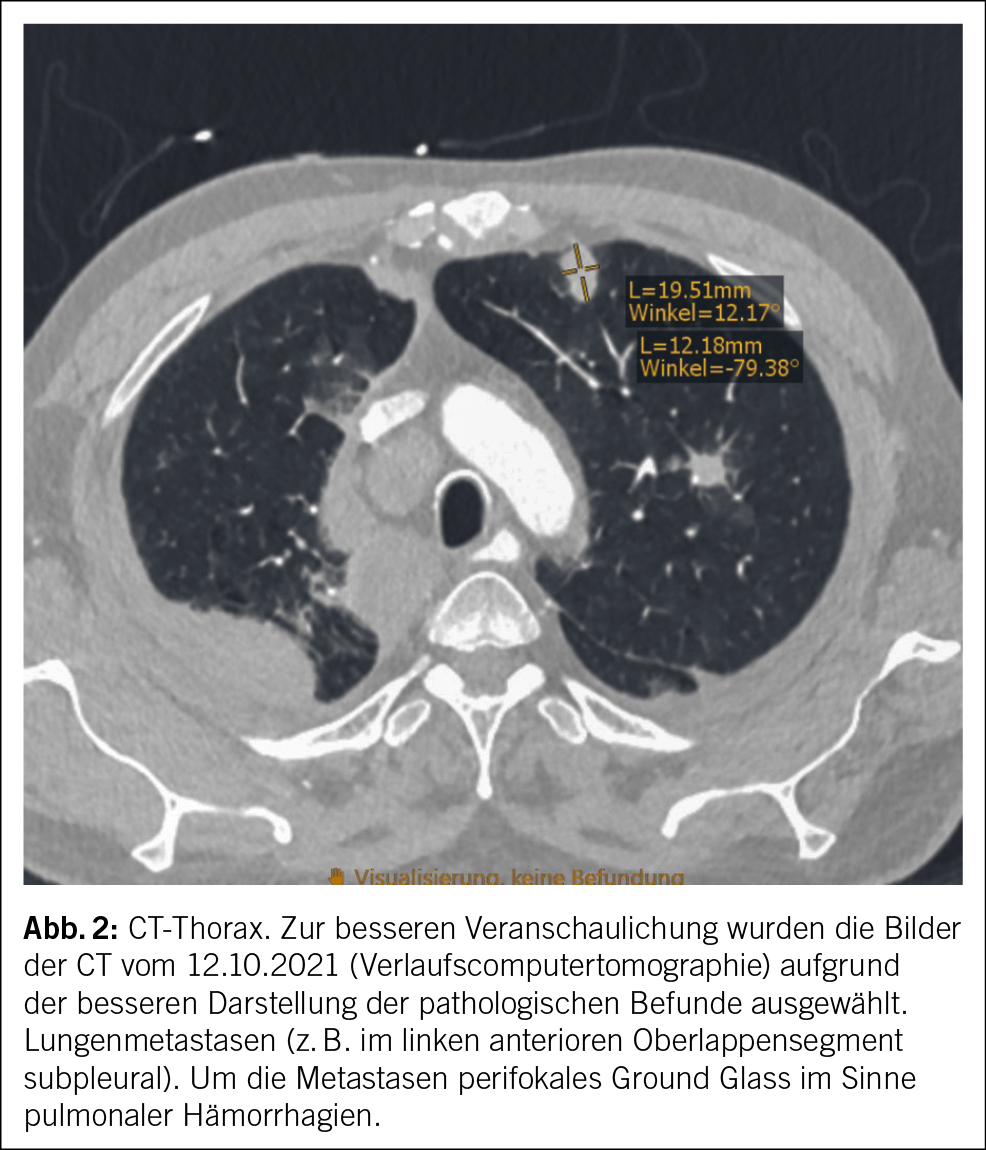
In der CT-Hals/-Thorax/-Abdomen zur Infektfokussuche wurden einschmelzende, pathologisch vergrösserte Lymphknoten mediastinal rechts von max. 3 cm und eine Splenomegalie (Poldistanz 15 cm) festgestellt. Der grösste mediastinale Lymphknoten lag direkt neben dem Aortengraft. Pulmonale Infiltrate, Raumforderungen oder Zeichen einer interstitiellen Pneumopathie zeigten sich nicht. In der bronchoskopischen Lymphknotenpunktion fanden sich zytologisch einzelne mehrkernige histiozytäre Riesenzellen. Eigentliche Granulome oder maligne Zellen wurden nicht festgestellt. Mikrobiologisch konnte im Lymphknotenpunktat wenig normale Mundflora nachgewiesen werden, einer Kontamination entsprechend. Mikroskopie, PCR und Kultur für Mycobakterien blieben negativ. Eine hiläre oder mediastinale Lymphknotenpunktion – oder falls diese nicht konklusiv, eine Lymphknotenexzision – dient der zytologischen/histologischen und mikrobiologischen Abklärung bei unklarer mediastinaler Lymphadenopathie, primär zur Abklärung hinsichtlich Neoplasie inklusive Lymphom, Tuberkulose oder Sarkoidose.
