Die kardiale Amyloidose entsteht durch die Ablagerung fehlgefalteter Proteine in Form von Amyloidfibrillen. Die beiden häufigsten Formen mit Herzbeteiligung sind die Immunglobulin-Leichtketten-Amyloidose (AL) und die Transthyretin-Amyloidose (ATTR), die entweder als Wildtyp (ATTRwt) oder als hereditäre Variante (ATTRv) vorkommt. In den letzten Jahren hat sich im Bereich der kardialen Amyloidose viel getan: Ein besseres Verständnis der Pathophysiologie, Fortschritte in der Bildgebung und die Entwicklung neuer Therapien haben diese Krankheitsbilder stark verändert. Was einst als seltene Krankheit mit einer sehr ungünstigen Prognose galt, hat sich mittlerweile zu einer Krankheit entwickelt, die viel häufiger auftritt als bisher angenommen und für die zunehmend mehr Therapien zur Verfügung stehen.
Cardiac amyloidosis results from the deposition of misfolded proteins in the form of amyloid fibrils. The two most common subtypes affecting the heart are light-chain (AL) amyloidosis and transthyretin (ATTR) amyloidosis, which occurs either in the wild-type form (ATTRwt) or in a hereditary variant (ATTRv). In recent years, major advances have been made in the field of cardiac amyloidosis: a better understanding of the pathophysiology, progress in imaging, and the development of new therapies have profoundly changed this field. What was once considered a rare disease with a devastating prognosis has now developed into a disease that occurs much more frequently than previously thought and for which more treatment strategies are available.
Keywords: Kardiale Amyloidose, Transthyretin, Herzinsuffizienz
Die kardiale Amyloidose galt lange als seltene, kaum behandelbare Erkrankung mit schlechter Prognose. Durch Fortschritte in Diagnostik und Therapie hat sich das Bild jedoch gewandelt: Die Erkrankung wird heute häufiger und in früheren Stadien erkannt, und neue Wirkstoffe verändern den klinischen Verlauf nachhaltig. Diese Übersicht zeigt, wie sich Diagnostik, Bildgebung und Therapieansätze in den letzten Jahren weiterentwickelt haben und welche Herausforderungen nun im Vordergrund stehen.
