

Morbus Osler (hereditäre hämorrhagische Teleangiektasie, HHT) ist eine seltene, autosomal-dominant vererbte Multisystemerkrankung mit einer Prävalenz von etwa 1 : 5000 bis 1 : 8000. Charakteristisch sind wiederkehrende, spontane Blutungen (vor allem Epistaxis), mukokutane Teleangiektasien sowie arteriovenöse Malformationen (AVMs) in Organen wie Lunge, Leber, Gehirn und dem Gastrointestinaltrakt. Die Diagnose basiert auf den klinischen Curaçao-Kriterien und wird durch genetische Analysen (ENG, ACVRL1, SMAD4) gesichert. Das Erkrankungsbild ist heterogen und reicht von milden Verläufen bis zu schwerwiegenden Komplikationen wie Schlaganfall, Herzinsuffizienz oder chronischer Blutungsanämie. Die Therapie umfasst symptomorientierte lokale Massnahmen sowie moderne systemische Ansätze, beispielsweise mit Bevacizumab oder Pomalidomid. Um Komplikationen frühzeitig zu erkennen und die Prognose zu verbessern, ist eine lebenslange, interdisziplinäre Betreuung der Patienten erforderlich.
Hereditary hemorrhagic telangiectasia (HHT, Osler’s disease) is a rare autosomal-dominant multisystem disorder with a prevalence of approximately 1 : 5000 to 1 : 8000. The disease is characterized by recurrent, spontaneous bleeding (most commonly epistaxis), mucocutaneous telangiectasias, and arteriovenous malformations (AVMs) in organs such as the lungs, liver, brain, and gastrointestinal tract. Diagnosis is based on the clinical Curaçao criteria and confirmed by genetic analysis (ENG, ACVRL1, SMAD4). The clinical spectrum is highly variable, ranging from mild manifestations to severe complications such as stroke, heart failure, or chronic anemia due to bleeding. Treatment includes local symptomatic measures as well as novel systemic approaches, for example with bevacizumab or pomalidomide. Lifelong, interdisciplinary management is essential to enable early detection of complications and to improve long-term prognosis for patients.
Keywords: Hereditary hemorrhagic telangiectasia, arteriovenous malformation, epistaxis
Einleitung und Übersicht
Die Pathophysiologie des Morbus Osler beruht auf einer gestörten Angiogenese mit Ausbildung fragiler Gefässverbindungen (1). Verursacht wird sie überwiegend durch Mutationen in den Genen ENG und ACVRL1 sowie seltener in SMAD4 (2-4). Diese Gene codieren für zentrale Komponenten des TGF-β-Signalwegs, der eine Schlüsselrolle bei der Gefässentwicklung und -stabilität spielt (5-8). Eine gestörte Signalübertragung im TGF-β/BMP-Signalweg führt zu einer Überaktivität des vaskulären endo-thelialen Wachstumsfaktors (VEGF), wodurch Teleangiektasien und arteriovenöse Malformationen (AVMs) in verschiedenen Organen wie Lunge oder Leber entstehen (1, 5, 6). Trotz hoher Penetranz zeigt die Erkrankung ein breites Spektrum klinischer Ausprägungen. Die Diagnose stützt sich auf die Curaçao‑Kriterien und wird durch bildgebende sowie molekulargenetische Untersuchungen ergänzt (9–12). Diese ermöglichen eine präzise genetische Klassifikation und sind essenziell für die familiären Abklärungen und das individuelle Risikomanagement.
Diagnostik des Morbus Osler
Bei der Diagnose von Morbus Osler sollten bereits in der Anamnese und bei der körperlichen Untersuchung typische Manifestationen systematisch erfasst werden. Hierzu zählen rezidivierendes, oft spontan auftretendes Nasenbluten (Epistaxis), Teleangiektasien der Haut und Schleimhäute (v. a. an Lippen, Zunge, Fingerkuppen) sowie Hinweise auf viszerale arteriovenöse Malformationen (AVMs).
Die klinische Präsentation dieser AVMs variiert je nach betroffenem Organ: pulmonale AVMs können sich durch Dyspnoe, Zyanose oder paradoxe Embolien äussern; zerebrale AVMs durch Kopfschmerzen, epileptische Anfälle oder hämorrhagische Insulte; hepatische AVMs durch Zeichen einer High‑Output‑Herzinsuffizienz oder einer Leberfunktionsstörung (13–15).
Obwohl erste Symptome häufig schon in der Kindheit auftreten, wird die Diagnose in vielen Fällen erst verzögert gestellt – in Studien mitteleuropäischer Kohorten lag die mittlere Zeit zwischen Symptombeginn bis zur Diagnosestellung bei etwa 15 Jahren (16).