Eine PET-CT zeigte eine mediastinal konfluierende Läsion um den Graft der Aorta ascendens mit moderater Stoffwechselsteigerung sowie flau aktive, nicht vergrösserte Lymphknoten paratracheal rechts. Eine PET-CT ist für die Suche nach metabolisch aktiven Veränderungen zur Abklärung von möglichen Neoplasien und/oder Infektionen (inklusive Prothesen-Endokarditis bei negativer Echokardiographie) geeignet.
Es folgte eine thorakoskopische Exzision des grössten Lymphknotens, der intraoperativ zur Aorta deutlich adhärent war. Die Histologie zeigte eine ausgedehnte plasmazellreiche und fokal chronisch-granulierende und xanthomatöse Entzündung mit vielen mehrkernigen Riesenzellen im Bereich der Lymphknotenadhärenz an die Aorta (Abb. 2). Hinweise auf Malignität oder eine IgG4-assoziierte Erkrankung fanden sich nicht. Aufgrund der vorliegenden Befunde wurde die Verdachtsdiagnose einer chronischen Aortengraftinfektion gestellt.
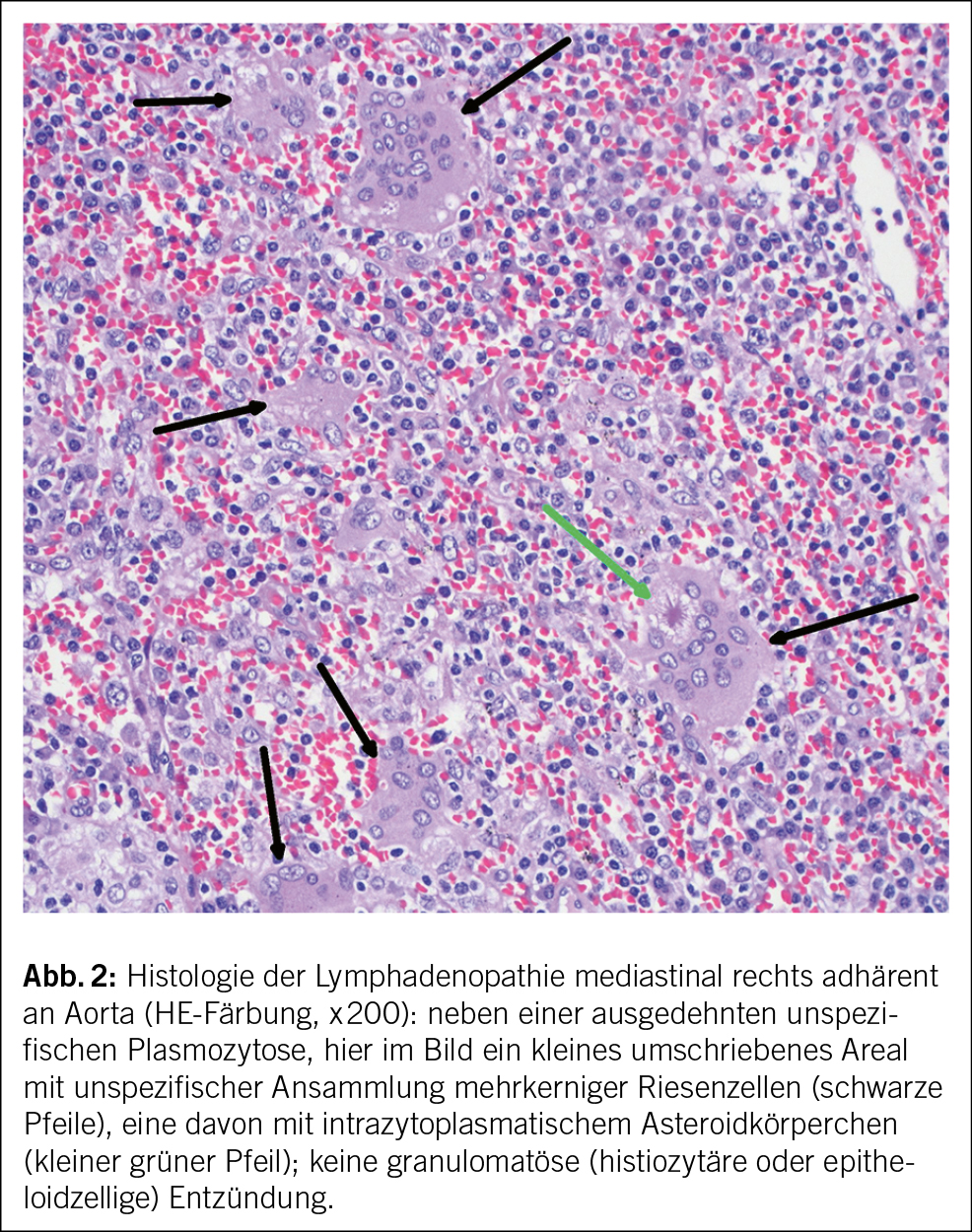
Gefässprotheseninfektionen werden überwiegend durch konventionell kultivierbare Erreger verursacht, zu ca. 80 % durch Staphylococcus aureus oder Koagulase-negative Staphylokokken. Enterokokken, Streptokokken, gramnegative Stäbchen (v. a. E. coli, Pseudomonas aeruginosa und Klebsiella spp.) und Pilze (v. a. Candida) sind deutlich seltener (1, 2, 3). Differenzialdiagnostisch muss bei «Kultur- negativen» Graftinfektionen