Veränderung der Studienpopulation
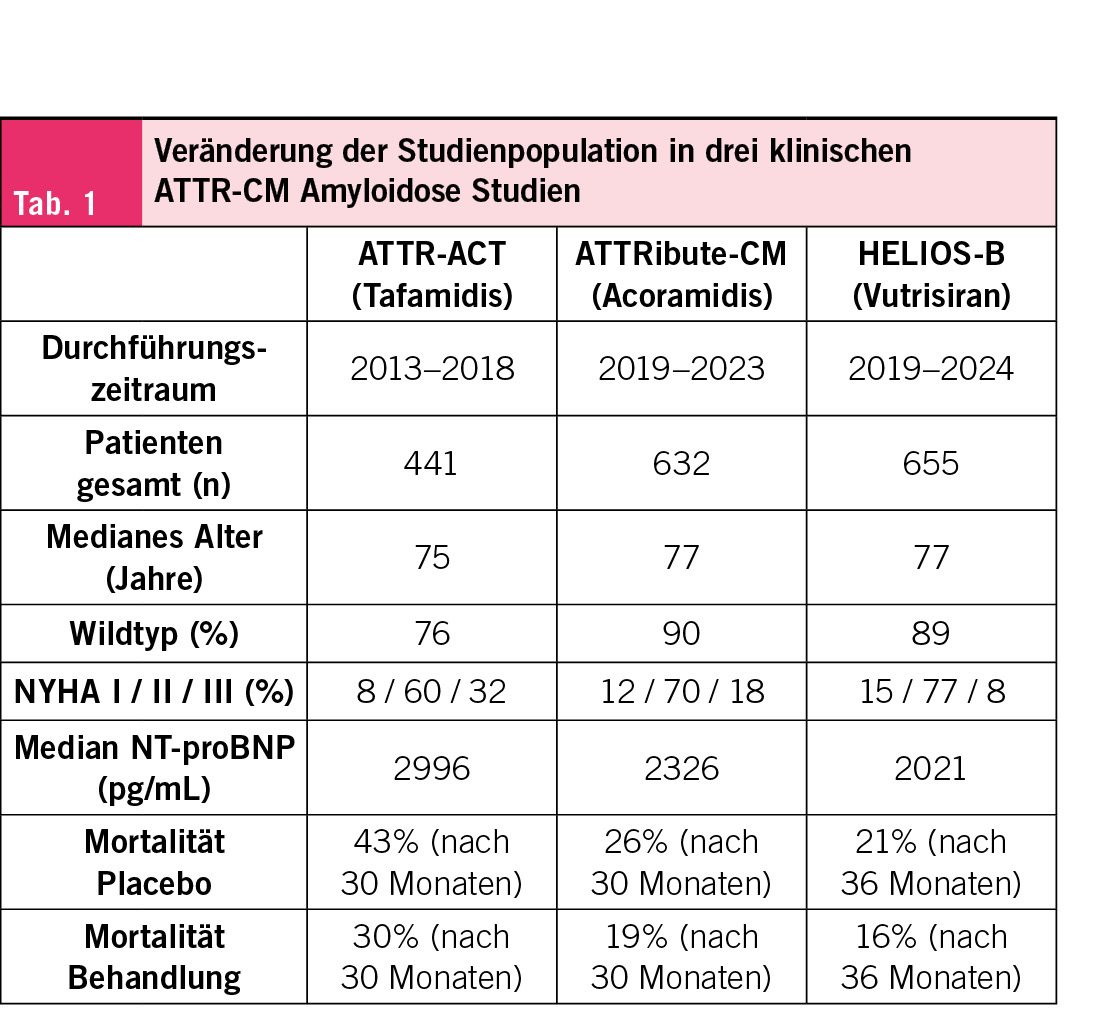
Die Patienten mit kardialer Amyloidose, die wir im klinischen Alltag sehen, haben sich in den vergangenen Jahren stark verändert. Dank des erhöhten Bewusstseins der Kardiologen für das Krankheitsbild und der verbesserten Diagnosemöglichkeiten wird die kardiale Amyloidose immer früher erkannt. Eine kardiale Amyloidose wird zunehmend bei der Abklärung von Patienten mit Herzinsuffizienz und erhöhter Wanddicke (über 12 mm) aber auch in anderen klinischen Szenarien (z. B. low-flow low-gradient Aortenstenose, hypertrophe Kardiomyopathie oder bei Herzinsuffizienz mit erhaltener Auswurffraktion) vermutet. Patienten mit beidseitigem Karpaltunnelsyndrom als Ausdruck von frühen Amyloid-Ablagerungen werden systematischer auf eine zugrundeliegende Amyloidose untersucht, und Zufallsbefunde bei Biopsien oder typische Muster im Herz-MRI tragen ebenfalls zur früheren Diagnose bei. Diese Veränderungen haben dazu geführt, dass Patienten mit kardialer Amyloidose heute meist in einem früheren Krankheitsstadium diagnostiziert werden. Dies widerspiegelt sich auch in den aktuelleren klinischen Studien, bei denen die Erkrankung weniger fortgeschritten ist als in früheren Studien. Wie Tab. 1 zeigt, unterscheiden sich die Studienpopulationen der grossen klinischen Studien. Neuere Studien haben zunehmend Patienten in früheren Krankheitsstadien mit niedrigerer Mortalität eingeschlossen. Nichtsdestotrotz: Die Studien bestätigen auch ein besseres Therapieansprechen in früheren Krankheitsstadien (z.B. NYHA Klasse 1 und 2 oder NT-proBNP < 2000 ng/L) (1).
