Anamnese
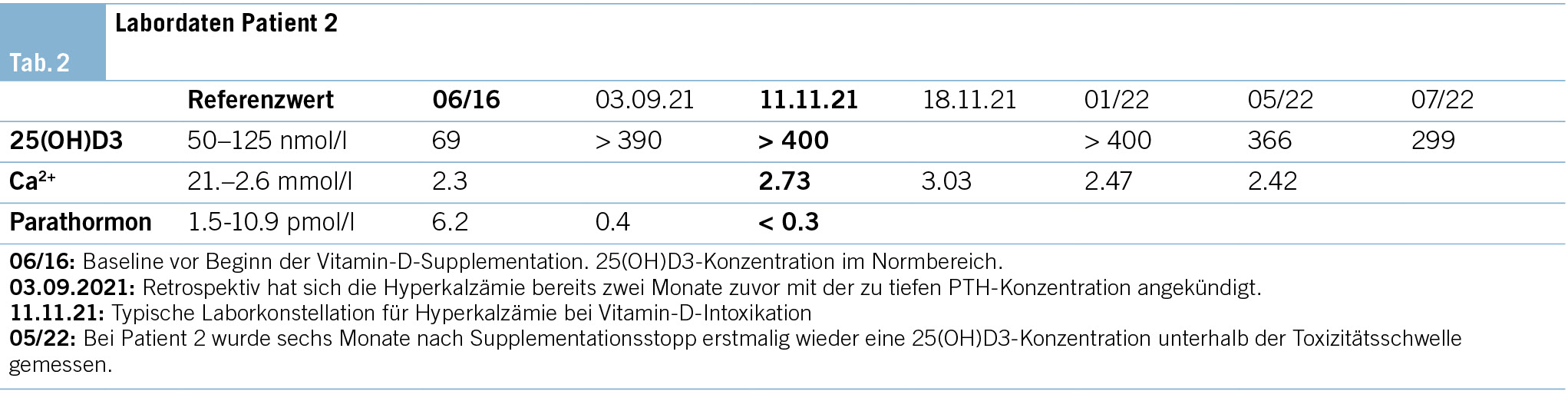
Ein 25-jähriger Patient stellt sich in der gastroenterologischen Sprechstunde vor, da er seit drei Wochen unter abendlichen Fieberepisoden mit Schüttelfrost leidet. Aufgefallen sei dem Patienten zudem eine vermehrte Müdigkeit. Vor allem morgens fühle er sich so erschöpft, dass er seinen Beruf als Elektriker nur noch schwer ausüben könne. Der Patient ist nicht hausärztlich angebunden. Er befindet sich fast drei Jahre in gastroenterologischer Betreuung aufgrund einer Therapie mit Infliximab im Rahmen einer Colitis ulcerosa, welche in Remission ist. Zu Nebenwirkungen der Behandlung sei es bisher nicht gekommen. Gastroenterologische Symptome verneint er ausdrücklich. Andere chronische Erkrankungen liegen nicht vor. Bezüglich der Reiseanamnese ist der Patient zwei Wochen, als die Symptome bereits bestanden, gemeinsam mit seiner Partnerin für 10 Tage nach Thailand gereist. Zuvor habe er sich nur in der Schweiz aufgehalten. Während des Auslandsaufenthaltes setzten sich die Fieberepisoden unverändert fort. Weitere Symptome präsentierten sich nicht. In der Umgebungsanamnese fand sich kein Hinweis auf andere erkrankte Personen oder eine mögliche Exposition. Der Sexualkontakt war ausschliesslich mit der langjährigen Partnerin. Im Haushalt sowie während des Urlaubes hatte der Patient keinen Tierkontakt.
Systemanamnese
Der Patient berichtet über einen leichten Gewichtsverlust von 2–3 kg bei einem BMI im unteren Normalbereich mit erhaltenem Appetit. Bei abendlichen Fieberepisoden gibt es keine weiteren Symptome wie Nachtschweiss, Atembeschwerden, Husten, Halsschmerzen, Bauchschmerzen, Durchfall, Hautveränderungen, Gelenkschmerzen oder neurologische Symptome. Auch kardiovaskulär und urogenital finden sich keine Auffälligkeiten. Hinweise auf einen Infektfokus liegen nicht vor. Die einzige Medikation in den letzten Monaten ist Infliximab (120 mg s.c., zweiwöchentlich) gewesen.
Körperliche Untersuchung
Der Patient ist schlank und befindet sich in gutem Allgemeinzustand. Die Haut zeigt ein unauffälliges Kolorit. Vergrösserte Lymphknoten sind nicht zu palpieren. Die Vitalparameter sind stabil. In der Auskultation des Herzens finden sich weder eine Tachykardie noch eine Arrhythmie bei unauffälligen Herztönen. Die Atemwege sind frei. Die Lunge ist beidseits vesikulär belüftet, ohne pathologische Atemgeräusche. Das Abdomen ist weich, ohne Abwehrspannung oder tastbare Organomegalie. Die Gelenke sind weder druckdolent noch überwärmt. Im neurologischen Status zeigt der Patient keine Auffälligkeiten. Bei der Untersuchung am frühen Nachmittag zeigt sich zudem eine normale Körpertemperatur bei anamnestisch abendlichen Fieberepisoden.
