Anamnese und Befunde
Spätabends brachten 2 Leiterinnen eines Skilagers eine 11-jährige Patientin auf die Notfallstation eines Regionalspitals. Sie wurde im Rollstuhl in die Koje geschoben, weil sie zu schwach zum Gehen war. Die Begleitpersonen berichteten über seit 2 Tagen stetig zunehmende Schwäche, Appetitlosigkeit und vermehrten Schlafbedarf. Am Eintrittstag einmalig Durchfall mit Einstuhlen und Einnässen. Die Patientin klagte über Bauchschmerzen, Übelkeit und einmaliges Erbrechen sowie Kopfschmerzen. Keine Dysurie. Fieber und Schüttelfrost in den vorangehenden Tagen wurden verneint, wobei im Skilager die Körpertemperatur nicht gemessen worden war.
Die Systemanamnese war sonst wenig ergiebig bis auf eine COVID-19-Erkrankung vor rund drei Wochen. Die Diagnose war mittels positiven Spucktests in der Schule gestellt worden. Der Krankheitsverlauf war milde über wenige Tage mit leichtem Fieber, Husten und Schnupfen mit kompletter Genesung. Die Umgebungsanamnese war positiv für eine Gastroenteritis-Exposition vor wenigen Tagen. Geimpft sei sie nach CH-Impfplan. Beide Eltern stammen aus Eritrea, die Reiseanamnese war unauffällig.
Status und Labor (Tabelle 1)
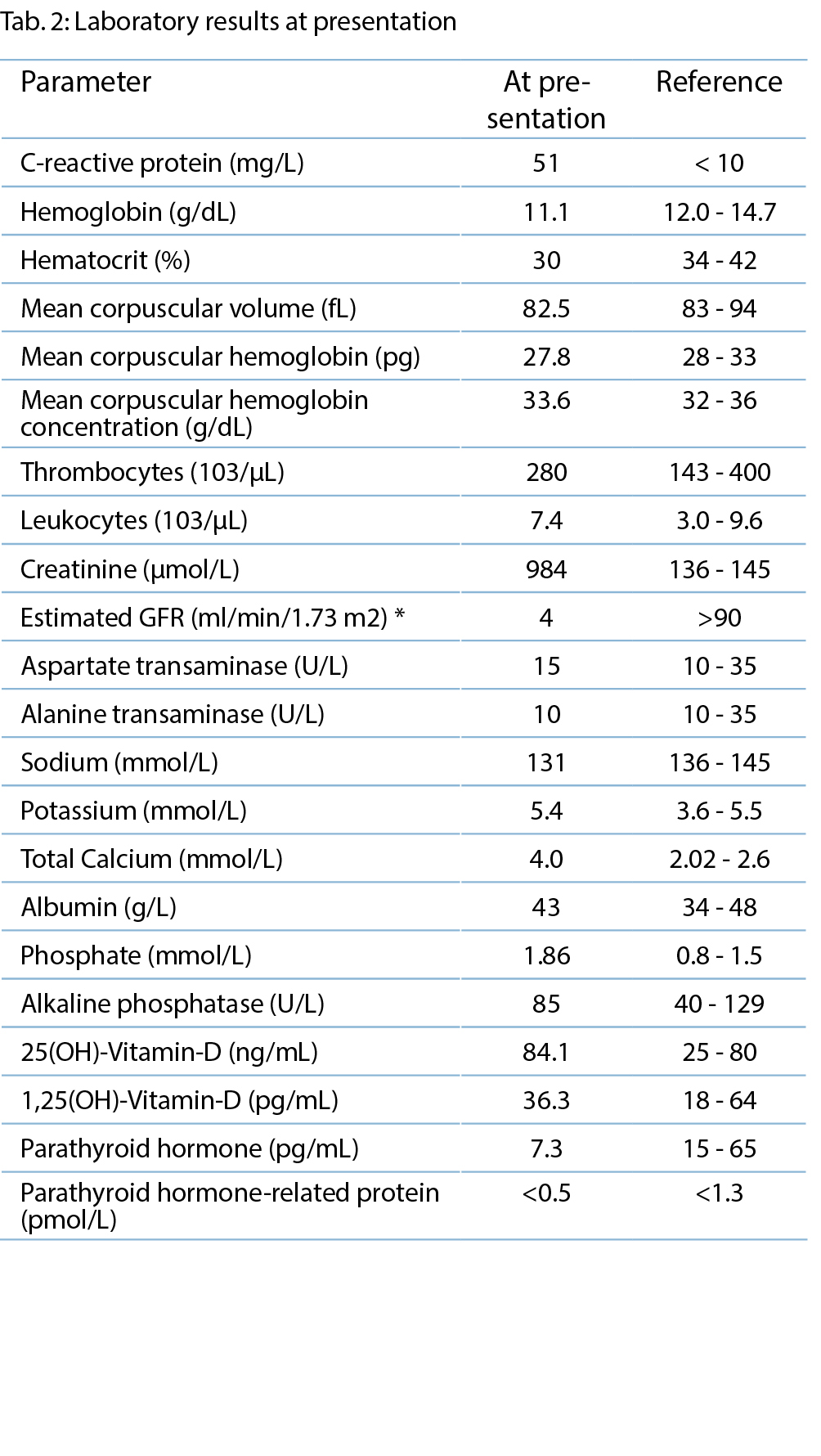
Auffällig tiefer BMI von 13.5 kg/m2. Blutdruck 102/70 mmHg, Puls 99 bpm, 97 % Sauerstoffsättigung bei Atemfrequenz von 15/min und 40.2° C Körpertemperatur. Bei der Lymphknotenpalpation fiel einzig submandibulär rechts eine druckdolente Schwellung auf (im Zentrumsspital später sonographisch als Lymphadenopathie identifiziert). Das Abdomen war weich, diffus druckdolent, v.a. periumbilikal betont, ohne tastbare Organomegalie oder Abwehrspannung. Neurologisch wirkte die Patientin in der Interaktion verlangsamt. Sie war somnolent, jedoch allseits orientiert. Die Meningismusprüfung war schmerzhaft, aber nicht eindeutig positiv. Die Sensibilität der Extremitäten war normal, die Kraft wurde bei M4 eingestuft, und die Hirnnervenprüfung war unauffällig. Das Gangbild war nicht prüfbar wegen allgemeiner Schwäche. Kardiopulmonale Untersuchung, Otoskopie, Integument und enoral, bis auf etwas trockene Schleimhäute mit unauffälligen Befunden.
Differenzialdiagnose