Eine strukturierte Abklärung ist vor allem bei Patienten mit unklaren Anämien, ausgeprägter Epistaxis oder positiver Familienanamnese indiziert. Grundlage der klinischen Beurteilung sind die Curaçao‑Kriterien (10). Diese umfassen vier Hauptmerkmale: spontane und rezidivierende Epistaxis, multiple Teleangiektasien an typischen Stellen, viszerale AVMs sowie eine positive Familienanam-nese bei einem Verwandten ersten Grades. Die Diagnose gilt als gesichert, wenn mindestens drei Kriterien erfüllt sind; bei zwei Kriterien ist sie möglich, bei weniger als zwei unwahrscheinlich.
Zur Abklärung viszeraler AVMs werden organspezifische bildgebende Verfahren wie Transthorakale Kontrast-Echokardiographie (TTCE), Magnetresonanz-Tomographie (MRT) oder Sonographie eingesetzt (12, 13, 17); weiterführende Einzelheiten hierzu sind im Abschnitt zum Screening dargestellt.
Die molekulargenetische Untersuchung ist zentral für die Diagnosesicherung und das Familienscreening. Bei der Mehrzahl der Betroffenen lassen sich Mutationen in den Genen ENG, ACVRL1 und seltener SMAD4 nachweisen (2–4). Der Nachweis einer krankheitsverursachenden Variante ermöglicht eine eindeutige molekulare Klassifikation und eine gezielte prädiktive Testung von Angehörigen ersten Grades. In Einzelfällen kann eine genetische Analyse bereits bei unklarer klinischer Präsentation entscheidend sein, etwa bei Kindern vor Auftreten der typischen Symptome oder bei isolierten Organmanifestationen.
Genetische Varianten und neue Klassifikation bei Morbus Osler
Die genetische Heterogenität bei Morbus Osler ist ausgeprägt, da mehrere Gene an der Krankheitsentstehung beteiligt sein können. Dennoch entfallen etwa 85–90 % aller Fälle auf Mutationen in ENG und ACVRL1 (2, 3). Auf dieser Grundlage wurde eine erweiterte molekulare Klassifikation vorgeschlagen, die dabei helfen soll, unterschiedliche klinische Schwerpunkte besser einzuordnen und ein gezielteres Screening vornehmen zu können (11, 12). Die bekannten Subtypen sowie ihre typischen Organmanifestationen sind in Tabelle 1 zusammengefasst und werden nachfolgend näher erläutert.
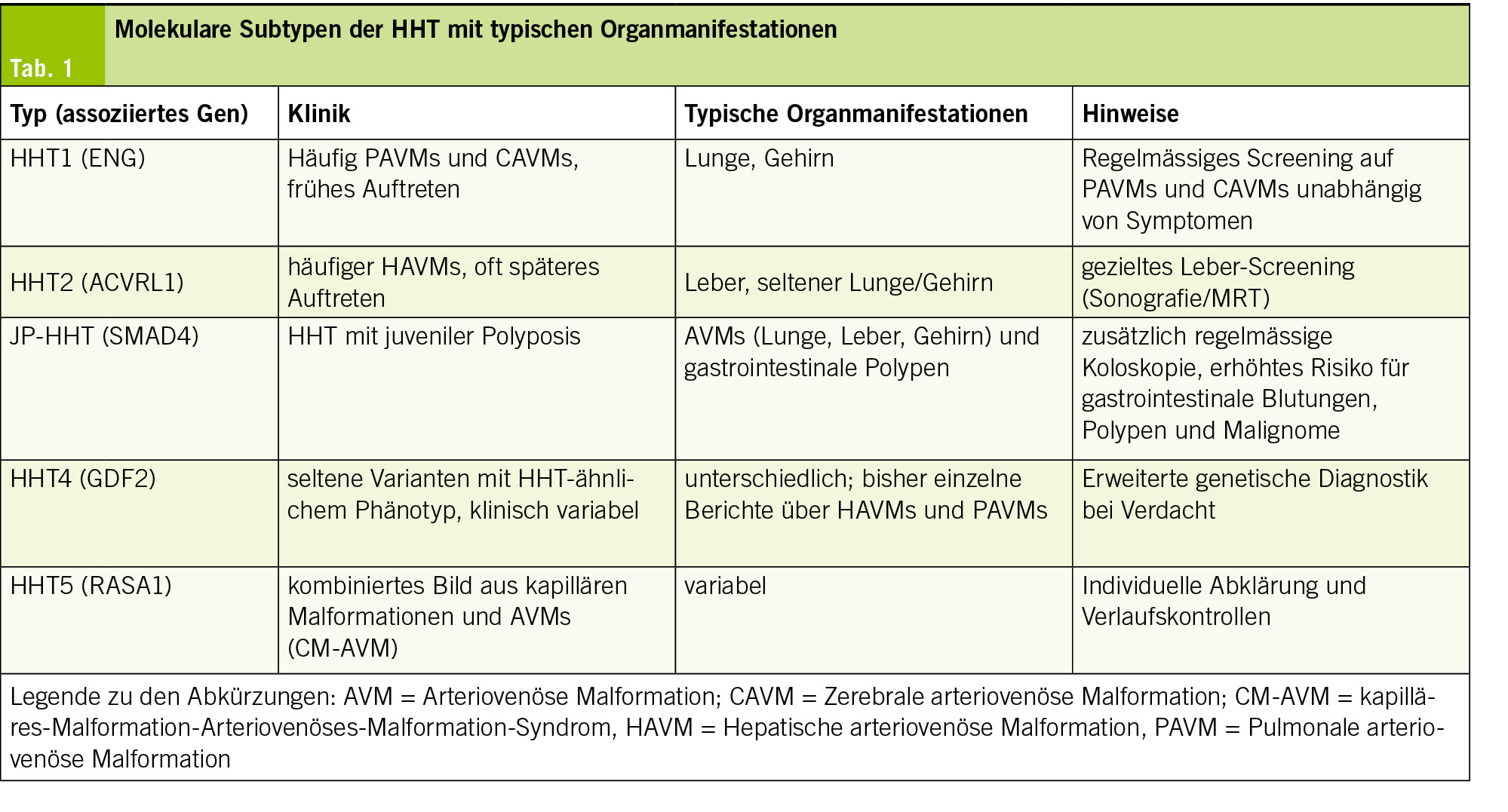
HHT Typ 1 (ENG‑assoziiert)
Mutationen im ENG‑Gen (Endoglin) sind für etwa 40–50 % aller Erkrankungen von Morbus Osler verantwortlich (2,7). Endoglin kodiert für einen endothelialen Co-Rezeptor im TGF‑β‑Signalweg, der für eine stabile Gefässentwicklung und -reifung essenziell ist. Defekte in diesem Gen führen zu einer gestörten Signalübertragung und resultieren in einer inadäquaten Angiogenese mit erhöhter Gefässfragilität. HHT1 ist oft mit einem frühen Krankheitsbeginn verbunden. Typisch sind eine ausgeprägte Epistaxis und ein erhöhtes Risiko für pulmonale AVMs (PAVMs) und zerebrale AVMs (CAVMs). Studien zeigen, dass über 50 % der ENG‑Mutationsträger PAVMs entwickeln (1, 19). Aufgrund der hohen Penetranz wird bei Nachweis einer ENG‑Mutation ein regelmässiges Screening von Lunge und Gehirn empfohlen, auch bei asymptomatischen Patienten (12, 17).