Fortschritte in der Bildgebung
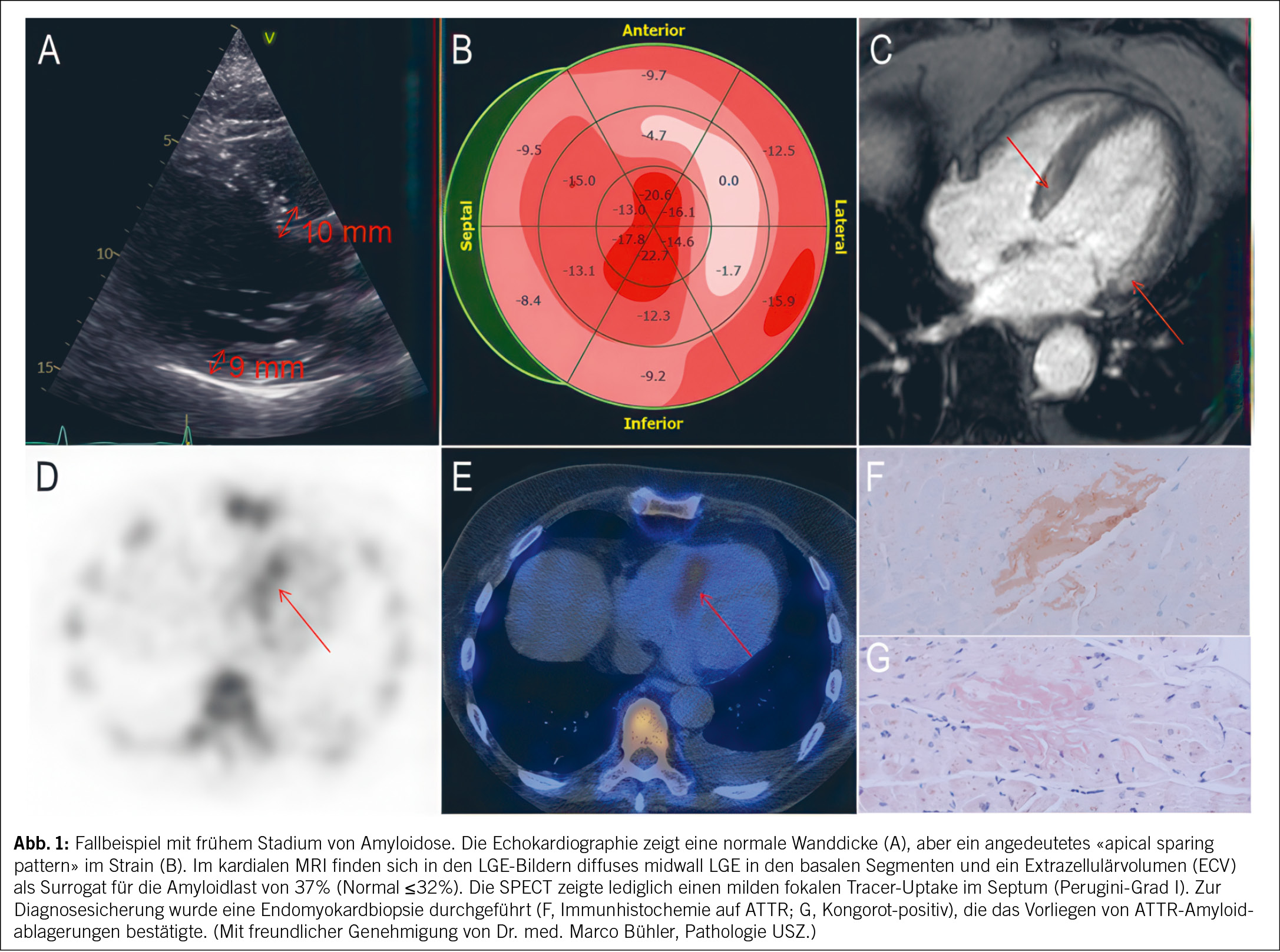
Fortschritte in der Bildgebung haben das Feld bereits grundlegend verändert. So ermöglicht die SPECT mit knochenaffinen Tracern (99mTc-DPD, -PYP, -HMDP) eine frühzeitige und verlässliche nicht-invasive Diagnose der kardialen ATTR-Amyloidose, wodurch eine Endomyokardbiopsie (EMB) in den meisten Fällen überflüssig geworden ist. Wenn man mittels Serum- und Urin-Immunfixation sowie freier Leichtketten im Serum eine AL-Amyloidose ausgeschlossen hat – und das ist zentral bei jedem Verdacht auf eine kardiale Amyloidose –, erhält man mit einem Perugini-Grad 2 oder 3 eine 100 %ige Spezifität zur Diagnostik der ATTR-Amyloidose (2). In sehr frühen Krankheitsstadien nimmt die Sensitivität jedoch leicht ab: Oftmals wird dann ein Perugini-Grad 1 gefunden, der weiter abgeklärt werden muss (z.B. mittels Herz-MRI oder EMB). Abb. 1 illustriert diese Herausforderung. Der exemplarische Fall zeigt frühe Hinweise für eine kardiale Amyloidose in der Bildgebung und, dass auch ein milder, fokaler Uptake in der SPECT/CT eine weiterführende Diagnostik erfordert. Diese Fälle repräsentieren oft frühe Amyloidablagerungen ohne manifeste Amyloidose, deren natürlicher Verlauf bislang wenig verstanden ist (3). Besonders bei sehr früher Erkrankung hat das Amyloid-PET eine höhere Sensitivität (4). Im Gegensatz zu den knochenaffinen SPECT-Radiotracern sind PET-Radiotracer speziell dafür entwickelt worden, direkt an Amyloidfibrillen zu binden. Dadurch ermöglichen sie eine sehr genaue Diagnose und eine Quantifizierung der Krankheitslast. Ursprünglich für die Alzheimer-Diagnostik entwickelt, werden diese Tracer zunehmend auch für die kardiale Amyloidose erprobt, sind jedoch für diese Indikation bislang noch nicht zugelassen. In