Die diffuse klinische Symptomatik war initial schwierig einzuordnen. Eine (beginnende) Meningitis/Enzephalitis war nicht sicher auszuschliessen. Im Labor fiel vor allem der deutlich erhöhte Kreatininkinasewert auf, welcher zusammen mit den erhöhten Entzündungsparametern differenzialdiagnostisch an eine Myositis oder Myokarditis im Rahmen einer systemischen Inflammation denken liess. Die dadurch ausgelöste Literaturrecherche führte bei bekannter kürzlicher COVID-19-Erkrankung zur Verdachtsdiagnose eines Pediatric Inflammatory Multisystem Syndrome Temporally Associated with SARS-CoV-2 (PIMS-TS). In Europa wird mehrheitlich der Begriff PIMS-TS verwendet, in der US-amerikanischen Terminologie meist die Bezeichnung MIS-C (Multisystem Inflammatory Syndrome in Children). PIMS-TS ist ein seltenes, aber mit hoher Morbidität einhergehendes, meist zwei bis sechs Wochen nach SARS-CoV-2-Infektion auftretendes hyperinflammatorisches Immunreaktionssyndrom mit Beteiligung verschiedener Organsysteme. Die vorangehende COVID-19-Erkrankung ist oft oligo- oder asymptomatisch. Das Syndrom betrifft vor allem Schulkinder (2, 10, 11). Häufigste klinische Zeichen sind hohes Fieber, gastrointestinale Symptome wie Abdominalschmerzen, Erbrechen und Diarrhoe, kardiovaskuläre Dysfunktion (eingeschränkte linksventrikuläre Funktion, Hypotonie, Schock) sowie neurologische Symptome wie Kopfschmerzen und Encephalopathie (1, 2, 4). Die Definitionskriterien umfassen diverse klinische und laboranalytische Parameter, welche je nach Verfasser (WHO, Centers for Disease Control and Prevention; Deutsche Gesellschaft Pädiatrische Infektiologie (DGPI); Schweizerische Gesellschaft für Intensivmedizin) leicht variieren (4, 6, 7).
Differenzialdiagnostische Überschneidungen finden sich mit häufigeren anderen Systemischen Inflammatorischen Syndromen, so mit dem Kawasaki-Syndrom, dem toxischen Schocksyndrom (TSS) und dem Haemophagozytose Lymphohistiocytose Makrophagen Aktivierungssyndrom (SHLH/MAS).
Das Kawasaki-Syndrom ist die häufigste Ursache von in der Kindheit erworbenen Herzerkrankungen, wobei im Gegensatz zum PIMS-TS primär Vorschulkinder betroffen sind. Klinisch-pathologisch stehen ein Exanthem/Enanthem, cervikale Lymphknotenschwellung und die Bildung von Koronaraneurysmata im Vordergrund, im Labor finden sich eine neutrophile Leukozytose und Thrombozytose (1).