HHT Typ 2 (ACVRL1‑assoziiert)
HHT Typ 2 beruht auf Mutationen im ACVRL1‑Gen, einem Serin/Threonin‑Kinase‑Rezeptor im TGF‑β‑Signalweg (3). Diese führen zu einer verminderten Kinaseaktivität und somit zu einer gestörten Signalübertragung. Klinisch manifestiert sich HHT2 häufiger mit hepatischen AVMs (HAVMs), die zu High‑Output‑Herzinsuffizienz oder biliären Komplikationen führen können (15, 20). Im Vergleich zu HHT1 treten die Symptome oft später auf und das Risiko für zerebrale AVMs scheint geringer zu sein. Aufgrund der Leberbeteiligung wird ein gezieltes Leber‑Screening (Sonografie und ggf. MRT) empfohlen (18).
JP‑HHT (SMAD4‑assoziiert)
Mutationen im SMAD4‑Gen verursachen eine seltene Sonderform der HHT, die als Juvenile‑Polyposis‑HHT‑Overlap‑Syndrom (JP‑HHT) bezeichnet wird. Diese Patienten erfüllen nicht nur die klassischen Kriterien der hereditären hämorrhagischen Teleangiektasie, sondern zeigen zusätzlich klinische Merkmale einer juvenilen Polyposis (4, 21). SMAD4 kodiert für ein intrazelluläres Signalmolekül des TGF‑β/BMP‑Signalwegs, das als Transkriptionsfaktor die Regulation der Gefässreifung und die Homöostase epithelialer Gewebe steuert (8). Neben den für Morbus Osler typischen Teleangiektasien und AVMs in Lunge, Leber und Gehirn treten bei SMAD4‑Mutationsträgern multiple juvenile Polypen im Gastrointestinaltrakt auf. Diese Polypen verursachen häufig bereits im Kindes- oder jungen Erwachsenenalter chronische Blutungen und Eisenmangelanämie und erhöhen langfristig das Risiko für gastrointestinale Malignome (12, 22). Daher werden neben dem Screening auf AVMs auch regelmässige Koloskopien empfohlen, um Polypen rechtzeitig zu behandeln und Komplikationen zu vermeiden.
HHT Typ 4 (GDF2‑assoziiert)
Seltene pathogene Varianten im GDF2‑Gen (Growth Differentiation Factor 2, kodiert für BMP9) wurden in den letzten Jahren als weitere Ursache eines HHT‑ähnlichen Phänotyps identifiziert. BMP9 ist ein Ligand des ALK1‑Rezeptors und spielt eine zentrale Rolle im TGF‑β/BMP‑Signalweg, insbesondere bei der Regulation der Angiogenese und Gefässhomöostase (8). Die bisher bekannten Fälle zeigen eine variable Klinik. In publizierten Einzelfallberichten und Fallserien wurden insbesondere hepatische und pulmonale AVMs beschrieben, die sich klinisch durch Dyspnoe, Hypoxie oder Zeichen einer High‑Output‑Herzinsuffizienz äussern können (23). Bei klinischem Verdacht auf Morbus Osler, jedoch ohne Nachweis einer Mutation in ENG, ACVRL1 oder SMAD4, sollte eine erweiterte genetische Diagnostik auf das GDF2-Gen in spezialisierten Zentren erwogen werden (9, 12).
