


Im letzten Jahrzehnt hat sich ein zunehmendes Bewusstsein für den Zusammenhang zwischen kardiovaskulärem (CV) Risiko und erhöhten Lipoprotein(a)-Spiegeln, Lp(a), sowohl aus epidemiologischen als auch aus genetischen Studien herauskristallisiert (1, 2, 3). Dieser Beitrag gibt einen Überblick über die derzeitigen Erkenntnisse zu Bedeutung und Umgang mit Lp(a) und Möglichkeiten der Lp(a)-Senkung.
Lipoprotein (a), Lp(a), ist das Produkt der kovalenten Bindung von Low Density Lipoprotein (LDL) und einem weiteren Protein, welches Apolipoprotein (a) genannt wird (4, 5). Apo (a) weist wie Plasminogen 5 Kringel-Einheiten auf, unterscheidet sich aber im Kringel IV von Plasminogen, welches einen Aminosäureaustausch dort aufweist, wo die Spaltung von Plasminogen in Plasmin erfolgt (6). Lp(a) besitzt deshalb keine Plasminaktivität und wirkt potentiell thrombogen. Aufgrund des LDL-Anteils (apo B) wirkt es zudem atherogen.
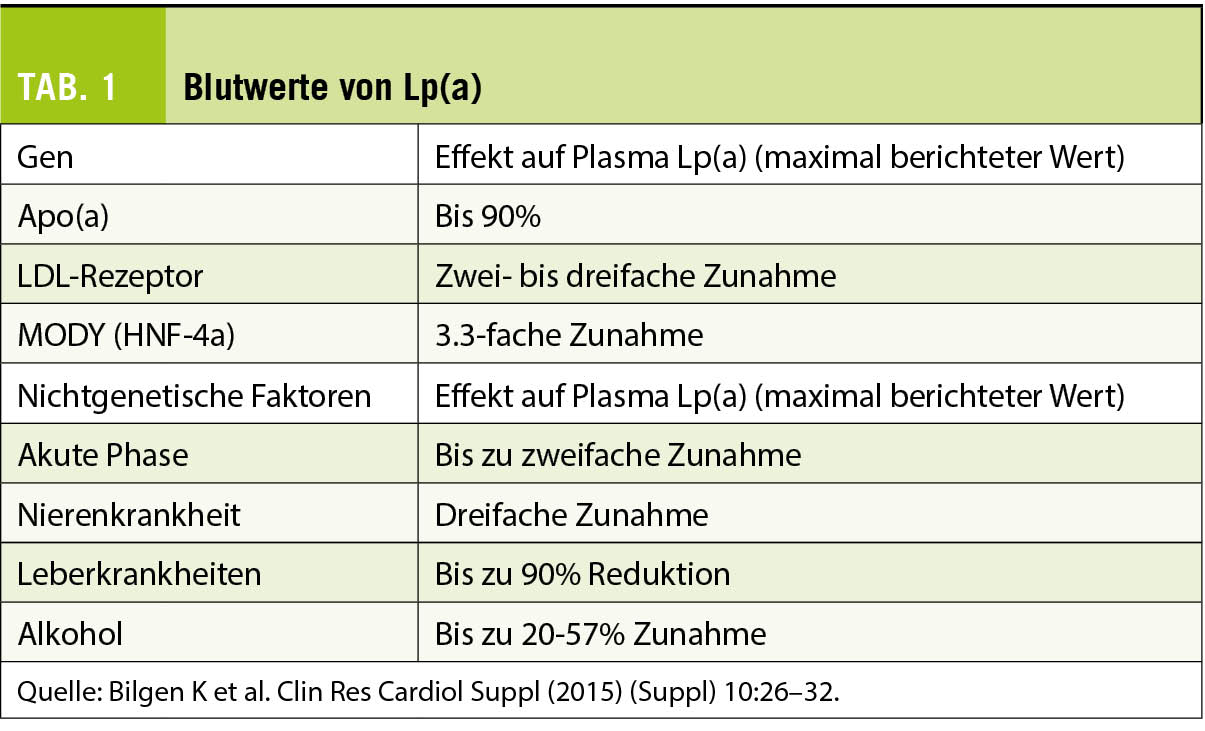
Lp(a)-Plasmawerte werden hauptsächlich genetisch durch den LPA-Genlokus kontrolliert, der durch einen umfangreichen Grössenpolymorphismus von apo(a) gekennzeichnet ist. Dieser Grössenpolymorphismus wird durch eine variable Anzahl verschiedener KIV-2-Wiederholungen verursacht. Die Grösse von Lp(a) ist sehr variabel und reicht von 300 bis 800 kDa. Personen mit kleinen apo(a)-Isoformen haben im Vergleich zu solchen mit grossen apo(a)-Isoformen höhere mediane Lp(a)-Spiegel. Die KIV-2-Kopienzahlvariationen allein erklären 19-77% der Variation der Lp(a)-Spiegel (7). Die Blutwerte von Lp(a werden vor allem durch die Genetik beeinflusst (Tab. 1).