Patienten mit PIMS-TS benötigen in den meisten Fällen eine intensivmedizinische Behandlung zur Kreislaufstabilisierung und (seltener) zur Beatmung. Medikamentös wurden bisher, je nach Schweregrad, Kortikosteroide, Immunglobuline und Biologika (v.a. Interleukin-1- und -6-Antagonisten; TNF-alpha-Antikörper) eingesetzt. In der kürzlich publizierten Studie der Swissped RECOVERY Trial Group zeigte sich kein signifikanter Unterschied der Hospitalisationsdauer zwischen der Behandlung mit Methylprednisolon allein im Vergleich zur intravenösen Immunglobulingabe (8). Trotz hoher Morbidität ist die Mortalität bei adäquater Behandlung gering, und es resultieren – soweit bisher bekannt – in der Regel keine langfristigen Folgeschäden (3). Analog dem PIMS-TS/MIS-C wurde bei Erwachsenen das MIS-A (A = adults) beschrieben, welches insgesamt mit schlechteren Verläufen assoziiert ist (1–3).
Diagnose, Therapie und Verlauf
Die Patientin wurde zur Antipyrese und Analgesie (Ibuprofen), Volumensubstitution (Ringeracetat) und Überwachung stationär aufgenommen. In der Nacht war sie teilweise desorientiert, febril (max. 39.9° C) und leicht hypoton (minimal 80/48 mmHg), reagierte jedoch gut auf die etablierte Therapie.
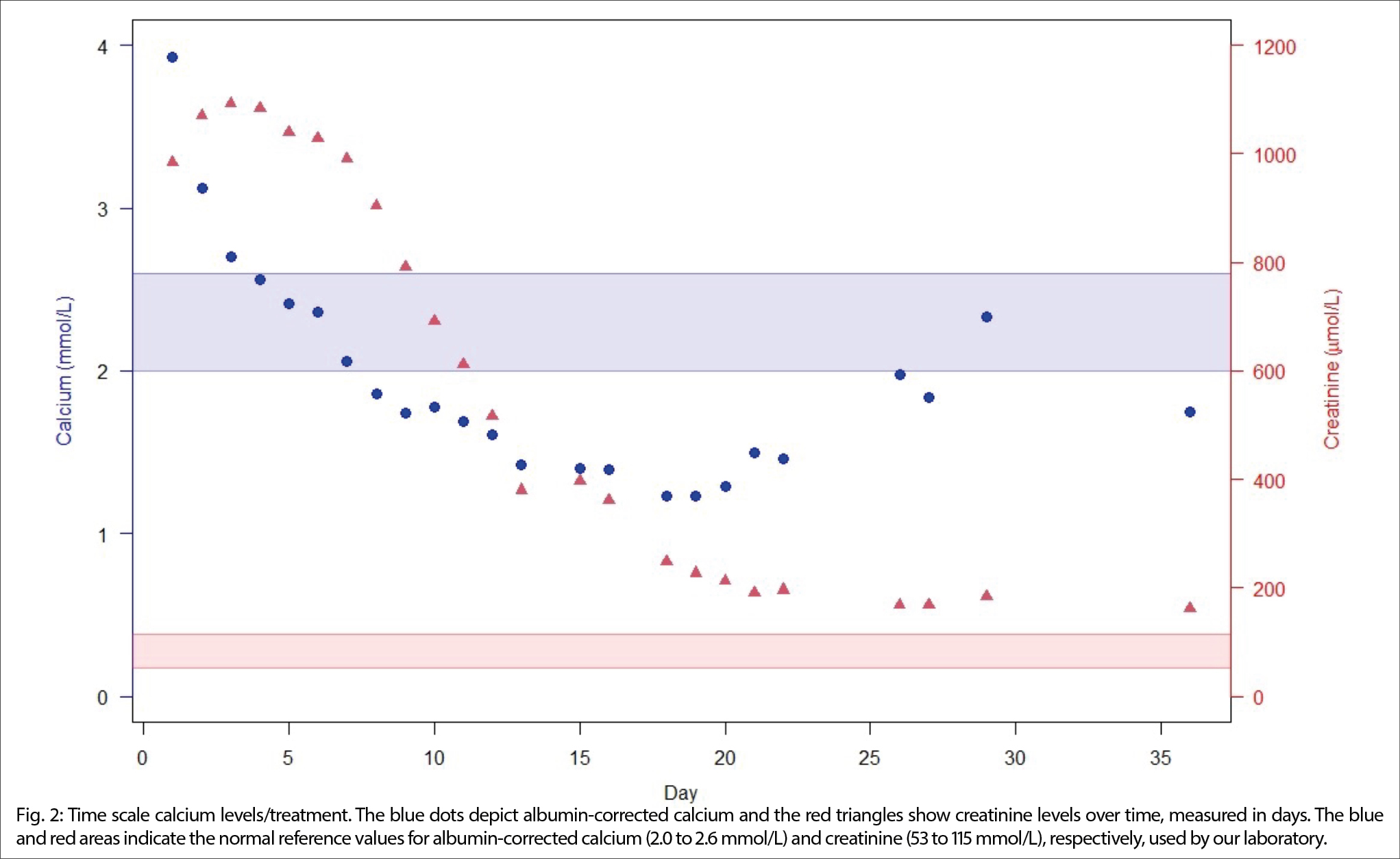
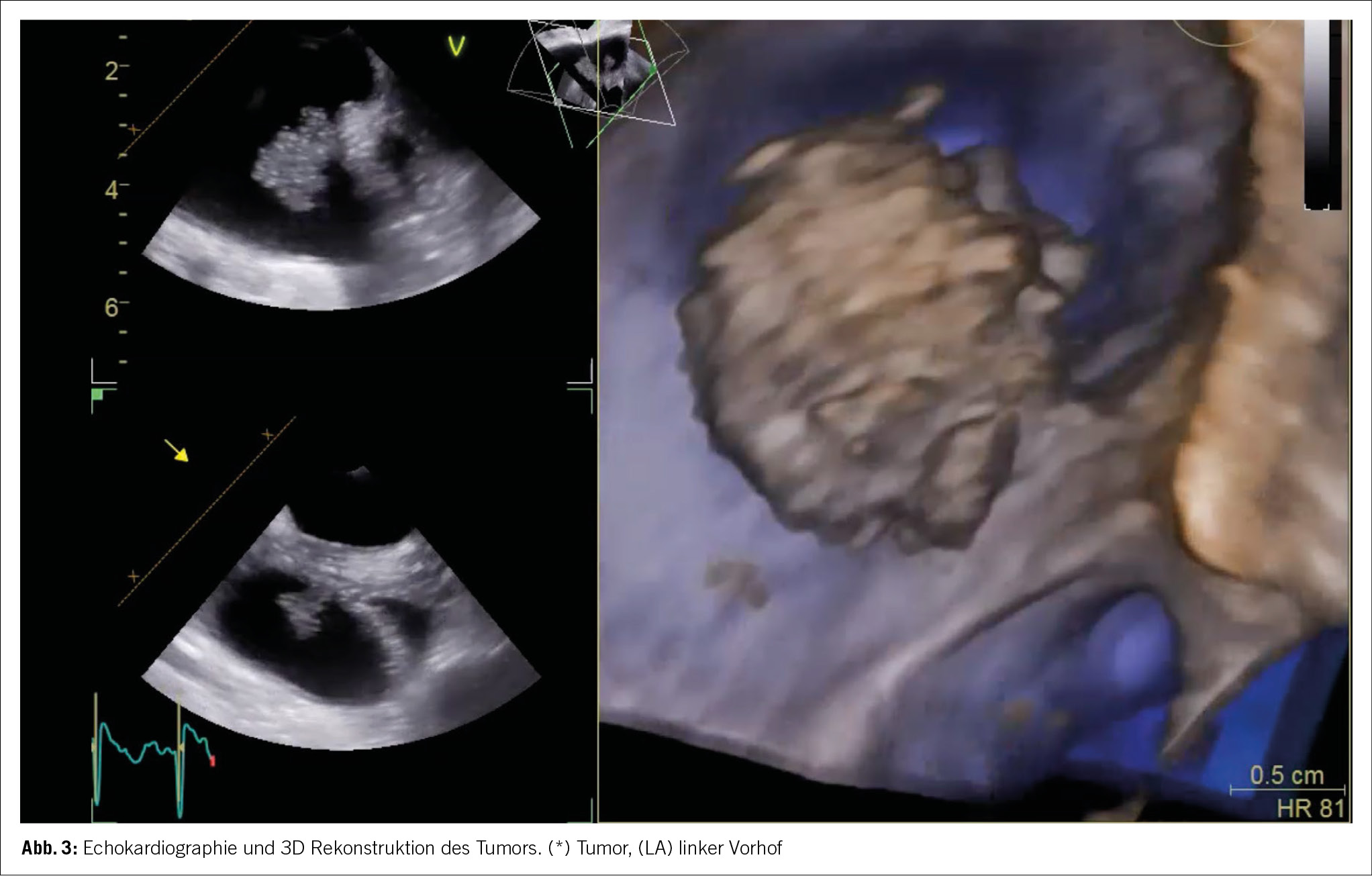
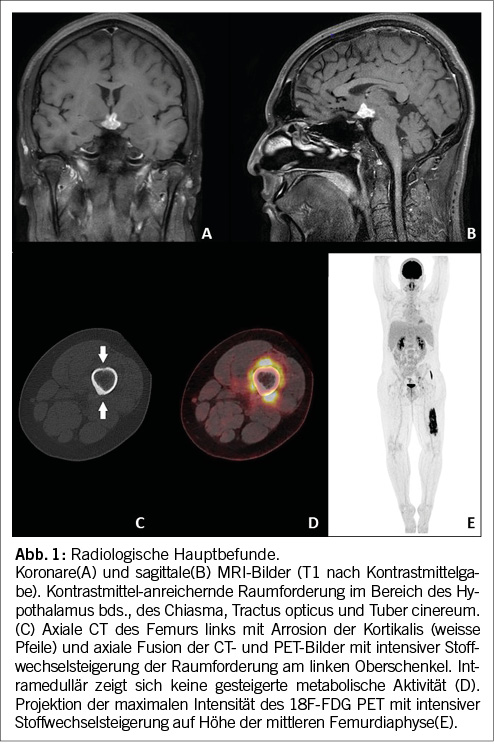
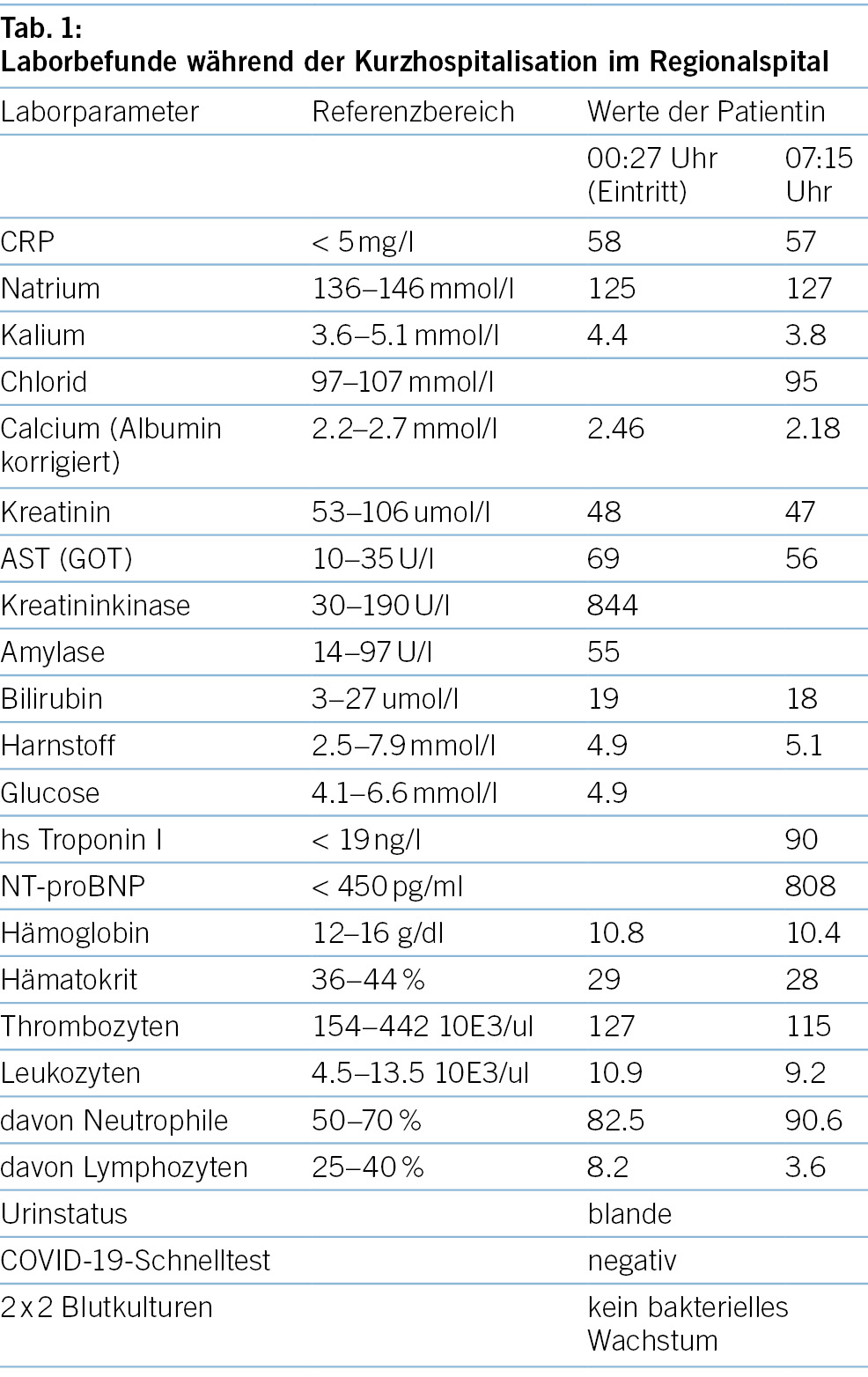
Aufgrund der Gesamtkonstellation wurde rasch die Verdachtsdiagnose eines PIMS-TS mit kardialer, gastrointestinaler, zentralnervöser und hämatologischer Beteiligung gestellt. Zur Diagnosestellung wurden die Kriterien der Schweizerischen Gesellschaft für Intensivmedizin verwendet (4). Im Folgelabor am nächsten Morgen erhärteten die erhöhten Werte von hs Troponin I und NT-proBNP, die charakteristische zunehmende Thrombo- und Lymphopenie und auch eine Hypochloridämie und Hypokalzämie die Verdachtsdiagnose (Tabelle 1). Eine transthorakale Echokardiographie zeigte einen kleinen Perikarderguss (Abbildung 1) und eine leichtgradig verminderte systolische linksventrikuläre Funktion (LVEF 52 %; Globaler Longitudinaler Strain –16 %).