HHT Typ 5 (RASA1‑assoziiert)
HHT Typ 5 wird mit Mutationen im RASA1‑Gen assoziiert, das für einen Regulator des RAS/MAPK‑Signalwegs kodiert und dadurch die Gefässentwicklung beeinflusst. Klinisch zeigt sich meist ein CM‑AVM‑Syndrom (Capillary-Malformation–Arteriovenous-Malformation-Syndrom), das durch multiple kapilläre Hautmalformationen und teils symptomatische AVMs gekennzeichnet ist. Anders als bei HHT1 oder HHT2 ist die Organbeteiligung weniger vorhersehbar. Die Literatur beschreibt heterogene Phänotypen von asymptoma-tischen Hautveränderungen bis hin zu schwerwiegenden viszeralen AVMs (24,25).
Die genetische Diagnostik sollte stets im Zusammenhang mit der klinischen Phänotypisierung erfolgen. Eine alleinige genetische Analyse ohne klinischen Kontext birgt die Gefahr von Fehlinterpretationen, insbesondere bei Varianten unklarer Signifikanz (VUS). Zudem ist zu berücksichtigen, dass bei etwa einem Drittel der Familien keine vollständige Penetranz besteht und sporadische Mutationen, insbesondere bei SMAD4‑assoziierten Formen, auftreten können (4, 7, 9). Die molekulare Diagnostik ist inzwischen ein integraler Bestandteil des Managements von Morbus Osler, da sie eine differenzierte Risikoeinschätzung, die gezielte Prävention potenziell lebensbedrohlicher Komplikationen und eine fundierte Familienberatung ermöglicht. Mit der Weiterentwicklung genomischer Technologien ist zudem mit der Identifizierung weiterer Gene und Signalwegkomponenten zu rechnen, die zur Pathogenese der HHT beitragen (17, 23, 26).
Sekundäre Organschädigungen bei Morbus Osler
Nach der Diagnose eines Morbus Osler (HHT) ist eine systematische Abklärung potenzieller Organbeteiligungen unerlässlich, da die Erkrankung durch AVMs in verschiedenen Organsystemen gekennzeichnet ist. Die häufigsten Manifestationen betreffen die Lunge, das zentrale Nervensystem (ZNS), die Leber sowie den Gastrointestinaltrakt. Auch wenn diese Organbeteiligungen in vielen Fällen lange asymptomatisch sind, können sie jedoch zu erheblichen Morbiditäten führen. Daher ist ein strukturiertes und frühzeitiges Screening essenziell, idealerweise bereits bei klinischem Verdacht auf Morbus Osler oder Nachweis von spezifischen molekulargenetischen Veränderungen (12, 17).
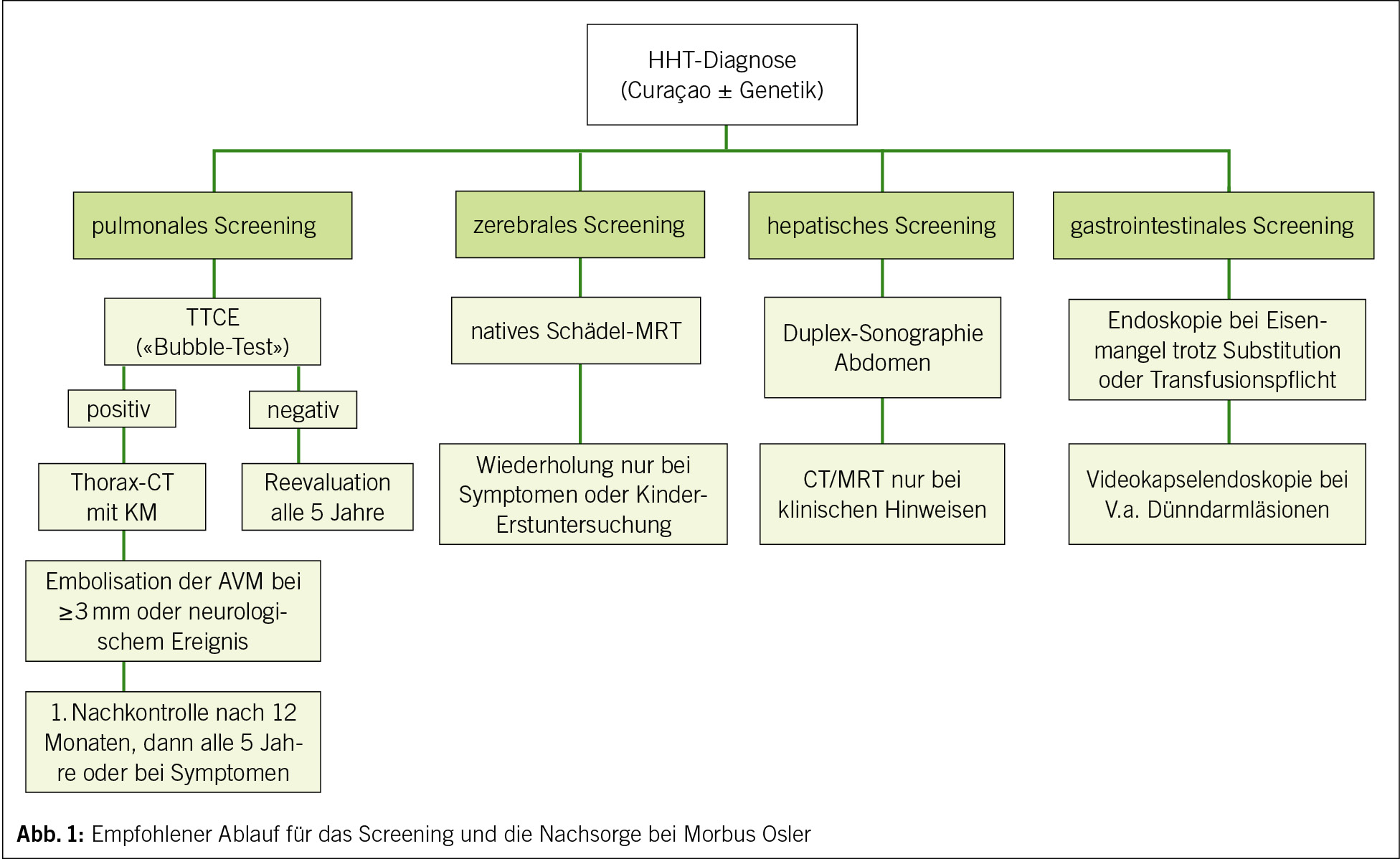
