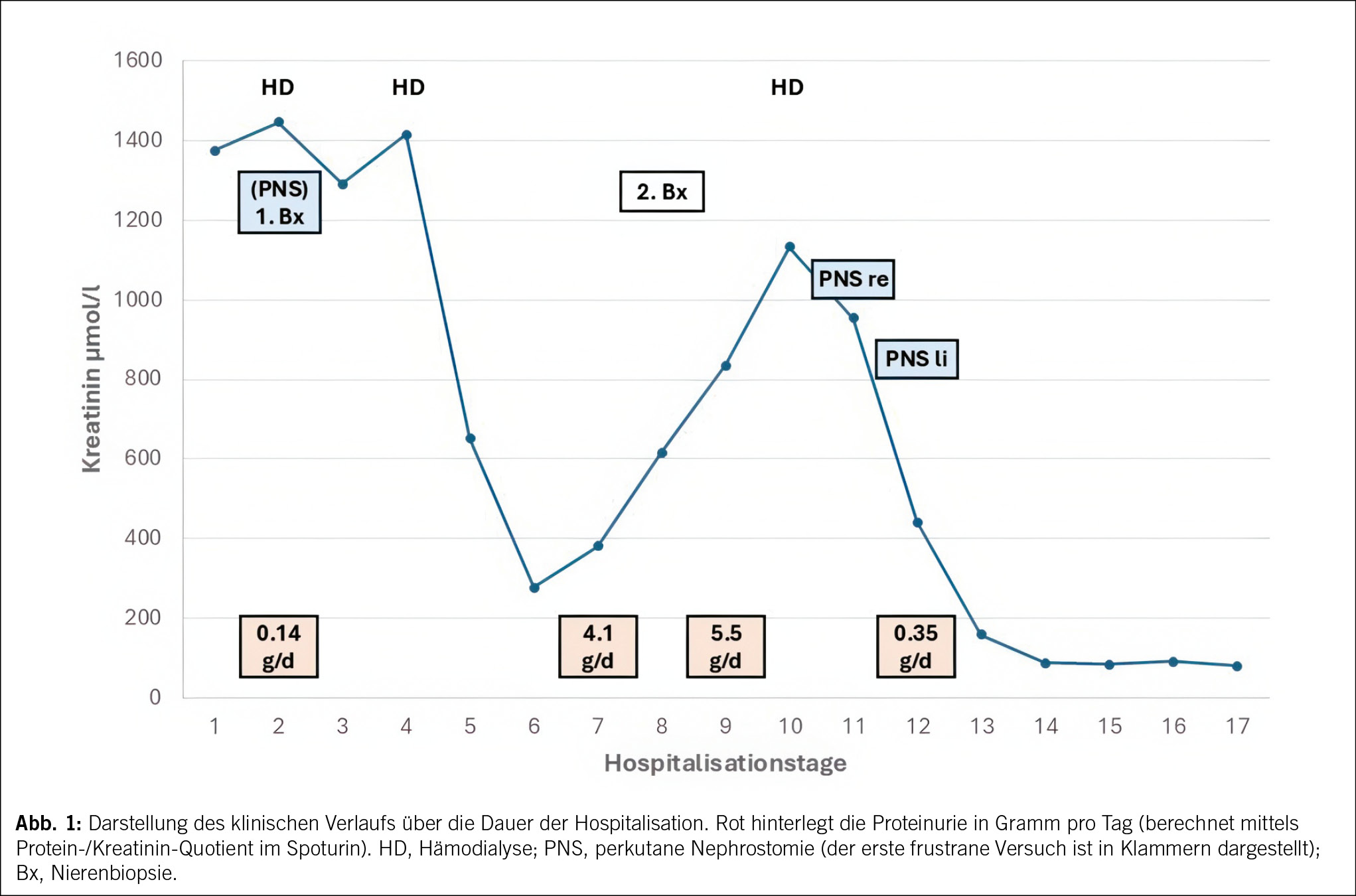
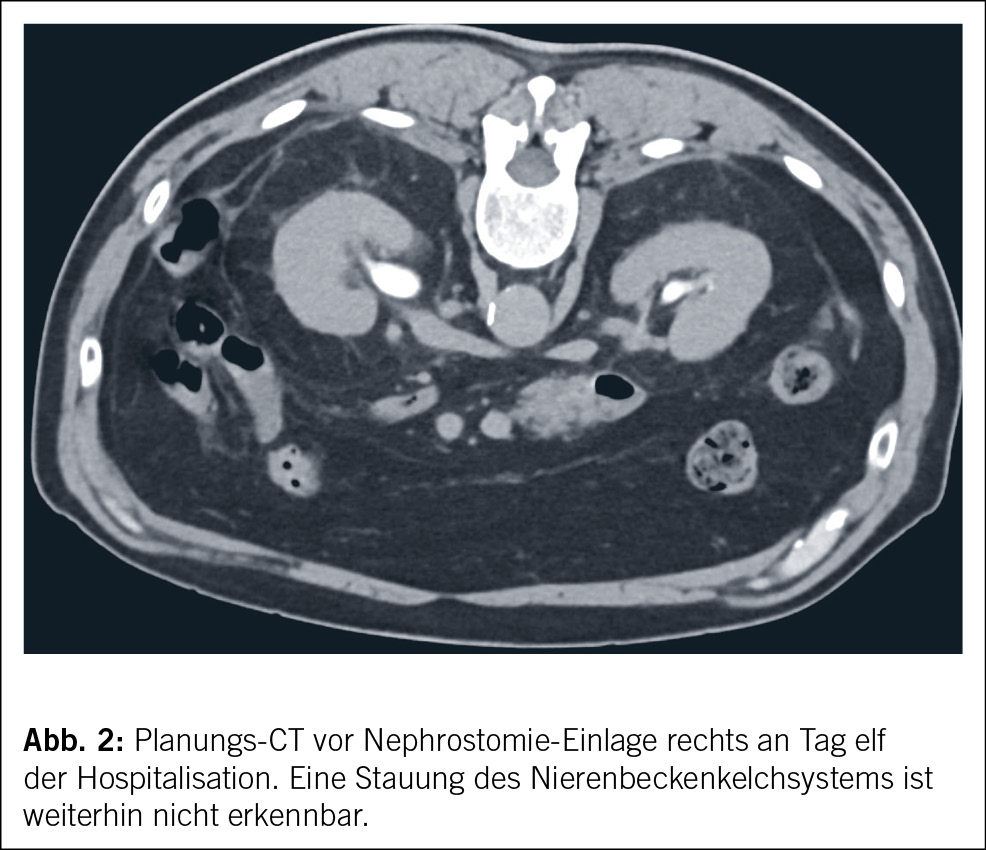
Anamnese und Befunde
Eine 44-jährige Frau ohne Vorerkrankungen litt an grippalen Symptomen; Aspirin hätte nur passager Linderung gebracht. Aufgrund zusätzlicher Adynamie und Tagesmüdigkeit erfolgte eine hausärztliche Vorstellung. Im Urinstix zeigte sich eine Leukozyturie, unter Vermutung eines Harnwegsinfekts erfolgte eine 3-tägige Antibiotikatherapie mit Ciprofloxacin. In der Verlaufskontrolle zeigte sich ein erhöhtes Kreatinin von 200 µmol/l (Referenzbereich: < 95 µmol/l), zudem kam es zu einer Rötung beider Augen, sodass eine Hospitalisation erfolgte.