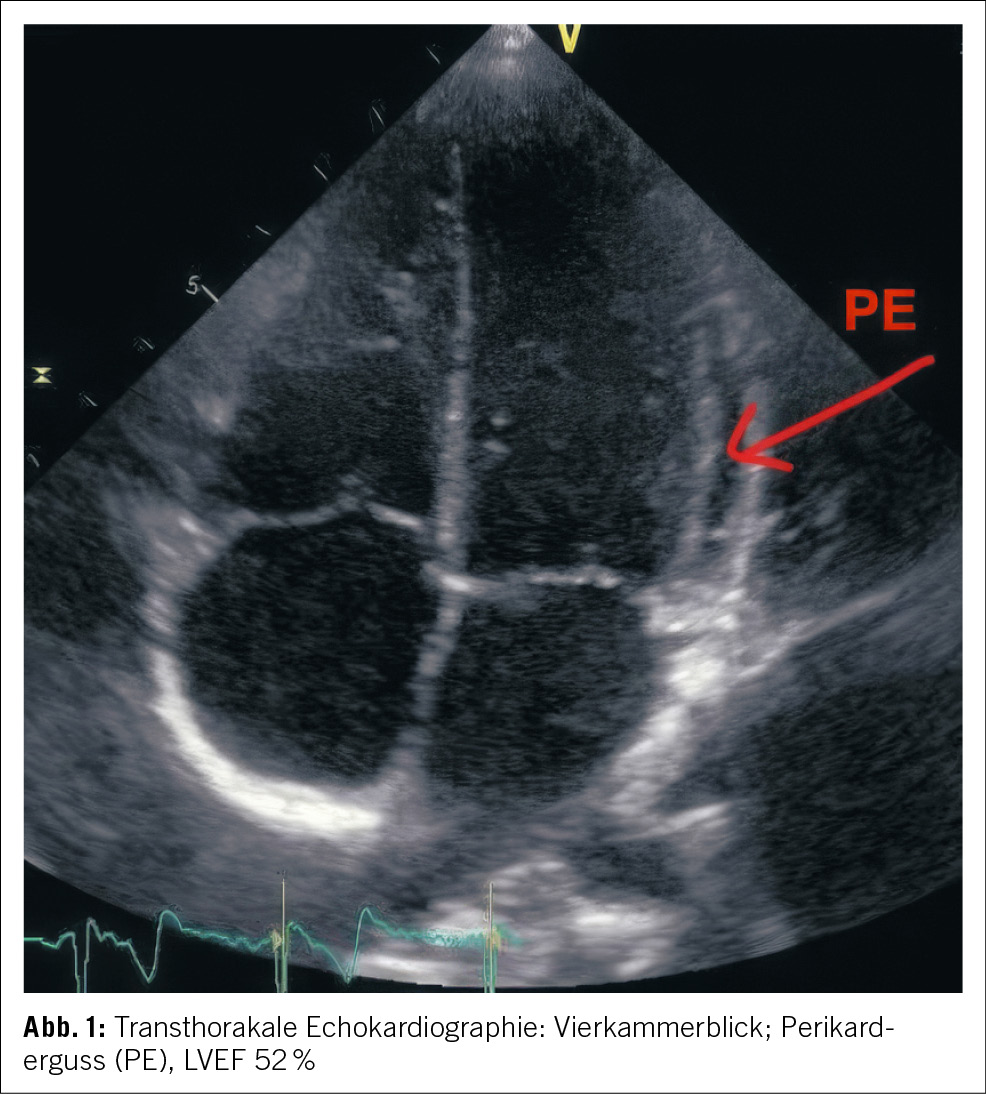
Es erfolgte die notfallmässige Verlegung ins pädiatrische Zentrumsspital zur weiteren Diagnostik und Therapie. Dort wurde die Elfjährige auf die pädiatrische Intensivstation aufgenommen. Weitere Abklärungen zeigten zusätzlich einen Pleuraerguss, Aszites sowie eine Gerinnungsstörung (erhöhter Spontan-INR, erhöhte D-Dimere und Fibrinogen, verlängerte aPTT). Eine Liquorpunktion ergab eine geringe gemischte Pleozytose (56 Leukozyten/ul; davon mononukleär 32/ul und 24/ul polynukleär); Glukose, Laktat und Protein waren im Normbereich.