zwei Phase-3-Studien werden die Genauigkeit und der Nutzen von 124I-Evuzamitide (REVEAL, NCT06788535) und 18F-Florbetaben (CArdiag, NCT05184088) aktuell getestet. Neben der Diagnostik spielt die Bildgebung eine immer wichtigere Rolle beim Monitoring des Therapieansprechens. Während die Echokardiographie als kostengünstiges und breit verfügbares Verfahren strukturelle und funktionelle Veränderungen abbilden kann, zeigen sich Veränderungen hier frühestens nach 18 Monaten. Demgegenüber können das kardiale MRI oder das Herz-PET bereits frühe Veränderungen unter Therapie visualisieren (5, 6).
Technologische Entwicklungen
In den vergangenen Jahren wurde die künstliche Intelligenz (KI) trainiert und validiert, um eine kardiale Amyloidose im EKG, der Echokardiographie, dem kardialen MRI, der Szintigraphie und in der Pathologie mit hoher diagnostischer Genauigkeit zu erkennen (7). So konnte in einer kürzlichen Studie die kardiale Amyloidose mittels KI im EKG und in der Echokardiographie um bis zu 3 Jahre früher erkannt werden (8). Das kann den Patienten potentiell einen schnelleren Zugang zur Therapie ermöglichen und die Krankheit früher stabilisieren. Neben den KI-basierten Verfahren zeigen auch automatisierte Auswertungen von elektronischen Gesundheitsakten Potenzial. So konnte ein Scoring-System, das unter anderem auf ICD-10-Diagnosen und Laborwerten basiert, Patienten mit erhöhtem Risiko für kardiale Amyloidose identifizieren (9). Trotz dieser vielversprechenden Ergebnisse ist die Evidenzlage für den Einsatz solcher KI-gestützten und datenbasierten Tools bislang gering (10). Es gibt noch keine prospektiven Studien und auch kaum Daten zur Umsetzung im klinischen Alltag. Diese Lücken müssen geschlossen werden vor einer breiten klinischen Einführung dieser Tools.