Der Grössenpolymorphismus geht mit Schwierigkeiten bei der Etablierung eines allgemein akzeptierten Messverfahrens einher (9). Erstens geben die verfügbaren Assays die Ergebnisse in Masse (mg/dL) anstelle der Konzentration in nmol/L an, und eine direkte Umrechnung ist aufgrund der variablen Anzahl von Wiederholungseinheiten in verschiedenen apo(a)-Isoformen nicht möglich. Zweitens wurde von absoluten Unterschieden in Lp(a)-Messungen für einzelne Proben mit bis zu fast 80 mg/dL berichtet (10). Drittens sind die Lp(a)-Werte unterschiedlich, wenn die verwendeten Proben frisch sind oder über längere Zeit eingefroren wurden.
Lp(a)-Plasma-Werte oberhalb 30-50 mg/dL oder oberhalb 75-125 nmol/L gehen mit einem erhöhten Atherosklerose-Risiko einher (11). Ob ein klinisch relevanter Effekt auf das Thromboserisiko besteht, gilt als umstritten. Das Herzinsuffizienzrisiko ist wahrscheinlich Folge des erhöhten Risikos für KHK und Aortenklappenstenose. Pathophysiologische, epidemiologische und genetische Studien haben überzeugende Evidenz für die Kausalität von Lp(a) als Risikofaktor für die Entstehung von Myokardinfarkt, Schlaganfall, periphere arterielle Verschlusskrankheit, Herzinsuffizienz und kalzifizierte Aortenklappen-Erkrankungen erbracht (11 - 13).
Lp(a) ist auch mit dem Risiko für plötzlichen Herztod (SCD) assoziiert, wie die prospektive Studie Kupio Ischemic Heart Disease gezeigt hat (14). In dieser Studie wurden bei 1881 Männern im Alter zwischen 42 und 61 Jahren nach medianem Follow-up von 24.7 Jahren 141 SCDs registriert. Die Lipoprotein(a)-Werte waren logarithmisch-linear assoziiert mit dem Risiko für SCD. Die HR für ein SCD pro Standardabweichung (3.56-fach erhöht) war 1.24 (1.05-1.47; P = 0.013).
Lp(a) und residuales kardiovaskuläres Risiko
Die Beziehung zwischen LDL-C und kardiovaskulärem Risiko ist unbestritten, ebenso wie der Nutzen einer lipidsenkenden Behandlung. Es zeigt sich aber, dass trotz Erreichung des LDL-C-Zielwerts ein kardiovaskuläres Restrisiko persistiert. Dieses Restrisiko aufgrund der Lipide ist durch einen Anstieg triglyceridreicher Lipoproteine, tiefem HDL-C, qualitativen Veränderungen der LDL-Partikel und durch erhöhtes Lp(a) gekennzeichnet (15).
In der JUPITER-Studie (16) betrug der erreichte LDL-C-Wert 1.6mmol/l, aber Patienten mit Lp(a) > 50 mg/dL) hatten eine Inzidenzrate von 1.20 vs. 0.99 bei Patienten mit Lp(a) < 10 mg/dl. In der LIPID-Studie (17) betrug der erreichte LDL-C-Wert 2.9 nmol/l. Bei Lp(a)-Werten über 73 mg/dL wurde eine Zunahme von 23% in MACE registriert. Diese Daten unterstützen die unabhängige Rolle von Lp(a) in der Erhöhung des Risikos für kardiovaskuläre Ereignisse trotz optimaler Statintherapie.