Bei einer 15-jährigen Jugendlichen wurde aufgrund eines geröteten schmerzhaften Auges links eine Uveitis anterior diagnostiziert und topisch mit Steroiden begonnen. Anamnestisch litt die Patientin seit Längerem an Müdigkeit und Abgeschlagenheit. Wegen Rückenschmerzen hatte sie gelegentlich Ibuprofen eingenommen. Aufgrund eines erhöhten Kreatinins erfolgte die Zuweisung an die Kindernephrologie.
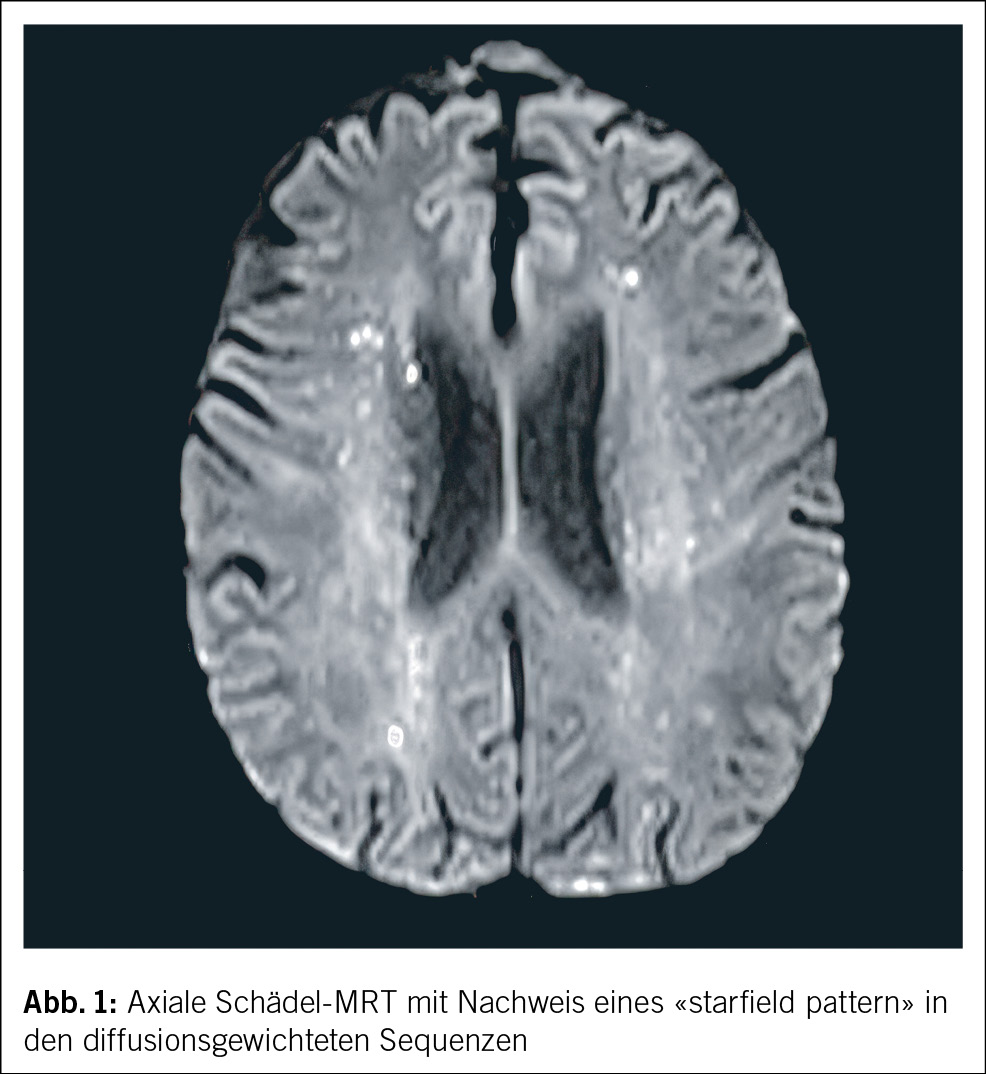
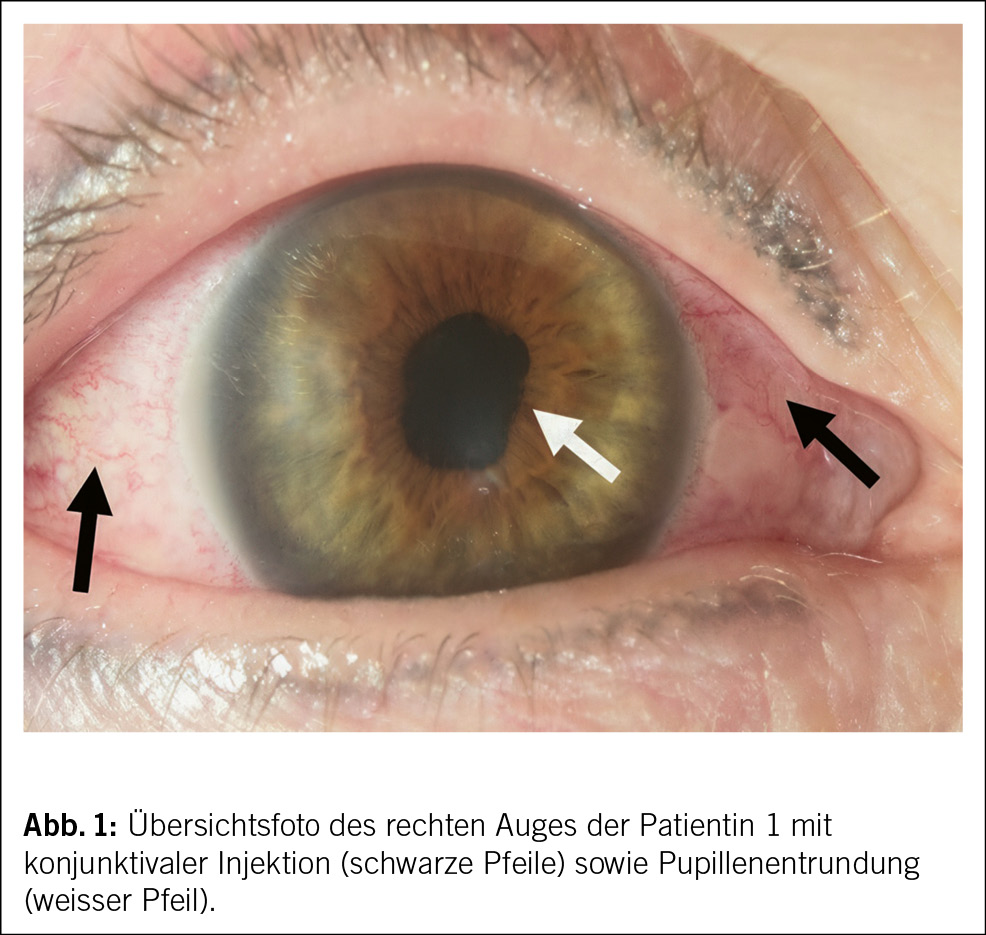
Der Status bei der 44-jährigen Patientin war bis auf die geröteten Augen unauffällig. Das Kreatinin lag bei 226 µmol/l. Die Elektrolyte waren normwertig. Das Urinsediment zeigte eine Leukozyturie, der Protein-Kreatinin-Quotient lag bei 26 mg/mmol (Referenzbereich: < 11 mg/mmol), der Albumin-Kreatinin-Quotient bei 2 mg/mmol (Referenzbereich: < 3 mg/mmol), zudem bestand eine euglykäme Glukosurie. Eine Urinkultur blieb steril. Die Nierensonographie war bis auf vergrösserte Nieren beidseits unauffällig. Virale Serologien (HIV, Hepatitis B und C) sowie immunologische Marker (ANA, ANCA, anti-GBM) waren negativ. Das Röntgenbild zeigte keine mediastinale Lymphadenopathie. Eine ophthalmologische Beurteilung diagnostizierte eine bilaterale Uveitis anterior (Abb. 1).