Neue Therapieansätze
Aktuelle Behandlungen der kardialen Amyloidose setzen bei den Amyloidvorläuferproteinen an. Bei der AL-Amyloidose sind dies Therapien, die gegen Plasmazellen im Knochenmark gerichtet sind und somit die amyloidogenen Leichtketten reduzieren. Bei der ATTR-Amyloidose zielen die Therapien dagegen auf das Transthyretin-Protein ab. Im Folgenden liegt der Fokus auf der ATTR-Kardiomyopathie (CM), da die AL-Amyloidose in enger Kooperation mit der Hämatologie behandelt wird und spezifischen Therapieprinzipien folgt. Für die ATTRwt-Amyloidose sind aktuell in der Schweiz Tafamidis und Vutrisiran zugelassen – letztere Substanz wird jedoch noch nicht vergütet. Aktuell befinden sich mehrere Medikamente im Zulassungsverfahren oder in Phase 3 klinischen Studien. Bei der ATTR-Amyloidose werden drei Therapieprinzipien unterschieden: Stabilisatoren, Suppressoren und Depletoren (Tab. 2).
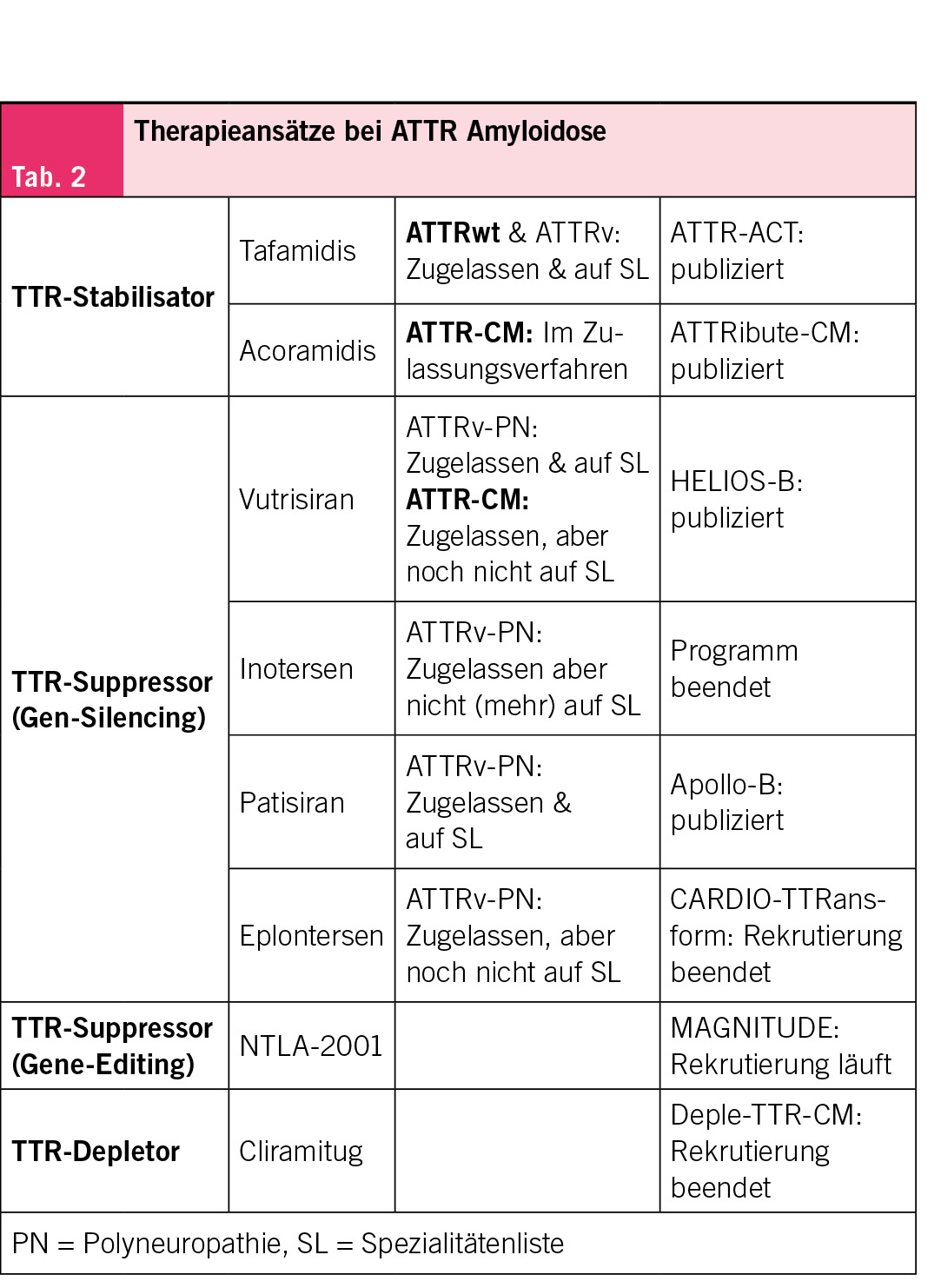
Als Stabilisator bereits zugelassen ist Tafamidis (11). Acoramidis ist ein weiterer Stabilisator, der in der Phase-3-Studie (ATTRibute-CM) die Mortalität und kardiovaskuläre Ereignisse reduziert hat und in der Schweiz nun im Zulassungsverfahren ist (12). Zu den Suppressoren zählen Gen-Silencing-Therapien wie Patisiran und Vutrisiran (small interfering RNA). In der Phase-3-Studie (HELIOS-B) zeigte Vutrisiran bei ATTR-CM eine Reduktion von Mortalität und kardiovaskulären Ereignissen und