an Infektionen durch Coxiella burnetii, Tropheryma whipplei, Bartonella henselae und (nicht tuberkulöse) Mycobacterien gedacht werden (4). Die häufigste Ursache für einen fehlenden kulturellen Erregernachweis ist eine vorgängige antibiotische Therapie. Eine empirische Antibiotikatherapie ist bei einem Patienten mit chronischen, unspezifischen Symptomen und ordentlichem Allgemeinzustand nicht gerechtfertigt und kann die weitere Erregersuche erheblich erschweren. Eine empirische Therapie sollte nur bei akuter und vital bedrohlicher Verschlechterung des Patientenzustandes in Betracht gezogen werden.
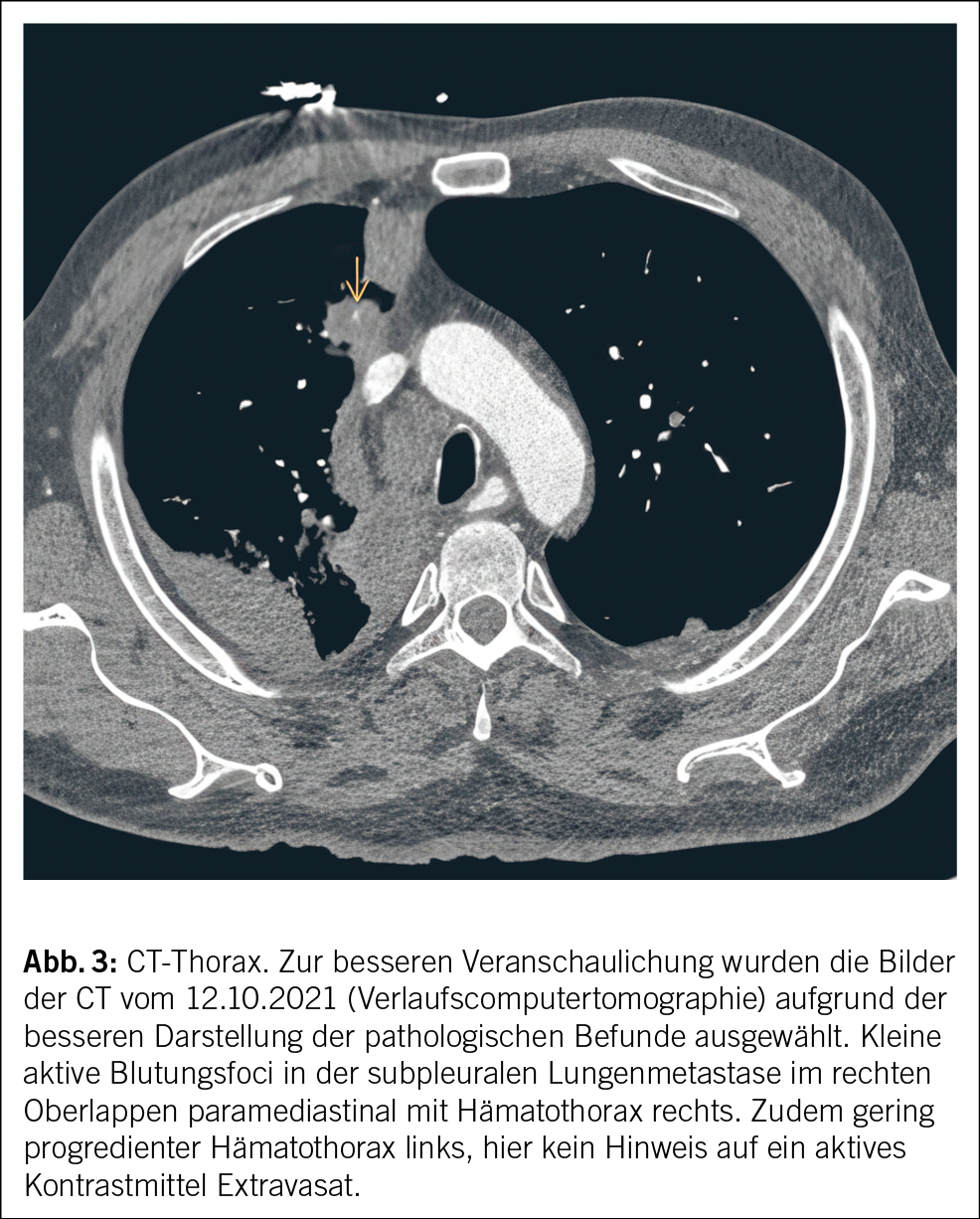
Eine genaue Expositionsanamnese kann helfen, die Wahrscheinlichkeit seltener Ursachen einer Graftinfektion einzugrenzen. Unser Patient besitzt eine Katze, sodass eine Infektion mit Bartonella henselae denkbar wäre. Auf gezieltes Nachfragen gibt er ausserdem an, dass er mehrmals pro Jahr in Südfrankreich in einem Ferienhaus verweile und gelegentlich lokale Rohmilchprodukte von einem Hofladen mit Schafen konsumiere. Damit besteht eine erhöhte Expositionswahrscheinlichkeit gegenüber Coxiella burnetii. Eine bakterielle Breitspektrum-PCR aus einem normalerweise sterilen Material kann bei hoher Erregerquantität zur Diagnose führen. Eine Breitspektrum-PCR ist aber deutlich weniger sensitiv als eine Erreger-spezifische PCR. Bei unserem Patienten