Bei der 15-jährigen Patientin war der Status bis auf eine Augenrötung ebenfalls unauffällig. Das Kreatinin lag bei 103 µmol/l, Blutzucker und Elektrolyte waren normwertig. Im Urinstix zeigte sich eine Glukosurie; eine Leukozyturie lag nicht vor. Im Spoturin fand sich ein Protein-Kreatinin-Quotient von 103 mg/mmol, der Albumin-Kreatinin-Quotient betrug 13 mg/mmol. Die Nierensonographie war bis auf eine etwas verminderte kortikomedulläre Differenzierung unauffällig. Auf serologische Abklärungen wurde verzichtet.
Differenzialdiagnostische Überlegungen
Bei geröteten Augen und systemischen Beschwerden wie Müdigkeit sollten neben lokalen Infektionen und Allergien auch systemische Erkrankungen berücksichtigt werden. Zu den Differenzialdiagnosen zählen infektiologische (wie Tuberkulose, Lues, Borreliose, Chlamydien, Viren) und immunologische (wie Spondyloarthropathien, Sarkoidose, juvenile idiopathische Arthritis) Erkrankungen.
Befunde wie tubuläre Proteinurie, euglykäme Glukosurie und sterile Leukozyturie sind typisch für eine tubuläre Dysfunktion und deuten, zusammen mit erhöhtem Serumkreatinin, auf eine interstitielle Nephritis hin. Häufige Ursachen sind Medikamente (wie NSAR, Antibiotika), Infektionen (wie Mykobakteriosen, Leptospiren) und Autoimmunkrankheiten (wie Sarkoidose, Sjögren-Syndrom, Lupusnephritis). Wenn sowohl eine Nierenschädigung als auch eine Uveitis zeitlich assoziiert, sollte auch an das TINU-Syndrom (Tubulointerstitielle Nephritis und Uveitis) gedacht werden.
Weitere Abklärungsschritte
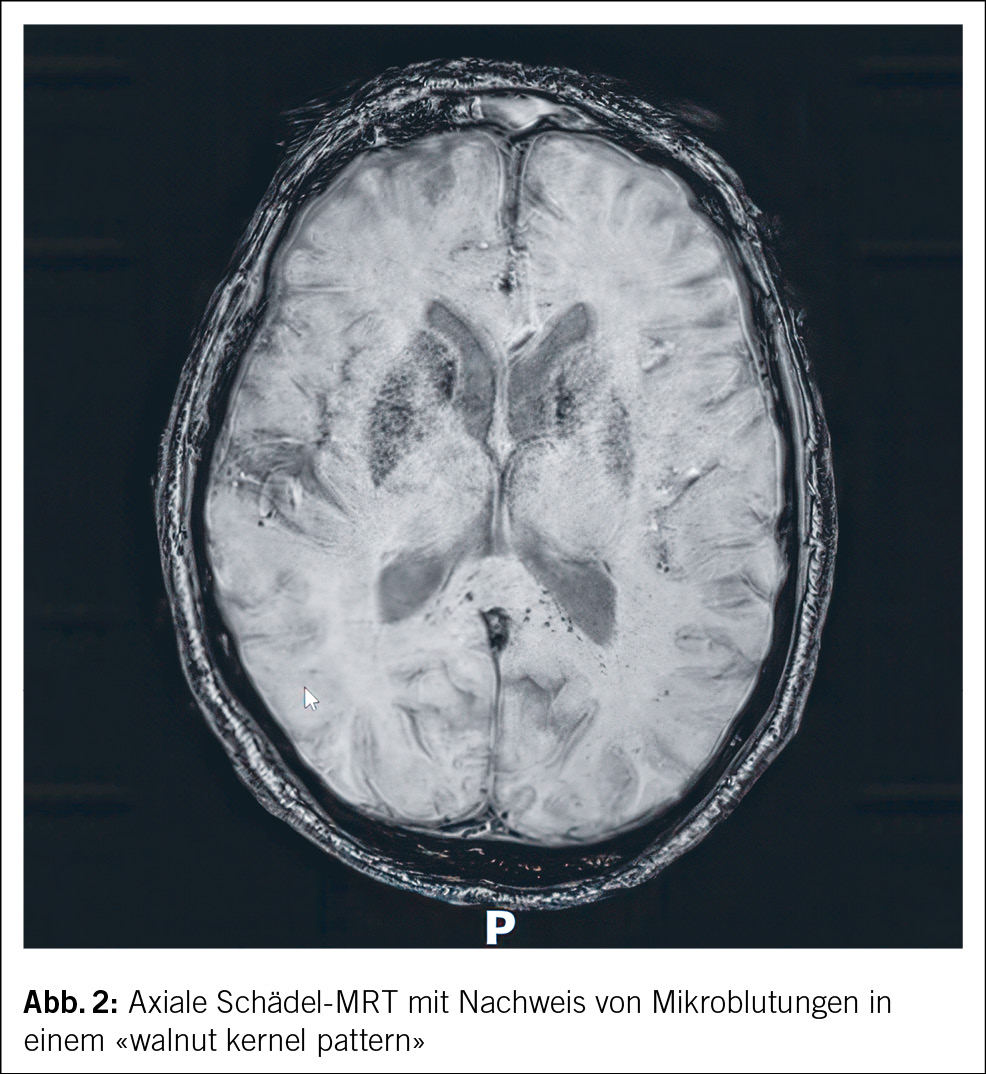
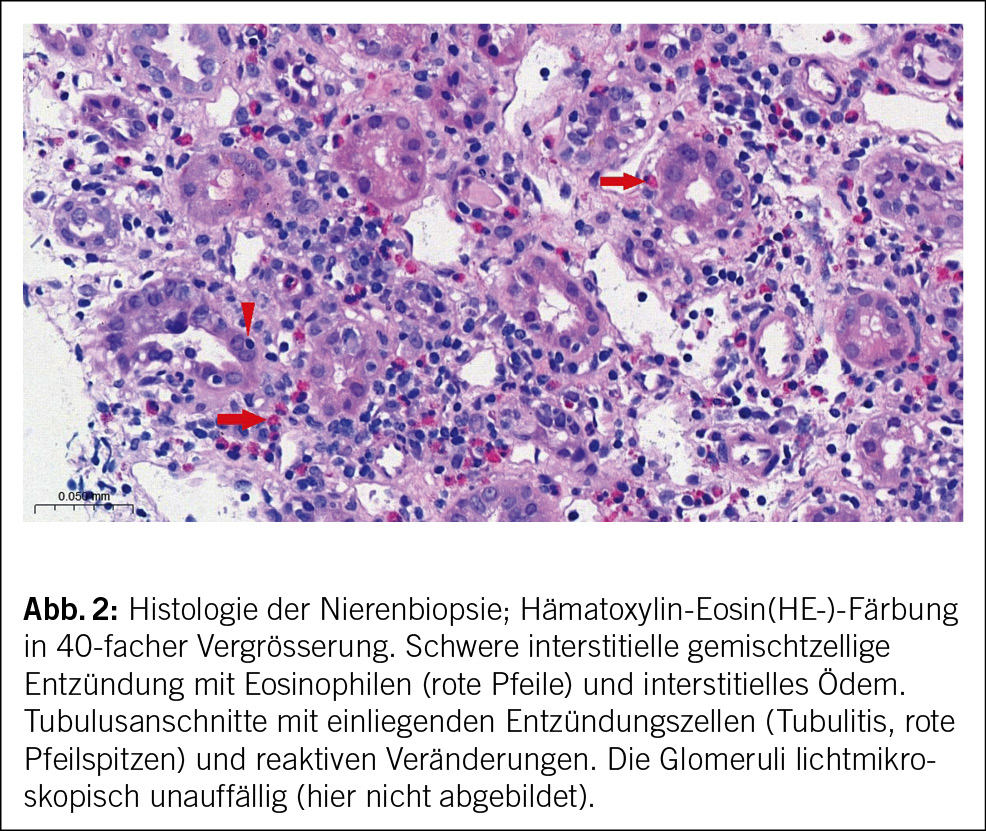
In beiden Fällen erfolgte eine Nierenbiopsie. Bei der 44-jährigen Patientin zeigte die Biopsie eine akute interstitielle Nephritis (AIN) ohne Granulome und unauffällige Glomerula (Abb. 2). Die Biopsie der 15-jährigen Patientin ergab ebenfalls eine tubulointerstitielle Entzündung mit fokaler Tubulitis.