Laboruntersuchung
Die Laboruntersuchungen ergeben ein normwertiges Differenzialblutbild ohne erhöhte Entzündungswerte sowie unauffällige Leber- und Nierenwerte bei unveränderten Elektrolyten. Der Urinstatus ist unauffällig. Urin- und Blutkulturen führen zu keinem Erregernachweis. Serologische Tests bezüglich viraler Hepatitiden, HIV und eine Quantiferon-Testung liegen bereits im Rahmen des Screenings bei Infliximab-Therapie vor und wurden risikobasiert wiederholt.
Bildgebung
Ein Röntgen des Thorax und eine Abdomensonographie zeigen keinen Hinweis auf einen Infektfokus. Auf eine Computertomographie wird zunächst aufgrund der Strahlenbelastung beim jungen Patienten in gutem Allgemeinzustand verzichtet. Eine Echokardiographie wird bei fehlendem Anhalt auf eine kardiale Genese und wiederholt negativen Blutkulturen nicht durchgeführt.
Notfallmässige Vorstellung
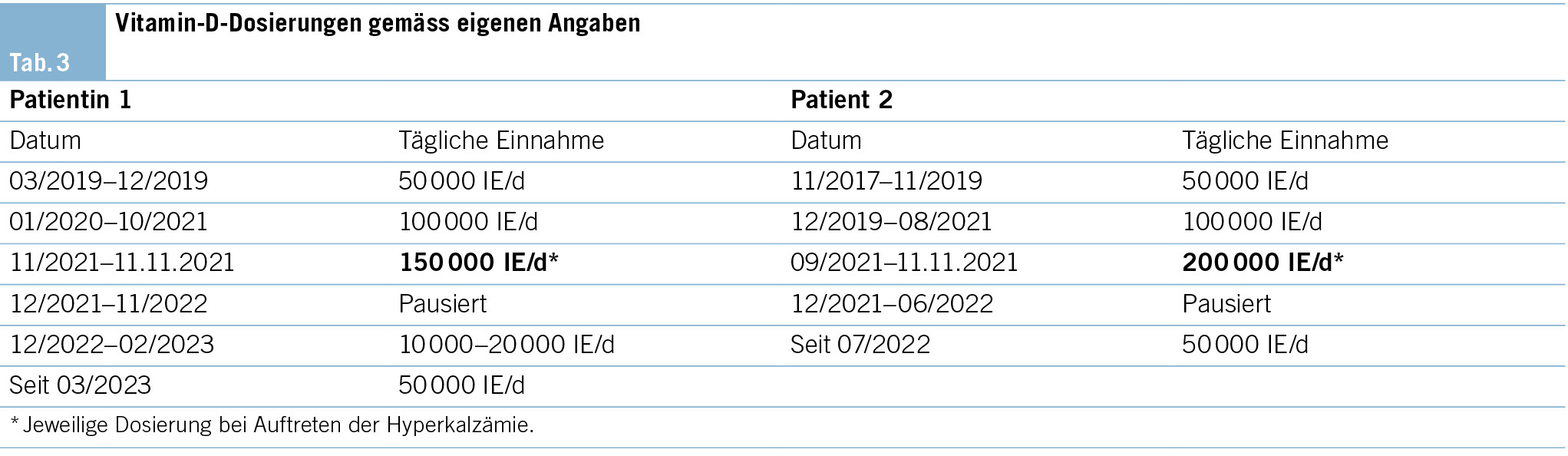
Eine erneute Vorstellung des Patienten in der gastroenterologischen Sprechstunde zur Besprechung der oben genannten Ergebnisse ist geplant, da weiterhin kein konkreter Hinweis auf die Ursache des Fiebers vorliegt. Kurz vor dem vorgesehenen Termin stellt sich der Patient jedoch notfallmässig auf der Notfallstation des angegliederten Spitals vor. Dabei berichtet er zusätzlich über Rückenschmerzen im Bereich der Brustwirbelsäule, die bis in den Hinterkopf und beide Schläfen ausstrahlen. Ein erinnerliches Trauma verneint der Patient. Die Kraft und Sensibilität sind unverändert und ohne Auffälligkeiten. Darüber hinaus klagt der Patient über eine seit Kurzem postprandiale Übelkeit nach jeder Nahrungsaufnahme. Ob diese Beschwerden im Zusammenhang mit den bisherigen Fieberepisoden stehen, bleibt unklar.