war die bakterielle Breitspektrum-PCR sowie eine spezifische PCR für Bartonellen aus Lymphknotengewebe negativ. Serologien für Bartonella und Coxiella sind bei chronischen Infektionen aussagekräftig. Die Serologie auf Coxiella burnetii fiel stark positiv aus (Phase I IgG 1 : 262 144 [< 1 : 16 Titer] und Phase II IgG 1 : 262 144 [< 1 : 16 Titer]) und ist vereinbar mit einem chronischen Q-Fieber. Die Erreger-spezifische PCR für Coxiella burnetii aus dem thorakoskopisch entnommenen Lymphknotengewebe fiel positiv aus und beweist die aktive Graftinfektion durch diesen Erreger. Ein Quantiferon- oder Tspot-TB-Test ist hier wenig hilfreich, da diese weder einen Ausschluss einer aktiven Infektion mit Mycobacterium tuberculosis erlauben (nur als Test auf latente TBC geeignet) noch Infektionen mit nicht tuberkulösen Mycobakterien detektieren. Im Verlaufs-PET-CT nach 3 Monaten, noch vor Start der antibiotischen Therapie – in erster Linie mit Frage nach Progression der Infektion, Graftintegrität sowie Herzklappenbeteiligung –, war die metabolisch aktive mediastinale Weichteilmasse grössenprogredient (Abb. 3), und in der TEE erschien die Aortenwurzel verdickt und inhomogen aufgetrieben im Sinne einer beginnenden Abszedierung. Die Integrität des Grafts war erhalten und die Funktion der mechanischen Aortenklappenprothese einwandfrei. Zeichen einer Klappen-Endokarditis gab es keine.