Diagnose
Bei beiden Patientinnen wurde ein TINU-Syndrom (Tubulointerstitielle Nephritis und Uveitis) diagnostiziert, basierend auf der zeitlichen Korrelation zwischen der Uveitis anterior und der interstitiellen Nephritis.
Verlauf
Bei der 44-jährigen Patientin wurde eine hoch dosierte Steroidtherapie (1 mg pro Kilogramm Körpergewicht) etabliert. Parallel erfolgte eine topische Therapie mit steroidhaltigen Augentropfen. Die Augensymptome sowie die Kopfschmerzen zeigten sich darunter zügig vollständig regredient. Die tubuläre Proteinurie zeigte sich bereits nach drei Wochen vollständig regredient. Die Nierenfunktion erholte sich über vier Monate partiell auf eine eGFR um 60 ml/min/1.73m2. Die Steroide wurden schrittweise bis auf 5 mg reduziert, dann aber von der Patientin selbstständig abgesetzt. Das Serumkreatinin zeigte sich anschliessend auf leicht erhöhtem Niveau stabil, jedoch fanden sich erneut Hinweise auf eine proximal-tubuläre Dysfunktion (euglykäme Glukosurie, tubuläre Proteinurie, grenzwertige Hypokaliämie); die Patientin lehnte eine erneute Therapie jedoch ab und entzog sich weiteren Kontrollen.
Bei der 15-jährigen Patientin erfolgte zuerst eine Behandlung mit steroidhaltigen Augentropfen über die ambulante Ophthalmologin. Nach der Vorstellung auf der Nephrologie wurde aufgrund der eindrücklichen Dynamik des Kreatinins eine Methylprednisolon-Stosstherapie mit nachfolgender peroraler Prednisontherapie etabliert. Innerhalb von Wochen verschwanden die Symptome vollständig, die Nierenfunktion sowie die Proteinurie normalisierten sich, und die Patientin ist gemäss behandelnden Kolleg/-innen drei Jahre später weiterhin asymptomatisch und hat eine normale Nierenfunktion.
Kommentar
Das TINU-Syndrom wurde 1975 erstmalig beschrieben als das Auftreten einer tubulointerstitiellen Nephritis (TIN) und einer Uveitis (U), in Abwesenheit anderer potenziell erklärender Erkrankungen. Wahrscheinlich handelt es sich um eine Autoimmunerkrankung; pathophysiologisch wird eine T-Zell-vermittelte, CD4+-dominante, verzögerte Hypersensitivitätsreaktion postuliert. Diese kann zur Bildung von entzündlichen, nicht verkäsenden Granulomen führen, was die Differenzierung von anderen granulomatösen Erkrankungen schwierig macht. Vermutet wird ein bislang unbekanntes Antigen, welches sowohl in den Nierentubuli als auch in der Uvea vorkommt. Ähnlich wie bei einem pulmorenalen Syndrom mit Antikörpern gegen dasselbe Antigen in der alveolären wie glomerulären Basalmembran kommt es zu einem Syndrom mit entzündlichen Vorgängen in den Nieren und Augen. Es wird vermutet, dass NSAR und Antibiotika ein TINU-Syndrom provozieren können, wobei diese Substanzklassen auch als Risikofaktoren für die klassische interstitielle Nephritis bekannt sind. Auch verschiedene Infektionen wurden mit dem Auftreten eines TINU-Syndroms in Verbindung gebracht. In Fallberichten werden vor allem respiratorische Infektionen und Virusinfekte diskutiert (z. B. Hantavirus, EBV, HIV) (1).
Gemäss einem Systematic Review wurden bis März 2020 kumuliert 592 TINU-Fälle beschrieben (2). Das mediane Alter betrug bei Diagnose 17 Jahre (Interquartilsabstand 13–46 Jahre) mit weiblicher Prädominanz. Meistens kam es zeitlich nach dem Auftreten einer Nephritis zu einer bilateralen Uveitis, welche meist einer Uveitis anterior entsprach. Bei pädiatrischen Patient/-innen wurde eine asymptomatische Uveitis gelegentlich erst diagnostiziert, wenn eine Nephritis zu einer ophthalmologischen Vorstellung führte. Eine plötzlich auftretende bilaterale Uveitis in pädiatrischen Patient/-innen war zudem in bis zu einem Drittel der Fälle mit der Diagnose einer tubulointerstitiellen Nephritis verbunden. Kinder und Jugendliche tendieren eher zu einer rezidivierenden Uveitis, während bei Erwachsenen das Risiko für eine akute Nierenfunktionseinschränkung und einen chronischen Nierenschaden im Vordergrund steht. Die Gründe für diese Diskrepanz im Phänotyp sind nicht abschliessend geklärt.