Differenzialdiagnostische Überlegungen
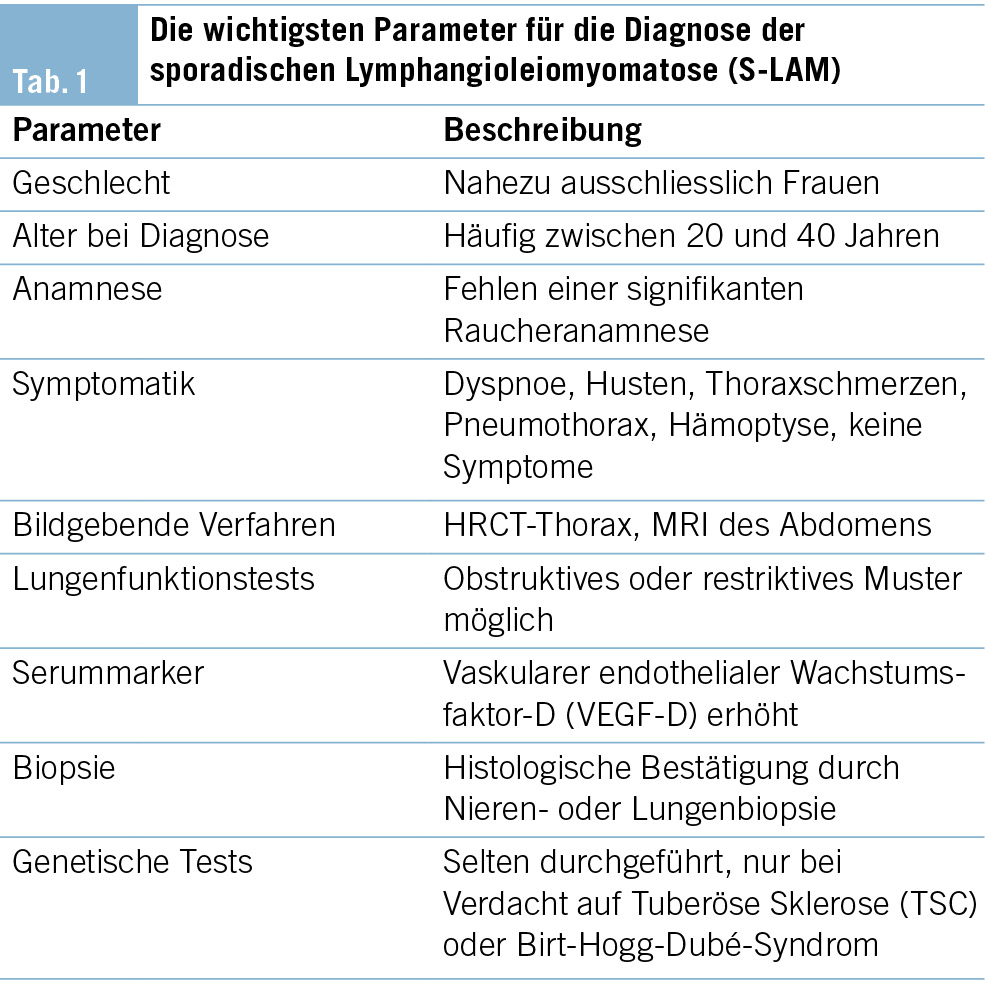
Als Leitsymptom zeigt sich ein Fieber unklarer Genese bei einem immunsupprimierten Patienten (vgl. Tab. 1).

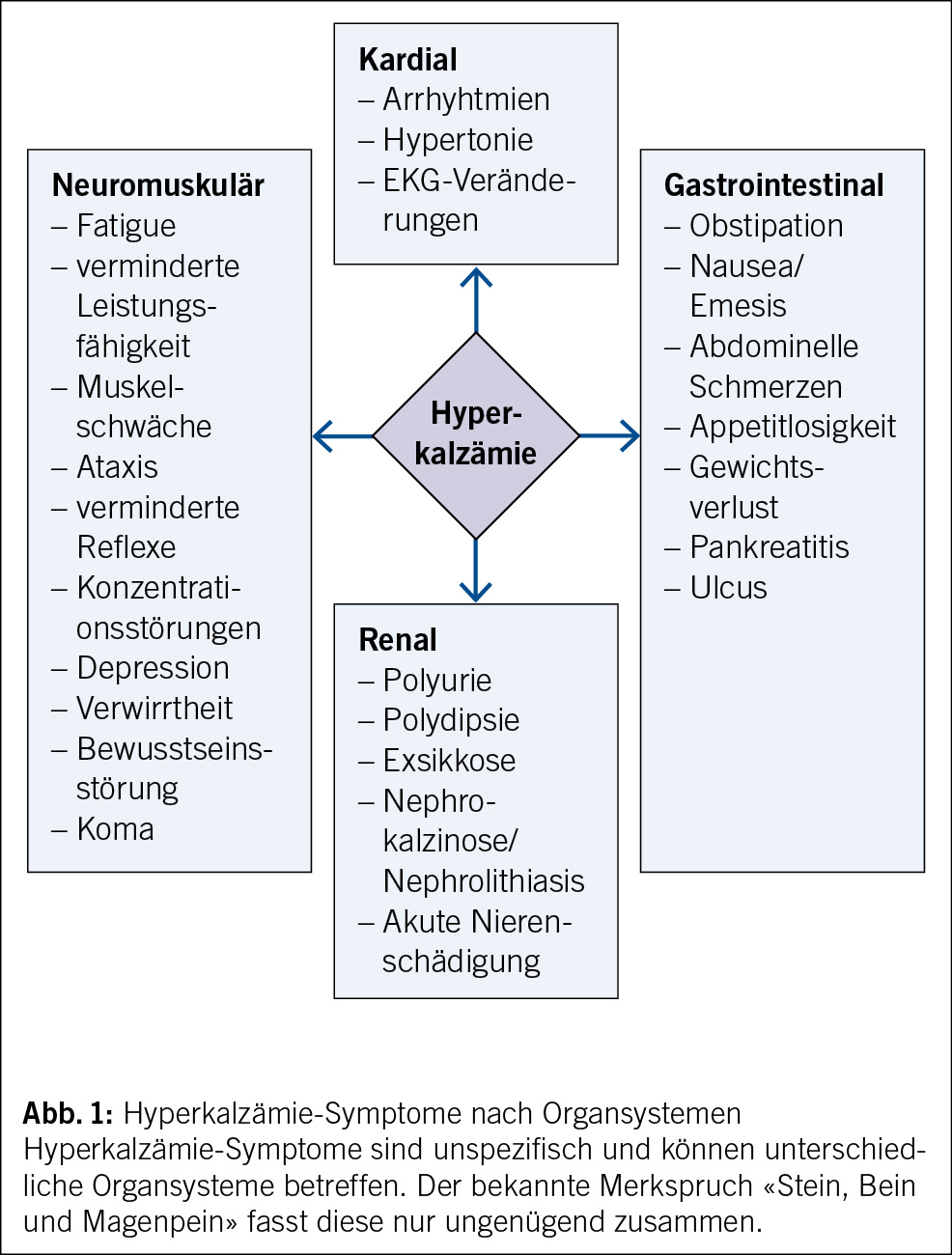
Definition von FUO
• Fieber über 38.3 °C zu mehreren Zeitpunkten
• Dauer des Fiebers mindestens 3 Wochen
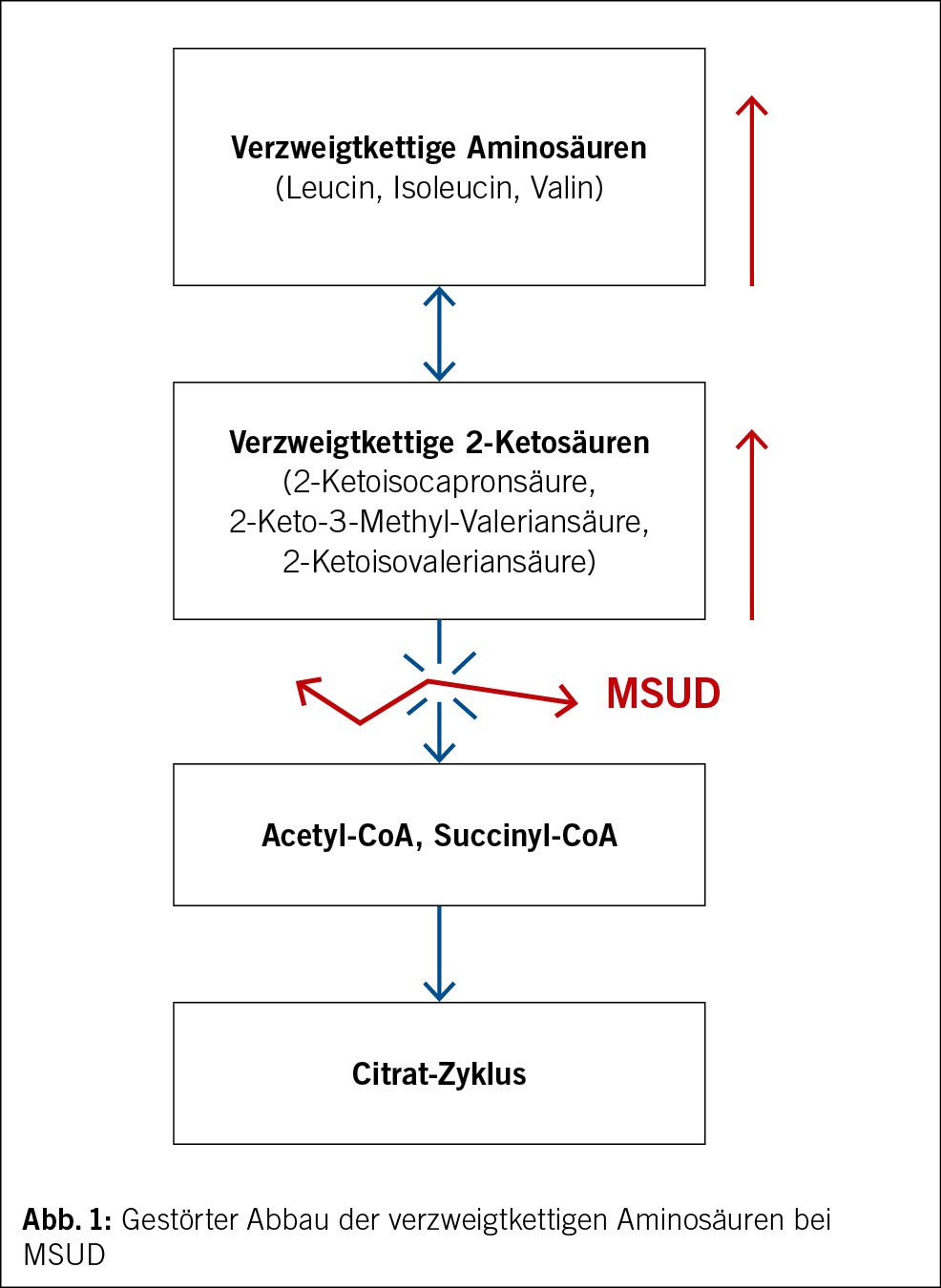