Management erhöhter Lp(a)-Werte
In den ESC/EAS-Leitlinien 2019 zum Management von Dyslipidämien wurde empfohlen, mindestens einmal im Leben eine Lp(a)-Messung in Betracht zu ziehen (da der Spiegel stark genetisch bedingt ist) (18). Das Ziel besteht darin, Personen mit einem sehr hohen Lp(a)-Spiegel (≥ 180 mg/dL oder ≥430 mmol/L) und damit einem sehr hohen ASCVD-Risiko (atherosklerotische kardiovaskuläre Erkrankung) zu identifizieren, welches ungefähr dem mit heFH (heterozygote Familiäre Hypercholesterinämie) verbundenen Risiko entspricht. Es wird ferner empfohlen, die Messung von Lp(a) bei ausgewählten Patienten mit einer Familienanamnese für vorzeitige ASCVD und zur Neuklassifizierung von Personen mit grenzwertigem (moderatem bis hohem) Risiko in Betracht zu ziehen. Während die amerikanischen Experten den anormalen Lp(a)-Spiegel als ≥ 50 mg/dL(≥ 100 nmol/L) definierten, wurde in den europäischen Leitlinien kein spezifischer Schwellenwert angegeben (18). Eine Senkung des Lp(a)-Spiegels kann zu einer Verringerung des Risikos für ASCVD führen. Wie jedoch Schätzungen aus Mendelschen Randomisierungsstudien zeigen, könnte die Senkung des Lp(a)-Spiegels zu einer geringeren Verringerung des KHK-Risikos führen als die gleich grosse Senkung von LDL-C (19, 20). Es wurde geschätzt, dass der Lp(a)-Spiegel um 65,7 mg/dL (95% CI 46,3-88,3) gesenkt werden müsste, um das Niveau der Reduktion des KHK-Ereignisrisikos (um 22%) zu erreichen, das mit einer Senkung von LDL-C um 38,7 mg/dL (ca. 1,0 nmol/L) verbunden ist (21).
In den Empfehlungen der (EAS) und der European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) wird unter dem Titel «Quantified atherogenic lipoproteins for lipid-lowering strategies» vorgeschlagen, Lp(a) zu messen, um eine korrekte Messung des LDL-C bei Patienten mit schlechtem Ansprechen auf eine LDL-C-senkende Therapie zu erhalten (22). Bei solchen Patienten und bei Patienten mit sehr tiefen LDL-C-Werten kann der Lp(a)-Cholesterinanteil wesentlich zum gemessenen oder berechneten LDL-C beitragen und kann der Grund für eine geringere als die erwartete Senkung des LDL-C mit Statinen sein. Die Korrektur von LDL-C mit Lp(a)-Werten, die in mg/dL oder nmol/L angegeben werden, kann wie folgt durchgeführt werden:
Lp(a) korrigiertes LDL-C (mg/dL) = LDL-C(mg/dL) – Lp(a)(mg/dL) x 0,30
Lp(a) korrigiertes LDL-C (mmol/L = LDL-C(mmol/dL) – Lp(nmol/dL)) x 0,0078
Lp(a)-Senkung mit PCSK9-Inhibitoren
In der FOURIER-Studie mit Evolocumab war das relative Risiko für CAD-Tod, nicht-tödlichen MI oder die Notwendigkeit einer sofortigen koronaren Revaskularisation über 2,2 Jahre Follow-up im obersten Quartil des Lp(a)-Spiegels am höchsten im Vergleich zum untersten Quartil) und hing nicht vom LDL-C-Spiegel ab (23). Nach 48 Wochen Evolocumab-Therapie war der Lp(a)-Spiegel im Vergleich zu Placebo signifikant um 26,9% gesenkt. Das Risiko betreffend den zusammengesetzten Endpunkt war bei Patienten mit einem Lp(a)-Spiegel über dem Median um 23% niedriger und bei Patienten mit einem Lp(a)-Spiegel unter dem Median nur um 7% reduziert. In