Die in Abbildung 1 dargestellte Übersicht zeigt den aktuellen Standard der Versorgung in vielen auf Morbus Osler spezialisierten Zentren: Beginnend mit der Diagnosesicherung folgen standardisierte Untersuchungen zur Erfassung pulmonaler, zerebraler, hepatischer und gastrointestinaler Manifestationen. Je nach Befund sind die Verlaufskontrollen in definierten Intervallen vorgesehen. Im Folgenden werden die organspezifischen Aspekte näher erläutert.
Pulmonale AVMs (PAVMs)
PAVMs sind bei bis zu 50 % der Betroffenen nachweisbar und können zu relevanten Komplikationen wie zentraler Zyanose, Schlaganfällen durch paradoxe Embolien oder septischen Infarkten führen (20). Das empfohlene Screening erfolgt mittels transthorakaler Kontrast-Echokardiographie (TTCE) mit Bubble-Test, idealerweise bereits im Kindesalter (27). Bei positivem TTCE‑Befund wird eine kontrastverstärkte Thorax‑CT (Computertomographie) zur genauen Lokalisation und Grössenbeurteilung durchgeführt. PAVMs ab einem Durchmesser von 2–3 mm oder mit relevanter Rechts-Links-Shuntfunktion sollten embolisiert werden, um Komplikationen zu vermeiden. Nach erfolgreicher Therapie ist eine Nachkontrolle nach 12 Monaten indiziert, anschliessend in Fünfjahresintervallen oder bei Symptomen (28, 29). Ist die TTCE unauffällig, spricht dies zunächst gegen das Vorliegen relevanter PAVMs. Da jedoch neue Läsionen im Verlauf entstehen können, wird eine Reevaluation in regelmässigen Intervallen empfohlen, in der Regel alle fünf Jahre oder bei neu auftretenden Symptomen (12). Aufgrund des erhöhten Risikos für septische Embolien wird bei Patientinnen und Patienten mit nachgewiesenen PAVMs eine konsequente Infektprophylaxe entsprechend einer antibiotischen Endokarditisprophylaxe bei invasiven Eingriffen oder zahnärztlichen Behandlungen empfohlen (12, 13).
Zerebrale AVMs (CAVMs)
CAVMs treten in etwa 10–20 % der Fälle auf (30). Die Manifestationen reichen klinisch von asymptomatischen Befunden über Kopfschmerzen bis zu Krampfanfällen oder hämorrhagischen Insulten (14). Ein einmaliges Screening mittels MRT des Schädels (mit oder ohne Kontrastmittel) ist bei neu diagnostiziertem Morbus Osler empfohlen, bevorzugt im Kindes- oder jungen Erwachsenenalter. Weitere Verlaufskontrollen erfolgen in der Regel nur bei neu aufgetretenen Symptomen oder Erstuntersuchung als Kind (12).