• Keine Diagnose nach 3 Tagen stationärer Untersuchung oder nach mindestens 2 ambulanten Untersuchungen (1) (Abb. 1)
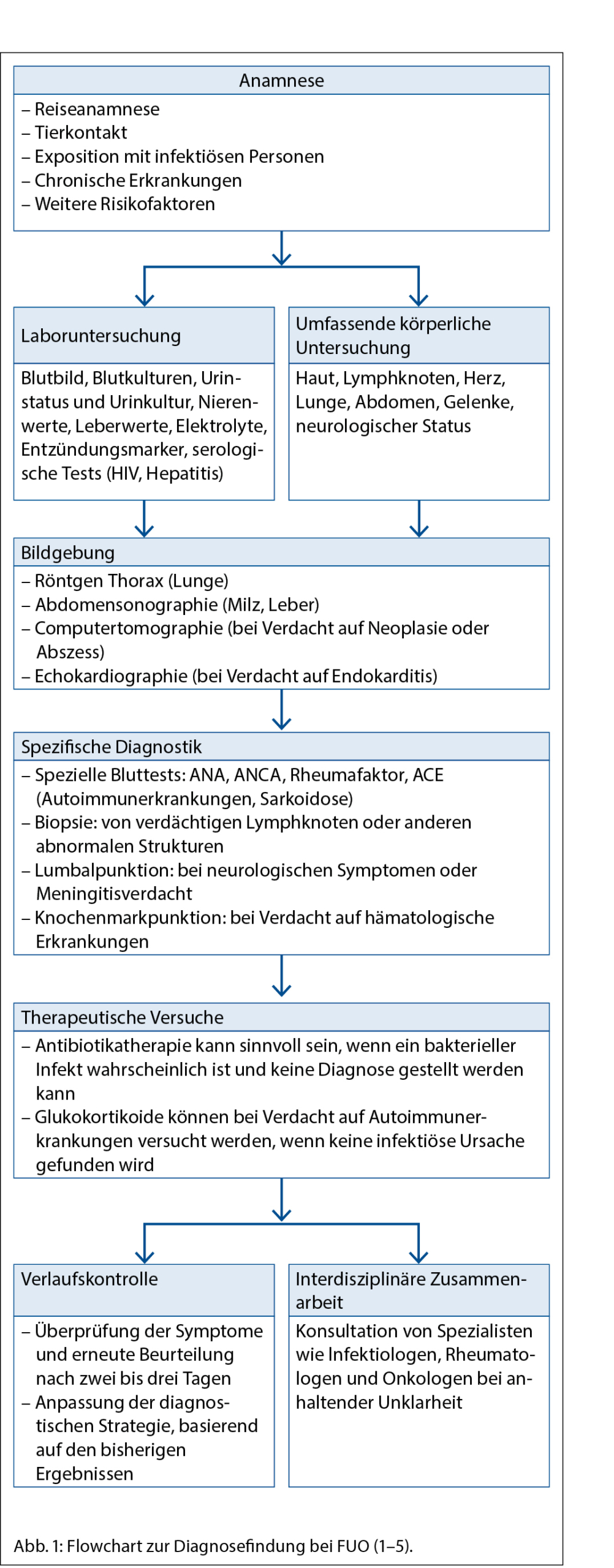
Spezifische Diagnostik
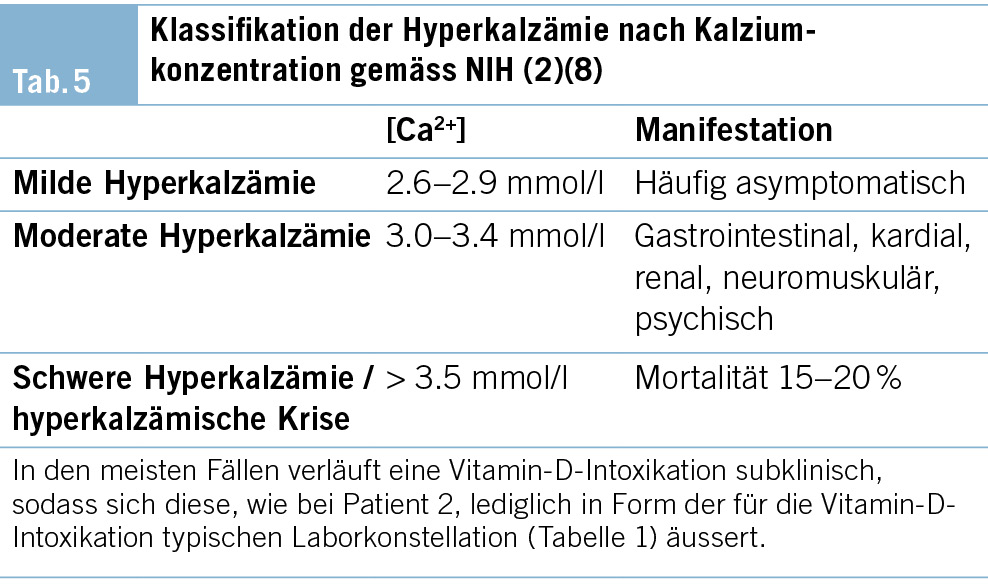
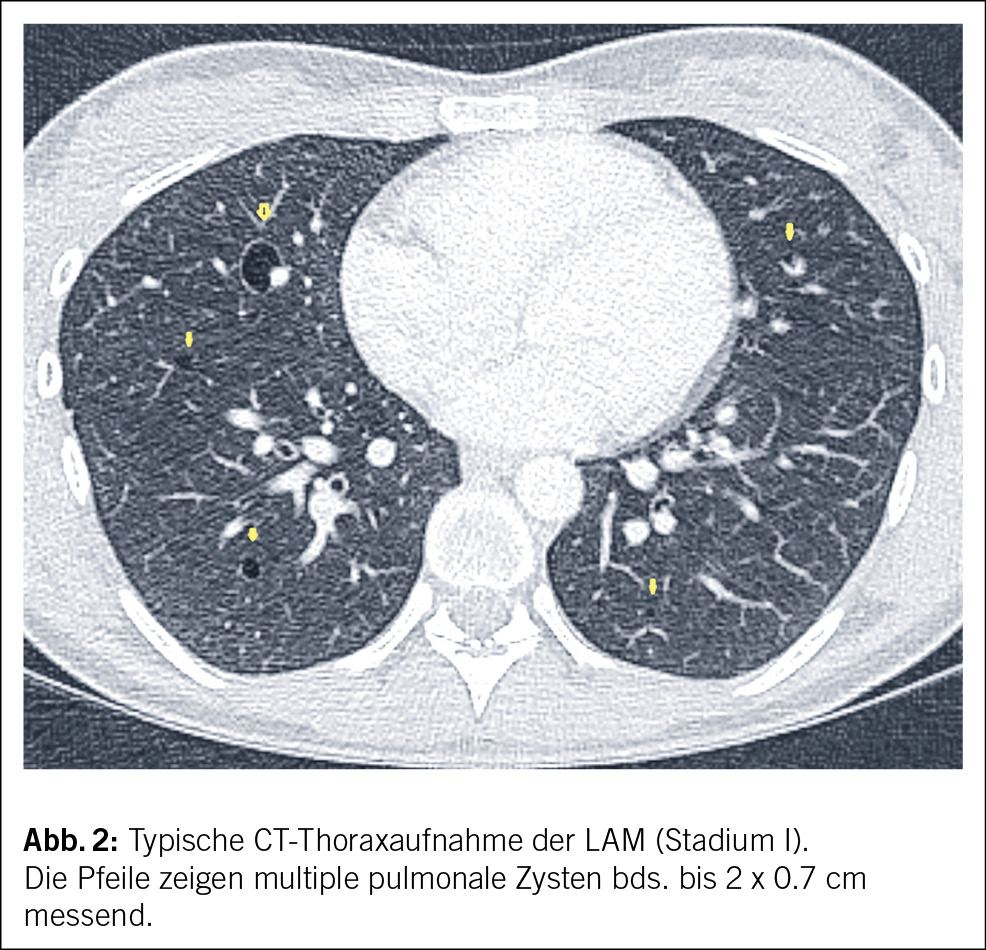
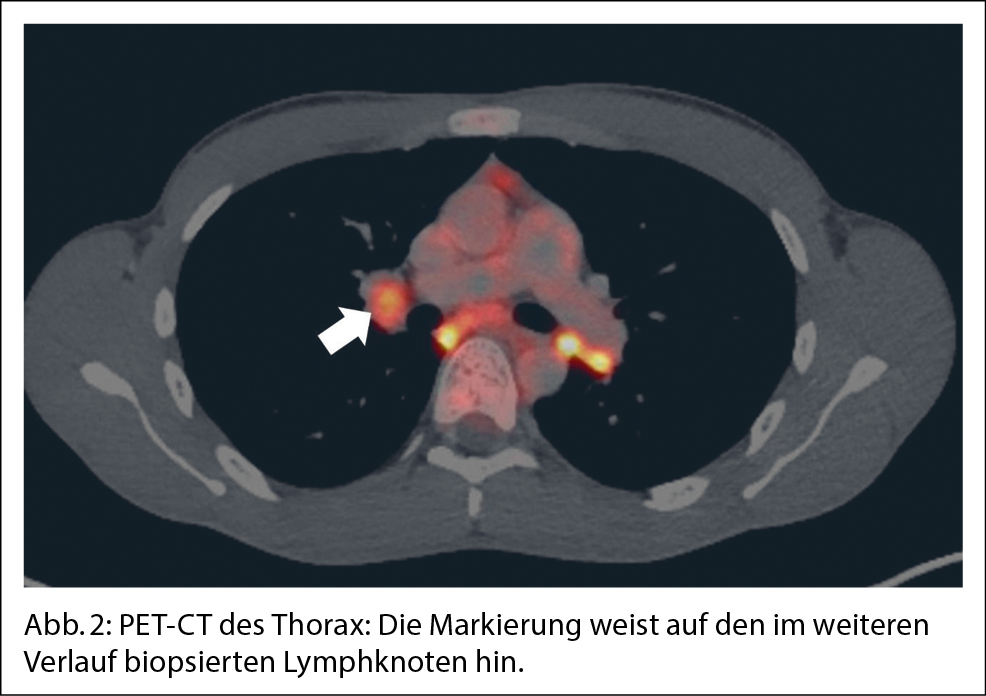
Der Patient wird stationär aufgenommen. Zum Ausschluss einer Spondylodiszitis oder Sakroiliitis mit Hinblick auf die neu aufgetretenen Symptome erfolgt eine MRT des Iliosakralgelenks und der Wirbelsäule, das einen altersentsprechenden Normalbefund ohne Hinweise auf entzündliche Prozesse zeigt. Auch die Hämatologie und Blutchemie bleiben während des stationären Aufenthaltes unauffällig. Zur Abklärung einer Autoimmunerkrankung sowie einer möglichen Sarkoidose werden spezielle Bluttests veranlasst. Es finden sich hierbei normwertige ANA- und ANCA-Titer sowie ein negativer Rheumafaktor und ACE im Normalbereich. In der ambulant durchgeführten PET-CT zeigen sich schliesslich multiple stark FDG-speichernde, perlschnurartige Noduli bzw. Lymphknoten rechts paravertebral/paraösophageal im unteren Mediastinum sowie linksbetonte FDG-avide Lymphknoten hilär, was primär mit einem Lymphom vereinbar erscheint (Abb. 2 und Abb. 3).