der Studie ODYSSEY OUTCOMES mit Alirocumab bei mit Statin-behandelten Patienten nach einem ACS waren die Ergebnisse ähnlich wie in der FOURIER-Studie (24). Die Lp(a)-Basiskonzentration in der Placebogruppe war ein Prädiktor für KHK-Tod, nicht-tödlichen MI, ischämischen Schlaganfall oder Hospitalisierung wegen instabiler Angina pectoris (major adverse CAD events, MACE) über 2,8 Jahre Follow-up (25). Jede Senkung des Lp(a)-Werts um 1,0 mg/dL unter Alirocumab war mit einer signifikanten Reduktion von MACE verbunden (HR 0,994, 95% CI 0,990-0,999). Dies galt auch für Senkung des mit Lp(a)-korrigierten LDL-C-Werts um 1,0 mg/dL. Die Autoren folgerten: «Die Basiskonzentrationen von Lipoprotein(a) und korrigiertem LDL-C sowie deren Senkung durch Alirocumab sagten das Risiko für MACE nach ACS voraus. Die Lp(a)-Senkung durch Alirocumab trägt unabhängig zur MACE-Reduktion bei, was nahelegt, dass Lipoprotein(a) ein unabhängiges Behandlungsziel nach ACS sein sollte.» (25). Die jüngste Post-hoc-Analyse der Phase-III-ODYSSEY-
Studien (ohne ODYSSEY-OUTCOMES) zeigte jedoch, dass die Senkung des Lp(a)-Basiswertes um 23,5 mg/dL mit Alirocumab (um 26,6% vs. 2,5% mit Placebo und um 21,4% vs. 0,0% mit Ezetimibe) nicht zu einer signifikanten Reduktion der schwerwiegenden koronaren Ereignisse (MACE) unabhängig von der Senkung des LDL-C-Wertes führte (26). Die Autoren folgerten, dass die Reduktion des MACE-Risikos durch eine gezielte Lp(a)-Senkung möglicherweise eine stärkere Lp(a)-Senkung mit wirksameren Therapien und/oder höheren Lp(a)-Anfangswerten erfordert.
Lp(a)-Senkung mit Antisense-Oligonukleotiden
Eine wirksamere Senkung von Lp(a) kann mit Antisense-Oligonukleotiden erzielt werden (27-29). Mit dem Antisense-Oligonukleotid AKCEA-APO(a)-LR (TQJ230; Pelacarsen) wurde eine mittlere Reduktion von Lp(a) um mehr als 80% erreicht (30). TQJ230 wird von Novartis weiterentwickelt und vermarktet. Die derzeit laufende HORIZON-Studie hat zum Ziel herauszufinden, ob TQJ230 sicher ist. Ausserdem möchte man untersuchen, ob TQJ230 das Risiko der Herz-Kreislauf-Erkrankten mit erhöhtem Lp(a)-Spiegel verringert, eine akute Verschlechterung zu erleiden.
Eine weitere Option ist die Lp(a)-Senkung durch RNAsilencing mit Olpasiran (Amgen 890) (31).
Copyright bei Aerzteverlag medinfo AG
riesen@medinfo-verlag.ch
Advisory Boards und Referentenhonorare Daiichi Sankyo, Amgen, Recordati, MSD und Sanofi.
1. Emerging Risk Factors Collaboration. Erqou S. et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–423.
2. Kamstrup P.R et al. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA 2009;301:2331–2339.
3. Saleheen D et al. Apolipoprotein(a) isoform size, lipoprotein(a) concentration, and coronary artery disease: A mendelian randomisation analysis. Lancet Diabetes Endocrinol. 2017;5:524–533.
4. Trieu V.N., McConathy W.J. A two-step model for lipoprotein(a) formation. J. Biol. Chem. 1995;270:15471–15474.
5. Vuorio A et al. Familial hypercholesterolemia and elevated Lp(a) : Double heritable risk and new therapeutic opportunities. J Intern Med 2020 ;287 :2-18.