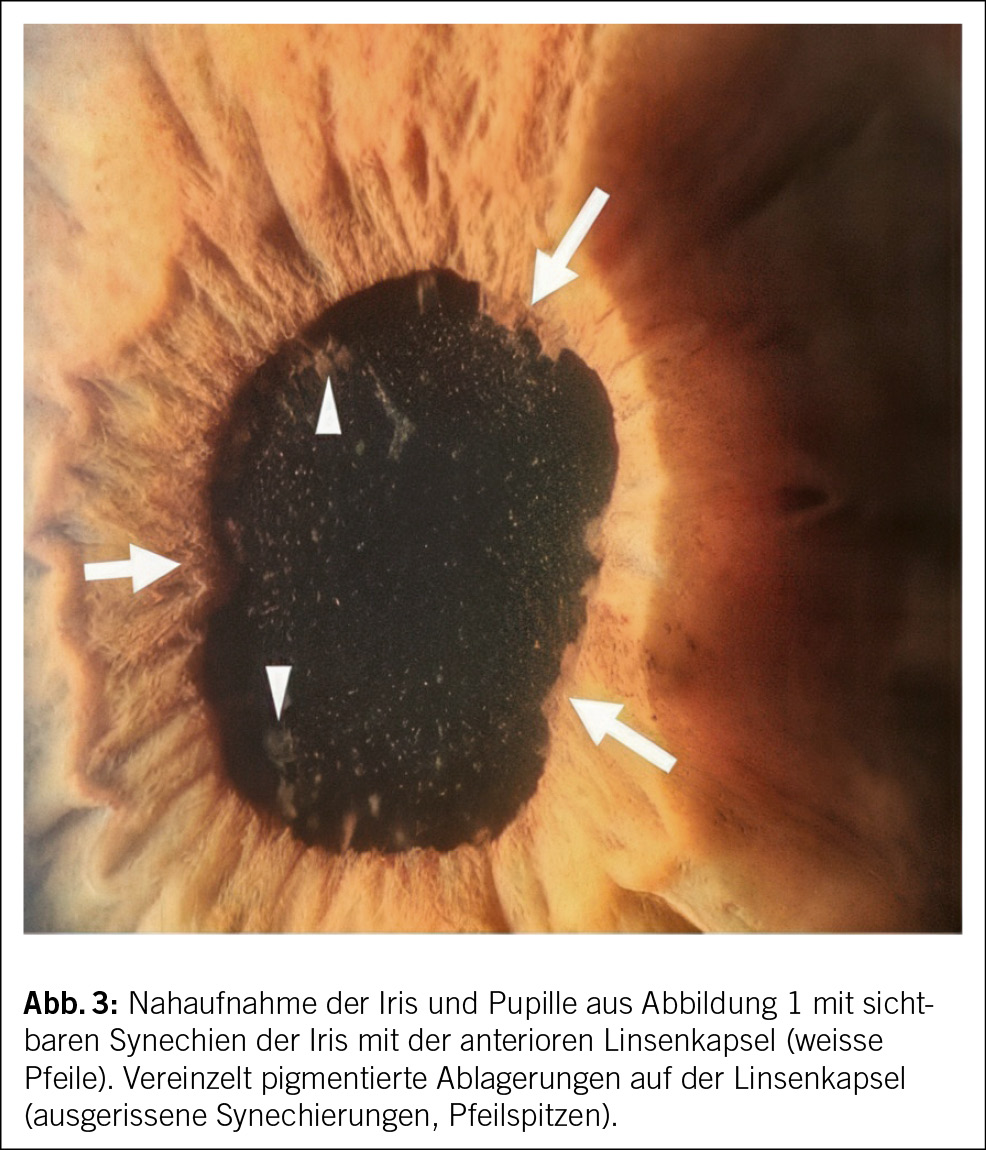
Das ophthalmologische Bild des TINU-Syndroms präsentiert sich als akut auftretende, nicht granulomatöse Uveitis. Klassische Symptome sind Augenrötung, -schmerzen und Photophobie, eine Visusverschlechterung kann auch vorliegen. Die Symptome treten akut innerhalb von Tagen auf und sind häufig direkt bilateral vorhanden. Okuläres wässriges Sekret oder morgendliche verklebte Augen, wie sie klassisch bei einer viralen Konjunktivitis der Fall sind, finden sich nicht. Bereits die makroskopische Beurteilung, wie sie auch in der Hausarztpraxis erfolgen kann, zeigt eine ausgeprägte konjunktivale Injektion. Eine entrundete Pupille (Abb. 1) kann als Folge eines Entzündungsreizes mit Verklebung der Iris mit der anterioren Linsenkapsel (Abb. 3) vorkommen und muss insbesondere bei Patient/-innen ohne vorherige intraokulare Operation an eine intraokulare Entzündung denken lassen. Weitere Befunde des Vorderkammerreizes sind ohne fachärztliche Untersuchung mittels Spaltlampe nicht zu eruieren. Diese Augenbefunde sind oftmals unspezifisch und können meist nicht eindeutig einer Ätiologie zugeordnet werden.