Gemäss CDC-Kriterien (5) liess sich beim Patienten ein chronisches Q-Fieber diagnostizieren, bei bildgebend bestätigter Infektion des Aortengrafts, Nachweis von Coxiella burnetii mittels PCR aus dem histologischen Material aus dem periaortalen Infektionsherd sowie serologisch Nachweis eines Phase-I-IgG-Titers von ≥ 1 : 800 im IFA («Indirekter Immunfluoreszenz-Antikörper-Assay»).
Definitive Diagnose
Chronische Infektion des Aortengrafts durch Coxiella burnetii.
Therapie und Verlauf
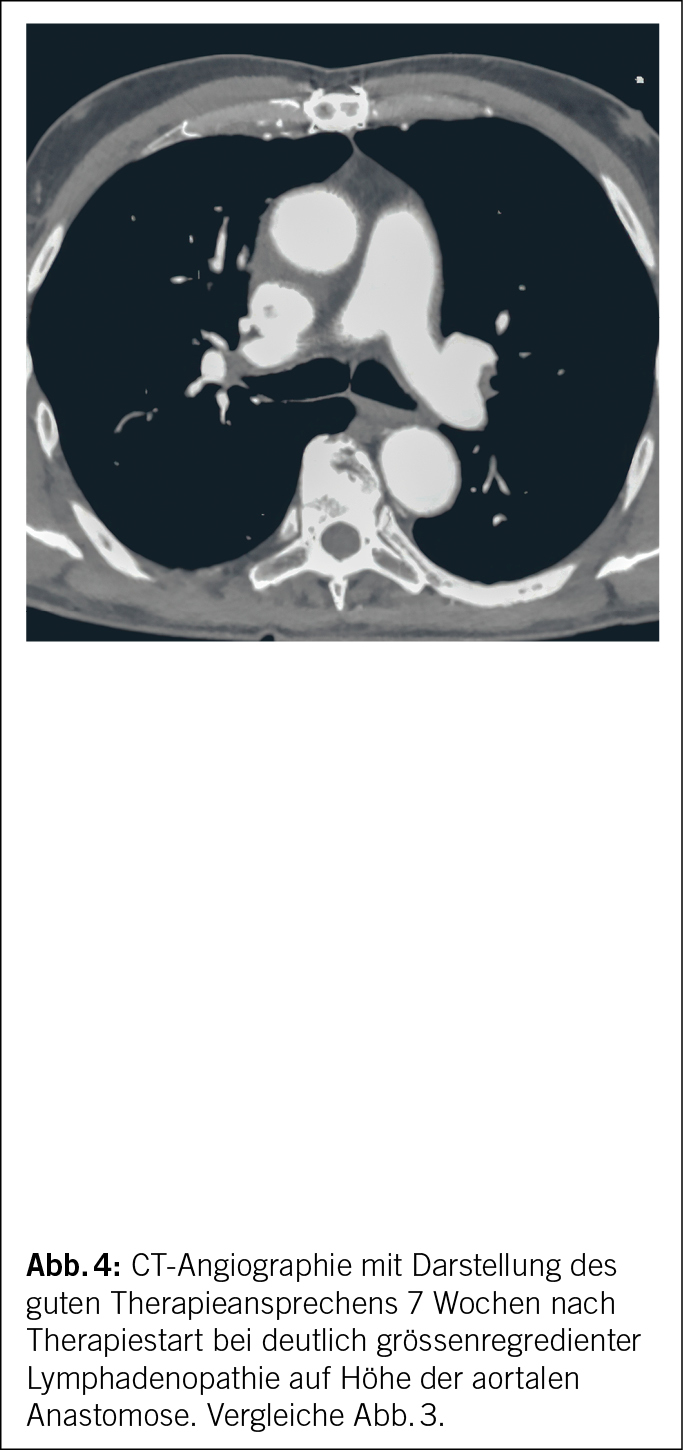
Die antibiotische Therapie der 1. Wahl des chronischen Q-Fiebers bei Erwachsenen ist eine Kombinationstherapie von Doxycyclin mit Hydroxychloroquin. Doxycyclin plus Chinolone wäre die Therapie der 2. Wahl (6). Eine operative Sanierung bei einwandfreier Aortenklappenfunktion und erhaltener Graftintegrität war bei unserem Patienten nicht indiziert. Eine symptomatische Therapie wäre nur bei akutem oligosymptomatischem Q-Fieber ohne Komplikationen beim ansonsten gesunden Patienten zu erwägen. Die mediastinale Lymphadenopathie zeigte sich 7 Wochen nach Therapiestart mit Doxycyclin plus Hydroxychloroquin grössenregredient (Abb. 4). Weiterhin normalisierten sich CRP, Blutsenkung und das periphere Blutbild.