Die Diagnose eines PIMS-TS wurde bestätigt. Die Patientin wurde katecholamin- (Noradrenalin und Adrenalin) sowie sauerstoffbedürftig (Maske mit Reservoir) und verweilte drei Wochen im Akutspital, davon zwei auf der Intensivstation. Die medikamentöse PIMS-TS-Therapie bestand in der Gabe von Kortikosteroiden, Immunglobulinen und Interleukin-1-Rezeptorantagonisten (Anakinra). Bei fraglichem Meningismus und angesichts der Schocksymptomatik erfolgte eine initiale empirische antibiotische Therapie mit Ceftriaxon, und Aciclovir (kein Erregernachweis), im weiteren Verlauf bei Verdacht auf eitrige Peritonitis Behandlung mit Meropenem und Metronidazol (kein Erregernachweis). Da die Patientin bei Spitalaustritt noch sehr geschwächt war, folgte ein Rehabilitationsaufenthalt. Inzwischen hat sich die Patientin komplett erholt. Eine Diagnose in Bezug auf den tiefen BMI wurde im Verlauf der Hospitalisation und Rehabilitation nicht gestellt.
Diskussion
Schwere COVID-19-Verläufe bei Kindern sind selten. Auch das PIMS-TS als Folgeerkrankung ist selten (7 Fälle auf 100 000 Personen <19 Jahre; (11)) und dadurch wenig bekannt. Deshalb ist, vor allem ausserhalb pädiatrischer Zentrumsspitäler, die Früherkennung des Syndroms schwierig, aber für die Prognose wichtig (12).
Bekannte Risikofaktoren für einen schweren Verlauf mit IPS-Bedürftigkeit sind Alter (5–12 Jahre), Ethnie («non-Hispanic black»), männliches Geschlecht, Adipositas, Abdominalschmerzen, Dyspnoe, verminderte LVEF, Myokarditis und erhöhte Werte für CRP, Troponin, NT-proBNP, Ferritin, D-Dimere, Thrombo- und Lymphopenie (10, 11). Unsere Patientin wies in den ersten 12 Stunden der Hospitalisation alle diese Risikofaktoren auf mit Ausnahme des Geschlechts, einer Adipositas und der sich erst später entwickelnden Dyspnoe. Ein zusätzliches Risiko der Patientin könnte der tiefe BMI gewesen sein. Hierzu gibt es jedoch keine Literatur. Die reduzierte Nahrungs- und Trinkmenge in den 2 Tagen vor der Hospitalisation akzentuierten sicherlich diesen Wert. Eine vorbestehende Essstörung war nicht bekannt, und das übliche stabile Gewicht vor Krankheitsbeginn wie auch nach vollständiger Genesung lag ca. 2 kg höher und somit im Bereich der 3. Perzentile für Industrieländer, wobei verlässliche Daten für Kinder mit dem ethnischen Hintergrund der Patientin fehlen.
Hauptziel dieser Kasuistik ist, bei im Winter weiterhin hoher endemischer Inzidenz von COVID-19 (https://idd.bag.admin.ch/diseases/COVID/overview) die «klinische Awareness» für das PIMS-TS zu erhöhen, damit, wie in unserem Fall, mittels erweiterter Diagnostik eine zeitnahe Diagnosestellung und dadurch rasch adäquate Therapiemassnahmen eingeleitet werden können.
Daten von Luxemburg zeigen, dass die meisten PIMS-TS-Fälle während der Omikron-Welle auftraten, die relative Inzidenz pro SARS-CoV-2-Infektion war jedoch am höchsten während der Delta-Welle (9).
Eine Fall-Kontroll-Studie zeigte, dass 92 % von 304 Patienten mit PIMS-TS nicht SARS-CoV-2 geimpft waren (5). Von den Nichtgeimpften benötigten 44 % intensivmedizinische Unterstützung, von den Geimpften keiner. Ob dies bereits genügend Grund ist, eine Impfung auch für diese Altersgruppe zu empfehlen, bleibt angesichts der Seltenheit der Erkrankung offen. Hierzu braucht es mehr valable Daten.
Am wichtigsten für die Praxis ist die «klinische Aware-ness», aufgrund der Assoziation von klinischer Symptomatik, Laborbefunden und der Anamnese einer kürzlich durchgemachten COVID-19-Erkrankung an dieses Syndrom zu denken. Da in der aktuellen epidemiologischen Situation bei Symptomen eines viralen Infekts in der betroffenen Altersgruppe oft keine spezifische Diagnostik, im Speziellen kein SARS-CoV-2 Test mehr durchgeführt wird, sollte bei entsprechender klinischer Präsentation anamnestisch immer nach einem wenige Wochen vorausgehenden Infekt gefragt und differenzialdiagnostisch ein PMS-TS in Betracht gezogen werden.
Abkürzungen:
BMI Body Mass Index
COVID-19 Corona Virus Disease 2019
LVEF Left Ventricular Ejection Fraction (linksventrikuläre Auswurffraktion)
MIS-C Multisystem Inflammatory Syndrome in Children
PIMS-TS Pediatric Inflammatory Multisystem Syndrome Temporally Associated with SARS-CoV-2
SARS-CoV-2 Severe Acute Respiratory Syndrome Corona Virus type 2
Medizinische Klinik, Ospidal CSEB
Via da l’Ospidal 280
7550 Scuol
gian.flury@cseb.ch
Es bestehen keine Interessenkonflikte.