Hepatische AVMs (HAVMs)
HAVMs sind bei bis zu 70–80 % der erwachsenen M. Osler-Patienten nachweisbar, verlaufen aber häufig asymptomatisch (20). Sie können jedoch zu High-Output-Herzinsuffizienz, portaler Hypertension oder biliären Komplikationen führen (15). Das Screening sollte mittels abdomineller Duplex‑Sonografie erfolgen, bei auffälligen Befunden oder klinischen Hinweisen ergänzt durch CT oder MRT (18). Eine prophylaktische Therapie ist nicht angezeigt, jedoch erfordert eine kardiale Belastung oder eine beginnende Leberfunktionsstörung engmaschige Überwachung (12).
Gastrointestinale Blutungen
Gastrointestinale Blutungen infolge von Teleangiektasien treten häufiger bei älteren M. Osler‑Patienten auf und führen oft zu chronischer Eisenmangelanämie. Bei therapieresistenter Anämie oder Verdacht auf okkulte Blutung ist eine obere und ggf. untere Endoskopie indiziert; bei Verdacht auf Dünndarmläsionen kann eine Kapselendoskopie hilfreich sein (22). In schweren Fällen kommen auch antiangiogenetische Therapien zum Einsatz, z. B. mit dem Anti-VEGF-Antikörper Bevacizumab (31).
Kardiale und vaskuläre Komplikationen
HAVMs und PAVMs können sekundäre kardiale Probleme verursachen, etwa chronische Hypoxie, High‑Output‑Herzversagen oder paradoxe Embolien (15,19). In Fällen mit pulmonalen oder hepatischen AVMs sollte daher eine echokardiographische Beurteilung des Herzens erfolgen. Bei nachgewiesenen Shunt‑bedingten Embolien ist eine kardiovaskuläre Sekundärprophylaxe in Erwägung zu ziehen (12).
Hämatologische Aspekte und Eisenmangelmanagement bei Morbus Osler
Insbesondere bei älteren Patientinnen und Patienten mit chronischer Eisenmangelanämie sind die Bestimmung der Eisenparameter, des Vitamin-B12- und Folsäurespiegels sowie ggf. weitere hämatologische Untersuchungen sinnvoll. Aufgrund häufiger gastrointestinaler Unverträglichkeiten können orale Eisenpräparate problematisch sein; in solchen Fällen bietet sich die parenterale Eisengabe als geeignete Alternative an. Der Hämoglobinzielwert sollte in der Regel über 100 g/l liegen, wobei Übertransfusionen zu vermeiden sind (12, 32).
Therapieoptionen bei Morbus Osler
Die Behandlung erfolgt bei Morbus Osler in erster Linie symptomorientiert und richtet sich nach den jeweils betroffenen Organen und der Schwere der Blutungsneigung. Neben der Kontrolle lokaler Blutungen spielt die Prävention von Komplikationen durch AVMs eine zentrale Rolle. Ein interdisziplinäres Vorgehen ist dabei essenziell, das HNO‑Medizin, Radiologie, Gastroenterologie, Kardiologie, Hepatologie und klinische Genetik umfasst (11, 12).
Epistaxis-Management
Nasenbluten ist das häufigste Symptom bei Morbus Osler und kann wesentlich zur Entwicklung einer Eisenmangelanämie beitragen. Die Leitlinien empfehlen ein stufenweises Vorgehen. Basismassnahmen umfassen die regelmässige Befeuchtung der Nasenschleimhaut sowie die Anwendung von pflegenden Nasensalben und Spülungen. Bei persistierender Epistaxis kommen interventionelle Verfahren wie Laserkoagulation (z. B. KTP‑ oder Nd:YAG‑Laser) oder Radiofrequenzablation zum Einsatz, die in Studien die Blutungsfrequenz signifikant reduzieren (33). Laserbehandlungen sind auch für Hautveränderungen möglich. Zusätzlich können in therapierefraktären Fällen topische Antiangiogenetika wie intranasales Bevacizumab eingesetzt werden (34). Bei besonders schweren Verlaufsformen werden chirurgische Massnahmen wie die Septodermoplastik oder als ultima ratio der permanente Verschluss der Nase (Young‑Operation) erwogen (35).
Systemische Therapien
Bei ausgeprägter oder transfusionspflichtiger Blutungsneigung werden zunehmend systemische Therapien eingesetzt. Bevacizumab ist ein monoklonaler Antikörper gegen VEGF-A, der das VEGF-Signaling in den Endothelzellen blockiert. Dadurch werden Gefässneubildung, Endothel-Proliferation, Migration und Gefässpermeabilität gehemmt. Die intravenöse Applikation von Bevacizumab konnte in mehreren Studien die Häufigkeit von Blutungen reduzieren, den Transfusionsbedarf senken und bei hepatischen AVMs Symptome eines High‑Output‑Herzversagens verbessern (36). Neuere Studien zeigen zudem vielversprechende Ergebnisse für Pomalidomid, ein Thalidomid-Derivat mit antiangiogenetischem Effekt. In randomisierten Studien konnte Pomalidomid nicht nur die Zahl transfusionspflichtiger Blutungen deutlich reduzieren, sondern auch die Schwere der Epistaxis signifikant senken und die Lebensqualität der Patienten verbessern. Anzumerken ist die orale Applikation von Pomalidomid, die in den bisherigen Studien mit einer guten Verträglichkeit assoziiert war. Diese Eigenschaften unterstreichen das Potenzial des Wirkstoffs als vielversprechende Ergänzung im therapeutischen Management schwerer, therapierefraktärer Blutungen bei Morbus Osler (37–39). Die Anwendung solcher Therapien erfolgt off label nach Einholung einer Kostengutsprache und die Indikation sollte in erfahrenen Zentren gestellt werden.