Diskussion
Coxiella burnetii sind gramnegative kokkoide Stäbchen und Auslöser des Q-Fiebers, einer weltweit verbreiteten Zoonose. Als Wirte fungieren Rinder, Schafe, Ziegen, Hunde, Katzen, einige Wildtiere sowie Zecken. Beim Menschen wird eine Infektion mehrheitlich durch das Einatmen von erregerhaltigem Staub oder direkten Kontakt mit infizierten Tieren verursacht. Seltener durch Kontakt oder Konsum von kontaminierten Lebensmitteln wie Rohmilchprodukte oder durch den Stich infizierter Zecken (7). Beim Menschen genügen für die aerogene Infektion 1 bis 10 lebensfähige Organismen, die als Sporenbildner auch in schwierigen Umweltbedingungen (Kälte, Wärme, Trockenheit) teilweise jahrelang überleben. Die Erkrankung verläuft bei bis zu 50 % der infizierten Patienten asymptomatisch oder mit milden Symptomen. Akute Manifestationsarten sind atypische Pneumonien, Hepatitiden, Meningoenzephalitiden oder Endo-/Myokarditiden. Chronisches Q-Fieber kann in 1–5 % der Patienten symptomatischen oder asymptomatischen akuten Infektionen folgen und kann sich noch Jahre nach einer Ansteckung manifestieren (8). Patienten mit Herzklappenerkrankungen, Gefässprothesen oder arteriellen Aneurysmata sowie Immunsupprimierte und Schwangere haben ein erhöhtes Risiko für chronisches Q-Fieber. Infektionen von Gefässprothesen und Aneurysmata gelten mit 9 % als zweithäufigste Form des chronischen Q-Fiebers nach der Endokarditis mit 78 % (9).
Patienten mit Q-Fieber-Graftinfektionen zeigen häufig Symptome wie Fieber, Gewichtsverlust, Bauchschmerzen und Müdigkeit (9).
Bei Coxiella-Graftinfektionen wird eine antibiotische Therapie über 1.5–2 Jahre empfohlen (5). Coxiellen sind intrazelluläre Bakterien, womit die Immunantwort primär T-Zell-mediiert ist und Antikörper somit schlechte Marker sind. Daher kann zum Therapiemonitoring und dem Festlegen der definitiven Therapiedauer, ausser dem klinischen und radiologischen Ansprechen, die Negativierung der PCR im Serum herangezogen werden (10). Retrospektiv konnte Letzteres bei unserem Fall aus noch asservierten Serumproben gezeigt werden: Während die PCR auf
C. burnetii aus einer Serumprobe 4 Monate nach Therapiebeginn positiv ausfiel, zeigt sich die PCR in einer Serumprobe 11 Monate nach Therapiebeginn negativ.
Eine rechtzeitige Entfernung einer infizierten Gefässprothese vor deren weitgehenden Destruktion kann das Patientenüberleben verbessern (5, 9, 11). Die Mortalität korrekt behandelter vaskulärer Q-Fieber-Infektionen liegt bei 25–33 % (9).
Ob die beim Patienten vorliegenden Hautveränderungen an beiden Unterschenkeln im Zusammenhang stehen mit dem Q-Fieber, ist nicht auszuschliessen, zumal kutane Veränderungen zumindest bei akuten Coxiellen-Infektionen beschrieben sind (12). Unter Therapie ist es bis anhin zu keiner Regredienz der purpurischen Hautveränderungen gekommen.