Nephrologisch findet sich typischerweise das Bild einer AIN mit steriler Leukozyturie, tubulärer Proteinurie und Nierenfunktionseinschränkung. Andere Zeichen der proximalen Tubulopathie wie euglykäme Glukosurie, Phosphaturie und metabolische Azidose können vorkommen. Auch das histologische Bild in der Nierenbiopsie entspricht dem einer AIN mit lymphozytärem Infiltrat und interstitiellem Ödem. Granulome können vorkommen, während Glomerula und Gefässe typischerweise unauffällig sind. Teilweise lassen sich Granulome auch in Lymphknoten und Knochenmark finden. Insbesondere die Abgrenzung zur Sarkoidose kann dann schwierig sein, wenn keine andere Organbeteiligung vorliegt. Es existieren keine gut validierten Laborparameter, die spezifisch sind für das TINU-Syndrom – BSG und CRP können erhöht sein, wie bei anderen Erkrankungen. Antikörper gegen modifiziertes C-reaktives Protein (mCRP, ein sowohl in Uvea und Tubuluszellen vorkommendes Antigen) scheinen beim TINU-Syndrom erhöht zu sein im Gegensatz zum Sjögren-Syndrom, zu medikamenteninduzierter interstitieller Nephritis und gesunden Kontrollen, jedoch ist dieser Test nicht kommerziell erhältlich. Eine reduzierte eGFR wird in 40 % der betroffenen Patient/-innen nach 12 Monaten beschrieben. Dabei ist zu berücksichtigen, dass teilweise wahrscheinlich eine zuvor unbekannte, bereits eingeschränkte Nierenfunktion vor der Diagnose der Nephritis bestanden hatte. Bei den pädiatrischen Patient/-innen besteht ein besseres Outcome mit einer reduzierten eGFR in 20 % der Betroffenen nach 12 Monaten.
Analog zur Behandlung einer akuten interstitiellen Nephritis wird eine Therapie mit Kortikosteroiden empfohlen. Die optimale Dosierung wurde bislang nicht in prospektiven Studien untersucht, bezüglich der Therapiedauer wird in Fallserien ein eher längeres Fenster (12–24 Monate) gewählt. Entsprechend werden steroidsparend Mycophenolat oder Azathioprin eingesetzt. Bezüglich Uveitis ist eine Behandlung mit lokalen steroidhaltigen Augentropfen indiziert. Beim TINU-Syndrom ist die Uveitis meist mild bis moderat ausgeprägt und spricht in der Regel gut auf eine Lokaltherapie an (3).
Abkürzungen
AIN Akute interstitielle Nephritis
ANA Antinukleäre Antikörper
ANCA Anti-Neutrophile cytoplasmatische Antikörper
Anti-GBM Anti-Glomeruläre Basalmembran
BSG Blutsenkungsgeschwindigkeit
CRP C-reaktives Protein
EBV Epstein-Barr-Virus
eGFR geschätzte glomeruläre Filtrationsrate
HIV Human Immunodeficiency Virus
mCRP modifiziertes C-reaktives Protein
TINU tubulointerstitielle Nephritis und Uveitis
Historie
Manuskript eingegangen: 30.09.2024
Angenommen nach Revision: 12.03.2025
Pascal Gantenbein 1,
sabelle Binet 1
Regula Laux 2
Sascha Mathias Jung 3
Annette Enzler-Tschudy 4
Christian Kuhn 1
1 Klinik für Nephrologie und Transplantationsmedizin, HOCH Health Ostschweiz, Kantonsspital St. Gallen, St. Gallen
2 Nephrologie, Ostschweizer Kinderspital, St. Gallen
3 Klinik für Ophthalmologie, HOCH Health Ostschweiz, Kantonsspital St. Gallen, St. Gallen
4 Institut für Pathologie, HOCH Health Ostschweiz, Kantonsspital St. Gallen, St. Gallen
Klinik für Nephrologie und Transplantationsmedizin
HOCH Health Ostschweiz, Kantonsspital St. Gallen
Rorschacher Str. 95/Haus 10
9007 St. Gallen
christian.kuhn@h-och.ch
Die Autorenschaft hat keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Okafor LO, Hewins P, Murray PI, Denniston AK. Tubulointerstitial nephritis and uveitis (TINU) syndrome: a systematic review of its epidemiology, demographics and risk factors. Orphanet J Rare Dis. 14. Juli 2017;12(1):128
2. Regusci A, Lava SAG, Milani GP, Bianchetti MG, Simonetti GD, Vanoni F. Tubulointerstitial nephritis and uveitis syndrome: a systematic review. Nephrol Dial Transplant. 1. Mai 2022;37(5):876–86.
3. Mackensen F, Smith JR, Rosenbaum JT. Enhanced recognition, treatment, and prognosis of tubulointerstitial nephritis and uveitis syndrome. Ophthalmology. Mai 2007;114(5):995–9.