Schwangerschaft bei Morbus Osler
Schwangerschaft stellt für Patientinnen mit Morbus Osler eine besondere Situation dar, da die physiologische hämodynamische Mehrbelastung zu einer Destabilisierung bestehender AVMs führen kann. Vor einer geplanten Schwangerschaft wird daher ein Screening auf pulmonale und zerebrale AVMs mit anschliessender Behandlung relevanter Befunde empfohlen, um das Risiko maternaler und fetaler Komplikationen zu senken (40, 41). Während der Schwangerschaft sollten Patientinnen in einem erfahrenen Zentrum interdisziplinär betreut werden. In ausgewählten Situationen kann eine Embolisation von AVMs auch im zweiten Trimenon durchgeführt werden, sofern der erwartete Nutzen für die Mutter das Eingriffsrisiko überwiegt (12).
Dieser Artikel wurde gemeinsam mit MUDr. Philippe Wöllenstein erstellt.
Copyright
Aerzteverlag medinfo AG
Rare disease Sprechstunde, Klinik
und Poliklinik für Innere Medizin
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
Rare disease Sprechstunde, Klinik
und Poliklinik für Innere Medizin
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
Rare disease Sprechstunde
Klinik und Poliklinik für Innere Medizin
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
Die Autorenschaft hat keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 2010;24(6):203-219.
2. McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasia type 1. Nat Genet. 1994;8(4):345-351.
3. Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary hemorrhagic telangiectasia type 2. Nat Genet. 1996;13(2):189-195.
4. Gallione CJ, Repetto GM, Legius E, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004;363(9412):852-9.
5. Guttmacher AE, Marchuk DA, White RI Jr. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;333(14):918-924.
6. Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. 2009;17:860–871.
7. Lesca G, Olivieri C, Burnichon N, et al. Genotype-phenotype correlations in hereditary hemorrhagic telangiectasia. Genet Med. 2007;9(1):14–22.
8. Bernabeu C, Lopez-Novoa JM, Quintanilla M. HHT: involvement of TGF-β signaling pathway. J Cell Mol Med. 2010;14(7):1347-56.
9. McDonald J, Bayrak-Toydemir P, Pyeritz RE. Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Front Genet. 2015;6:1.
10. Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000;91(1):66-67.
11. Faughnan ME, Palda VA, Garcia-Tsao G, et al. International guidelines for the diagnosis and management of HHT. J Med Genet. 2011;48(2):73-87.
12. Faughnan ME, Mager JJ, Hetts SW, et al. Second International Guidelines for the Diagnosis and Management of HHT. Ann Intern Med. 2020;173(12):989-1001.
13. Shovlin CL. Pulmonary arteriovenous malformations. Am J Respir Crit Care Med 2014;190:1217–1228.
14. Kim H, Pawlikowska L, McCulloch CE, et al. Brain AVMs and HHT: risk of hemorrhage. Neurology. 2015;85(20):1893-9.
15. Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with HHT. N Engl J Med. 2000;343(13):931-6.
16. Dupuis-Girod S, Bailly S, Buscarini E, et al. Prevalence of hereditary hemorrhagic telangiectasia and diagnostic delay. Orphanet J Rare Dis 2017;12:68.
17. Kilian A, Clancy MS, Olitsky S, et al. Screening for pulmonary and brain vascular malformations is the North American standard of care for patients with hereditary hemorrhagic telangiectasia (HHT): A survey of HHT Centers of Excellence. Vasc Med. 2021;26(1):53-55.
18. Harwin J, Sugi MD, Hetts SW, Conrad MB, Ohliger MA. The Role of Liver Imaging in Hereditary Hemorrhagic Telangiectasia. J Clin Med. 2020;9(11):3750.
19. Buscarini E, Leandro G, Conte D, et al. Screening for pulmonary arteriovenous malformations by contrast echocardiography: data from 105 patients with hereditary hemorrhagic telangiectasia and review of the literature. Chest. 1999;115(6):1728-32.
20. Buscarini E, Danesino C, Olivieri C, et al. Liver involvement in HHT: angiographic findings in 600 cases. J Hepatol. 2004;41(4):380-7.
21. McDonald J, Bayrak-Toydemir P, Pyeritz RE. Hereditary hemorrhagic telangiectasia: An overview of diagnosis, management, and pathogenesis. Genet Med 2011;13(7):607‑616.