6. McLean JW et al. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987 ;330(6144):132-7.
7. Schmidt K et al. Structure, function, and genetics of lipoprotein (a) J. Lipid Res. 2016;57:1339–1359.
8. Wilson D.P et al. Use of Lipoprotein(a) in clinical practice: A biomarker whose time has come. A scientific statement from the National Lipid Association. J. Clin. Lipidol 2019;13:374–392.
9. Scharnagl Het al. Comparison of lipoprotein (a) serum concentrations measured by six commercially available immunoassays. Atherosclerosis. 2019;289:206–213.
10. Erqou S et al, Emerging Risk Factors Collaborations. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke and non cardiovascular mortality. JAMA 2009;302:412-423.
11. Kamstrup PR et al. Extreme lipoprotein(a) levels and improved cardiovascular risk prediction. J Am Coll Cardiol 2013;61:1146-1156.
12. Nodestgaard BG et al. Lipoprotein (a) as a cardiovascular risk factor : current status. Eur Heart J 2010 ; 31 : 2844-2853.
13. Tsimikas SJ. Evidence that elevated lipoprotein (a) (Lp(a)) levels contribute to cardiovascular disease. JACC 2017 ;69 :692-711.
14. Kunutsor SK et al. Lipoprotein(a) and risk of sudden cardiac death in middle-aged Finnish men: A new prospective cohort study. Int J Cardiol. 2016 1;220:718-25.
15. Schwartz GG et al. Fasting triglycerides predict recurrent ischemic events in patients with acute coronary syndrome treated with statins. J Am Coll Cardiol, 2015; 65: 2267-227.
16. Khera AV, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation, 2014; 129: 635- 642.
17. Nestel PJ et al. Plasma lipoprotein(a) concentration predicts future coronary and cardiovascular events in patients with stable coronary heart disease. Arterioscler Thromb Vasc Biol 2013;33:2902-8.
18. Mach F et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. The task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European atherosclerosis society (EAS). Eur Heart J 2020;41:111-188.
19. Burgess S et al. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a) lowering therapies. A Mendelian randomization analysis. JAMA Cardiol 2018;3:619-627.
20. Lamina C, Kronenberg F. Estimation of the required lipoprotein(a) lowering therapeutic effect size for reduction in coronary heart disease outcomes. JAMA Cardiol 2019;4:575-579.
21. Kamstrup PR, Tybiaerg-Hansen A, Nordestgaard BG. Genetic evidence that lipoprotein(a) associates with atherosclerotic stenosis rather than venous thrombosis. Atheroscler Thromb Vasc Biol 2012;32:1732-1741.
22. Nordestgaard BG et al. Quantifying atherogenic lipoproteins for lipid lowering strategies: consensus- based recommendations from EAS and EFLM. Atherosclerosis 2020;294:46-61.
23. O’Donoghue ML et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Insights from FOURIER trial. Circulation 2019;139:1483-1492.
24. Bittner V et al. Effect of alirocumab on lipoprotein(a) and cardiovascular risk after acute coronary syndrome. J Am Coll Cardiol 2020;75(2) https://doi.org/10.1016/jack.2019.10.057.
25. Ray KK, Valleyo-Vaz AJ, Ginsberg HN, et al. Lipoprotein(a) reductions from PCSK9 inhibition and major adverse cardiovascular events: pooled analysis of alirocumab phase 3 trials. Atherosclerosis 2019 https://doi.org/10.1016/j.atherosclerosis.2019.06.896.
26. Santos RD et al. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein(a) in various populations with hypercholesterolemia: results of 4 phase III trials. Arterioscler Thromb Vasc Biol 2015; 35: 689-699.
27.Tsimikas S et al. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015; 386:1472-1483.
28. Graham MJ et al. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J Lipid Res, 2016; 57:340-351.
29. Viney NJ et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials Lancet 2016; 388: 2239-2253.
30. Tsimikas S et al. Lipoprotein(a) reduction in persons with cardiovascular disease N Engl J Med 2020; 382:244-255
31. Koren MJ et al. Abstract 13951: safety, tolerability and efficacy of single-dose Amg 890, a novel sirna targeting Lp(a), in healthy subjects and subjects with elevated Lp(a). Circulation. 2020;142:A13951.

der informierte @rzt
- Vol. 11
- Ausgabe 7
- Juli 2021