Historie:
Manuskript eingereicht: 19.02.2024
Nach Revision angenommen: 25.04.2024
1. Nakra NA, Blumberg DA, Herrera-Guerra A, Lakshminrusimha S. Multi-System Inflammatory Syndrome in Children (MIS-C) Following SARS-CoV-2 Infection: Review of Clinical Presentation, Hypothetical Pathogenesis, and Proposed Management. Children 2020;7:69. doi: 10.3390/children7070069. PMID: 32630212; PMCID: PMC7401880.
2. Radia T, Williams N, Agrawal P, Harman K, Weale J, Cook J, Gupta A. Multi-system inflammatory syndrome in children & adolescents (MIS-C): A systematic review of clinical features and presentation. Paediatr Respir Rev. 2021; 38:51-57. doi: 10.1016/j.prrv.2020.08.001. PMID: 32891582; PMCID: PMC7417920.
3. Feldstein LR, Tenforde MW, Friedman KG, et al. Overcoming COVID-19 Investigators. Characteristics and Outcomes of US Children and Adolescents With Multisystem Inflammatory Syndrome in Children (MIS-C) Compared With Severe Acute COVID-19. JAMA 2021; 325:1-14 . doi: 10.1001/jama.2021.2091. PMID: 33625505; PMCID: PMC7905703.
4. Schlapbach LJ, Andre MC, Grazioli S, et al. PIMS-TS working group of the Interest Group for Pediatric Neonatal Intensive Care (IGPNI) of the Swiss Society of Intensive Care and the Pediatric Infectious Diseases Group Switzerland (PIGS). Best Practice Recommendations for the Diagnosis and Management of Children With Pediatric Inflammatory Multisystem Syndrome Temporally Associated With SARS-CoV-2 (PIMS-TS; Multisystem Inflammatory Syndrome in Children, MIS-C) in Switzerland. Front Pediatr. 2021; 9:667507. doi: 10.3389/fped.2021.667507. PMID: 34123970; PMCID: PMC8187755.
5. Zambrano LD, Newhams MM, Olson SM, et al. Overcoming COVID-19 Investigators. Effectiveness of BNT162b2 (Pfizer-BioNTech) mRNA Vaccination Against Multisystem Inflammatory Syndrome in Children Among Persons Aged 12-18 Years – United States, July- December 2021. MMWR Morb Mortal Wkly Rep. 2022; 71 :52-58. doi: 10.15585/mmwr.mm7102e1. PMID: 35025852; PMCID: PMC8757620.
6. WHO Team – WHO International; Publications / Scientific Brief Multisystem inflammatory syndrome in children and adolescents with COVID; WHO/2019- nCoV/Sci_Brief/Multisystem_Syndrome_Children/2020.1 (2020 May 15); (cited 2022 December 06) Available from: Multisystem inflammatory syndrome in children and adolescents with COVID-19 (who.int)
7. Deutsche Gesellschaft Pädiatrische Infektiologie (DGPI) – Falldefinition bei Kindern und Jugendlichen: PIMS bzw. MIS-C; (cited 2022 December 06) Available from: PIMS-Survey: Pediatric Inflammatory Multisystem Syndrome (PIMS) in Deutschland » DGPI: Deutsche Gesellschaft für Pädiatrische Infektiologie
8. Welzel T, Atkinson A, Schöbi N et al for the Swissped RECOVERY Trial Group. Methylprednisolone versus intravenous immunoglobulins in children with paediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2 (PIMS-TS): an open-label, multicentre, randomised trial. Lancet Child Adolesc Health 2023; 7: 238- 48; d oi.org/10.1016/S2352-4642(23)00020-2
9. Ooms C, Mossong J, Vergison JA et al. Multisystem inflammatory syndrome in children during the first two years of the COVID-19 pandemic in Luxembourg. Front Pediatr 2023;11:1141074. d oi: 10.3389/ped.2023.1141074
10. Abrams JY, Oster ME, Godfred-Cato SE et al. Factors linked to severe outcomes in multisystem inflammatory syndrome in children (MIS-C) in the USA: a retrospective surveillance study. Lancet Child Adolesc Health 2021;5:323-31
11. Rhedin S, Lundholm C, Horne AC et al. Risk factors for multisystem inflammatory syndrome in children – A population-based cohort study of over 2 million children. The Lancet Regional Health Europe 2022;19:100443
12. Brisca G, Consolaro A, Caorsi R et al. Timely Recognition and Early Multi-Step Antiinflammatory Therapy May Prevent ICU Admission of Patients With MIS-C : Proposal for a Severity Score. Front Pediatr. 2021;9:783745; Doi: 10.3389/ped.2021.783745