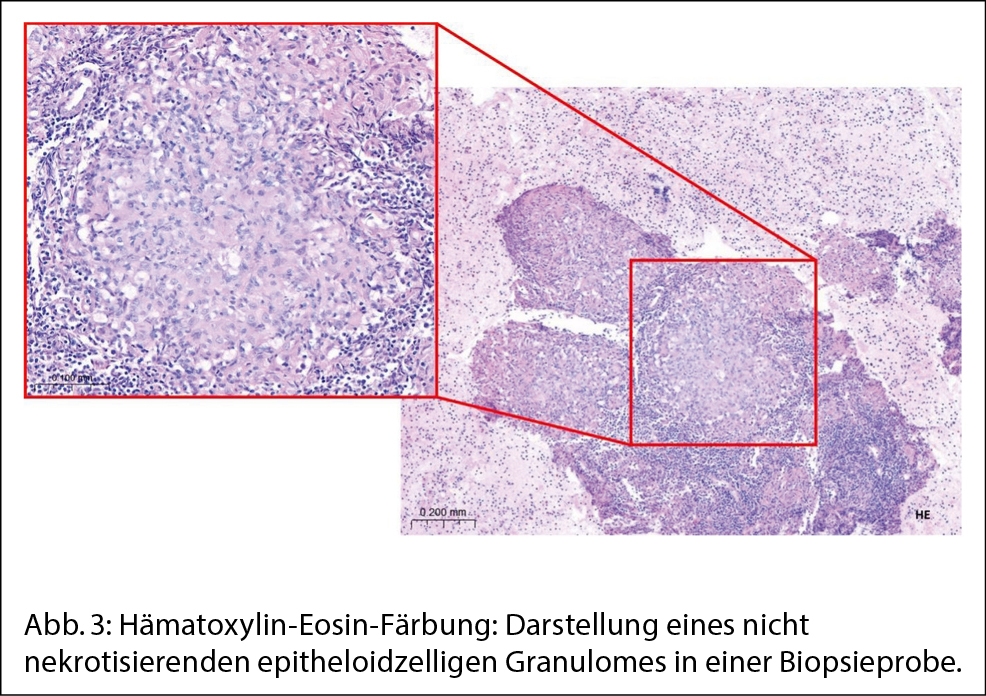
Zur weiteren Abklärung wird aufgrund von Lymphomverdacht eine endosonographisch gesteuerte Feinnadelbiopsie der mediastinalen Lymphknoten durchgeführt. Der histologische Befund ergibt eine granulomatöse Lymphadenopathie mit nicht nekrotisierenden epitheloidzelligen Granulomen. Nach Ausschluss einer infektiösen Genese und fehlendem Nachweis einer anderen Genese wird die Diagnose einer Sarkoidose als Ausschlussdiagnose gestellt.
Therapie
Der Patient wird nach Diskussion am Sarkoidoseboard durch die Pneumologie aufgeboten und erhält eine Behandlung mit oralen Glukokortikoiden. Im Verlauf zeigen sich die Fieberepisoden nicht mehr, und der Patient kann seine berufliche Tätigkeit wieder problemlos ausüben. Selbst nach schrittweisem Ausschleichen der Steroidtherapie bleibt der Patient symptomfrei. Ein erneutes Fieber tritt nicht mehr auf.