Kyriakos Marinakis 1, Annegret Meyer-Hari 2, Corina Dommann-Scherrer 3, Urs Karrer 4, Adrian Schmid 4
1 Klinik für Innere Medizin, Zentrum für Allgemeine Innere Medizin, Kantonsspital Winterthur
2 Klinik für Medizinische Onkologie und Hämatologie, Kantonsspital Winterthur
3 Institut für Pathologie, Kantonsspital Winterthur
4 Fachbereich Infektiologie und Spitalhygiene, Zentrum für Allgemeine Innere Medizin, Kantonsspital Winterthur
Author Contributions Statement
Wir bestätigen, dass alle Autoren an der Konzeption und dem Design des Manuskripts oder der Analyse und Interpretation der Befunde des Patienten beteiligt waren und mit der Veröffentlichung einverstanden sind.
Historie
Manuskript eingegangen: 05.04.2024
Angenommen nach Revision: 24.03.2025
Verdankung
Die Autoren danken den Kolleginnen und Kollegen der Radiologie und Nuklearmedizin des Kantonsspitals Winterthur für die computertomographischen Aufnahmen und deren Befundung.
Kantonsspital Winterthur
Zentrum für Intensivmedizin
Brauerstrasse 15, Postfach
8401 Winterthur
medpol@ksw.ch
Die Autorenschaft hat keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Stone PA, Back MR, Armstrong PA, Brumberg RS, Flaherty SK, Johnson BL,et al. Evolving microbiology and treatment of extracavitary prosthetic graft infections. Vasc Endovascular Surg. 2008;42:537–44.
2. Mayer D, Hasse B, Koelliker J, Enzler M, Veith FJ, Rancic Z, et al. Long-term results of vascular graft and artery preserving treatment with negative pressure wound therapy in Szilagyi grade III infections justify a paradigm shift. Ann Surg. 2011;254:754–9; discussion 760.
3. FitzGerald SF, Kelly C, Humphreys H. Diagnosis and treatment of prosthetic aortic graft infections: confusion and inconsistency in the absence of evidence or consensus. J Antimicrob Chemother. 2005;56:996–9.
4. Marjan Wouthuyzen-Bakker et al. Diagnosis and treatment of vascular graft and endograft infections: a structured clinical approach. International Journal of Infectious Diseases 126 (2023) 22-27.
5. Alicia Anderson et al. Diagnosis and management of Q fever—United States, 2013: Recommendations from CDC and the Q Fever Working Group. MMWR Recomm Rep. 2013 Mar 29;62 (RR-03): 1-30.
6. Maurin M, Raoult DQ. Fever. Clin Microbiol Rev 1999;12:518–53). A combination regimen is necessary to eradicate the organism because hydroxychloroquine raises the pH in the acidified phagosomal compartment and, in combination with doxycycline, has been shown to have in vitro bactericidal activity against C. burnetii.
7. Derrick EQ. Q fever, a new fever entity: clinical features, diagnosis and laboratory investigation. Med J Aust 1937;2:281–99.
8. Fenollar F, Fournier PE, Carrieri MP, Habib G, Messana T, Raoult D. Risks factors and prevention of Q fever endocarditis. Clin Infect Dis 2001;33:312–6.
9. Botelho-Nevers E, Fournier PE, Richet H, et al. Coxiella burnetii infection of aortic aneurysms or vascular grafts: report of 30 new cases and evaluation of outcome. Eur J Clin Microbiol Infect Dis 2007;26:635–40.
10. Buijs et al, The prognostic value of serological titres for clinical outcomes during treatment and follow-up of patients with chronic Q fever. Clin Microbiol Infect 2021 27: 1273-1278
11. Wegdam-Blans MC, Ter Woorst JF, Klompenhouwer EG, Teijink JA. David procedure during a reoperation for ongoing chronic Q fever infection of an ascending aortic prosthesis. Eur J Cardiothorac Surg 2012;42:e19–20.
12. Natalí Uribe Pulido et al, Acute Q Fever With Dermatologic Manifestations, Molecular Diagnosis, and No Seroconversion, Open Forum Infectious Diseases, 2021 6;8(10):ofab458.