22. Serra MM, Colombo L, Ravani A, et al. Management of GI bleeding in HHT. Dig Liver Dis. 2019;51(10):1399-1405.
23. Balachandar S, Graves TJ, Shimonty A, et al; Genomics England Research Consortium; Aldred MA, Shovlin CL. Identification and validation of a novel pathogenic variant in GDF2 (BMP9) responsible for hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations. Am J Med Genet A. 2022;188(3):959-964.
24. Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013 Dec;34(12):1632-41.
25. Wooderchak-Donahue WL, Johnson P, McDonald J, et al. Expanding the clinical and molecular findings in RASA1 capillary malformation-arteriovenous malformation. Eur J Hum Genet. 2018;26(10):1521-1536.
26. Biswas S, Li Y, Xie L, et al. Role of mTOR inhibition in HHT. J Clin Med. 2021;10(14):3098.
27. Canzonieri C, Maffeis R, Pagella F, et al. Screening of visceral AVMs in children and adolescents with HHT: recommendations. Orphanet J Rare Dis. 2021;16(1):289.
28. Lee DW, White RI Jr. Pulmonary AVM embolization in HHT: state of the art. CardioVasc Intervent Radiol. 2012;35(1):1-16.
29. White RI Jr, Lynch-Nyhan A, Terry P, et al. Pulmonary arteriovenous malformations: techniques and long-term outcome of embolotherapy. Radiology. 1988;169(3):663-9.
30. Maher CO, Piepgras DG, Brown RD Jr, et al. Cerebrovascular manifestations in HHT. Stroke. 2001;32(4):877-82.
31. Masood M, Coles M, Sifuentes H. Management of Refractory Gastrointestinal Bleeding in Hereditary Hemorrhagic Telangiectasia with Bevacizumab. Case Rep Gastrointest Med. 2021;2021:6685123.
32. Geisthoff UW, Seyfert UT, Kübler M, et al. Long-term management in HHT. Orphanet J Rare Dis. 2012;7:1.
33. Pagella F, Matti E, Colombo L, et al. Laser treatment of epistaxis in HHT: a meta-analysis. Rhinology. 2019;57(2):93-102.
34. Iyer VN, Apala DR, Pannu BS, et al. Intravenous bevacizumab for refractory HHT. Mayo Clin Proc. 2018;93(12):1811-22.
35. Chavan RN, Sharma RK, Shinde RS, et al. Young’s procedure for management of intractable epistaxis in HHT. Indian J Otolaryngol Head Neck Surg. 2018;70(3):474-8.
36. Dupuis-Girod S, Ginon I, Saurin JC, et al. Bevacizumab in severe hepatic vascular malformations in HHT. Liver Int. 2012;32(6):1043–1049.
37. Buscarini E, Botella LM, Pagella F, et al. Pomalidomide for Severe Bleeding in Hereditary Hemorrhagic Telangiectasia. N Engl J Med. 2019;381(6):557–566.
38. Pagella F, Colombo L, Grosso S, et al. Pomalidomide in HHT: long-term experience and management of adverse effects. Rhinology. 2023;61(2):153-159.
39. Al-Samkari H et al. Pomalidomide for Epistaxis in Hereditary Hemorrhagic Telangiectasia. N Engl J Med 2024;390:1234–1244.
40. Shovlin CL. Pregnancy and HHT. Thromb Res. 2020;191:S46-S51.
41. Kjeldsen AD, Vase P, Green A. HHT and pregnancy: risks and recommendations. Acta Otolaryngol. 2000;120(2):251-4.
42. Robert F, Desroches‑Castan A, Bailly S, et al. Future treatments for hereditary hemorrhagic telangiectasia. Orphanet J Rare Dis. 2020;15(1):4.
43. Geisthoff UW, Kübler M, Bieg B, et al. Epistaxis management in HHT. Rhinology. 2009;47(3):207-14.
44. Bose P, Holter-Chakrabarty J, et al. Antiangiogenic therapy for HHT. Curr Opin Hematol. 2020;27(3):129-136.
45. Kjeldsen AD, Vase P, Green A. Hereditary hemorrhagic telangiectasia: a population-based study of prevalence and mortality in Danish patients. J Intern Med. 1999;245:31–39.
46. Bayrak-Toydemir P, McDonald J, Markewitz B, et al. Genotype-phenotype correlation in HHT. J Med Genet. 2006;43(10):755-60.
47. Fuchizaki U, Miyamori H, Kitagawa S, et al. Hereditary hemorrhagic telangiectasia (HHT). J Hum Genet. 2003;48(10):585-92.
48. Silva BM, Hosman AE, Devlin HL, et al. The impact of HHT on psychological well-being and quality of life. Orphanet J Rare Dis. 2017;12(1):66.
49. Geisthoff UW, Seyfert UT, Kübler M, et al. Quality of life in hereditary hemorrhagic telangiectasia. Otolaryngol Head Neck Surg. 2007;136(5):726-31.
50. Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience, 1982–1997. Mayo Clin Proc. 1999;74(7):671-80.



der informierte @rzt
- Vol. 15
- Ausgabe 10
- Oktober 2025