Prognose bei ungeklärten FUO-Fällen
Die Prognose bei Patienten mit FUO ist in aller Regel günstig. Studienergebnisse deuten darauf hin, dass ein erheblicher Anteil der Patienten, bei denen trotz umfangreicher Diagnostik keine Ursache identifiziert werden kann, eine spontane Besserung oder vollständige Genesung ohne spezifische Behandlung erleben. Etwa 50–75 % der Patienten mit FUO berichten von einer spontanen Besserung des Fiebers oder über eine vollständige Genesung ohne eine spezifische Diagnosestellung. Diese Patienten zeigen häufig keine weiteren schwerwiegenden Symptome, und der klinische Verlauf bleibt stabil (6, 9). Ein kleinerer Anteil der Betroffenen, etwa 20–30 %, bleibt symptomatisch mit milden oder intermittierenden Fieberschüben, jedoch ohne Anzeichen einer ernsthaften Verschlechterung oder Progression. Diese Patienten haben oft eine relativ stabile Langzeitprognose, ohne dass das Fieber einen signifikanten Einfluss auf die Lebensqualität hat (11). Nur in seltenen Fällen, etwa 5–10 %, entwickeln sich ernsthafte Komplikationen, oder es kommt zu einer Verschlechterung des Gesundheitszustands. Diese Fälle können durch bisher unentdeckte maligne oder systemische Erkrankungen bedingt sein, die sich erst später manifestieren (5, 12). Zusammengefasst zeigen ungeklärte FUO-Fälle insgesamt eine positive Prognose, mit einem hohen Anteil an Patienten, die sich ohne spezifische Diagnose erholen. Dies unterstreicht die Bedeutung eines schrittweisen und systemischen diagnostischen Vorgehens sowie einer sorgfältigen Abwägung invasiver diagnostischer Massnahmen bei klinisch stabilen Patienten.
Roman Zimmermann 1, Szilveszter Pekardi 1, Julia Zimmermann 2, Alptug Doganci 3, Annette Enzler-Tschudy 4, Alexander Kueres-Wiese 1
1 Klinik für Gastroenterologie und Hepatologie, Health Ostschweiz Wil, Wil
2 Medbase St. Gallen, St. Gallen
3 Klinik für Innere Medizin, Universitätsspital Zürich, Zürich
4 Klinik für Pathologie, Health Ostschweiz Kantonsspital St. Gallen, St. Gallen
Abkürzungen
ACE Angiotensin-Converting-Enzym
ANA Antinukleäre Antikörper
ANCA Anti-Neutrophile cytoplasmatische Antikörper
FDG Fluordesoxyglucose
FUO Fieber unklarer Genese
HIV Human Immunodeficiency Virus
PET-CT Positronen-Emissions-Tomographie, kombiniert mit einer Computertomographie
Historie
Manuskript eingegangen: 21.01.2025
Angenommen: 19.03.2025
Fachassistenzarzt Gastroenterologie
Health Ostschweiz Standort Wil
Fürstenlandstrasse 32
9500 Wil
roman.zimmermann@h-och.ch
Die Autorenschaft hat keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Paltiel O, Steinfeldt R, Bashari A, et al. Fever of Unknown Origin (FUO): Diagnostic Strategies and Challenges. Clin Infect Dis. 2021;73(5):e124-e135. doi:10.1093/cid/ciaa1234.
2. Li H, Zhang W, Chen C, et al. Comprehensive History-Taking in FUO: A Case-Based Approach. J Intern Med Res. 2022;44(3):378-390. doi:10.1002/jimr.5678.
3. Paredes JP, Guerrero F, Molina R. Physical Examination in Fever of Unknown Origin: Key Findings. Med Clin Rev. 2023;101(2):45-58. doi:10.1016/j.medcr.2023.02.012.
4. Benson C, Patel S, Lawrence T. Laboratory Workup in FUO: A Stepwise Approach. Am J Clin Pathol. 2023;159(4):567-580. doi:10.1093/ajcp/aqac123.
5. Petersdorf RG, Beeson PB. Fever of unexplained origin: report of 100 cases. Medicine (Baltimore). 1961;40(1):1-30. doi:10.1097/01.md.0000104740.31928.61.
6. Knockaert DC, Vanneste LJ, Bobbaers HJ. Fever of unknown origin in 1980s. An update of the diagnostic spectrum. Arch Intern Med. 1992;152(1):51-55. doi:10.1001/archinte.1992.00400130069010.
7. Stojanovich L, Marinkovic G. Fever of unknown origin: a diagnostic challenge. Hematology/Oncology and Stem Cell Therapy. 2010;3(2):35-42.
8. Aringer M, Costenbader K, Pagnoux C, et al. Systemic Lupus Erythematosus. Ann Rheum Dis. 2008;67(3):306-317. doi:10.1136/ard.2007.080631.
9. Bleeker-Rovers CP, Vos FJ, de Kleijn EM, et al. A prospective multicenter study on fever of unknown origin: the yield of a structured diagnostic protocol. Medicine (Baltimore). 2007;86(1):26-38. doi:10.1097/MD.0b013e31802fe858.
10. Fleck M. Fieber ungeklärter Ursache – Differenzialdiagnosen und Abklärung. Fieber ungeklärter Ursache – Differenzialdiagnosen und Abklärung. 2013:1-10. Available from: https://www.thieme-connect.de
11. de Kleijn EM, Vandenbroucke JP, van der Meer JW. Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. Medicine (Baltimore). 1997;76(6):392-400. doi:10.1097/00005792-199711000-00004.
12. Mourad O, Palda V, Detsky AS. A comprehensive evidence-based approach to fever of unknown origin. Arch Intern Med. 2003;163(5):545-551. doi:10.1001/archinte.163.5.545.
Besonders empfohlene Literatur
1. Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Handlungsempfehlung nach der Leitlinie „Fieber unklarer Genese“. Available from: https://www.awmf.org
2. Peter ME, editor. Fever of unknown origin. 1st ed. New York: Springer; 2011
3. Fauci AS, Tino G, Zinner SH. Fever of unknown origin: Diagnosis and management. JAMA. 2019;321(8):781-792. Available from: https://jamanetwork.com