ist nun zugelasssen, aber noch nicht auf der Spezialitätenliste (13). Inotersen, ein weiterer Suppressor, wird in der Schweiz nicht mehr angewendet, während Eplontersen mit günstigerem Nebenwirkungsprofil für die ATTRv-PN bereits zugelassen ist. Zusätzlich läuft die Phase-3-Studie (CARDIO-TTRansform, NCT04136171) für den Einsatz von Eplontersen bei ATTR-CM. Andererseits zählt zu den Suppressoren auch ein Gen-Editing-Ansatz, der auf der CRISPR-Cas9-Technologie basiert und ebenfalls in einer Phase-3-Studie (MAGNITUDE, NCT06128629) getestet wird. Die dritte Klasse, die Depletoren, beinhaltet beispielsweise den monoklonalen humanen Antikörper Cliramitug, welcher auf die direkte Entfernung von Amyloidfibrillen abzielt und sich derzeit in einer Phase-3-Studie befindet (DepleTTR-CM, NCT06183931).
Die Therapie der AL-Amyloidose unterscheidet sich grundlegend. Hier hat die Einführung wirksamer Therapien, die gegen Plasmazellen oder reife B-Zellen gerichtet sind, die Behandlung deutlich verändert. Ein besonderer Fortschritt war der Einsatz monoklonaler Antikörper gegen das Oberflächenprotein CD38, wie Daratumumab, der mit relativ wenigen Nebenwirkungen die Prognose der Patienten deutlich verbessert hat. Zusätzlich wurde der antifibrilläre Antikörper Anselamimab in einer Phase-3-Studie (CAEL-101) geprüft, konnte jedoch die primären Endpunkte nicht erreichen, sodass der klinische Nutzen derzeit unklar bleibt (14, 15).
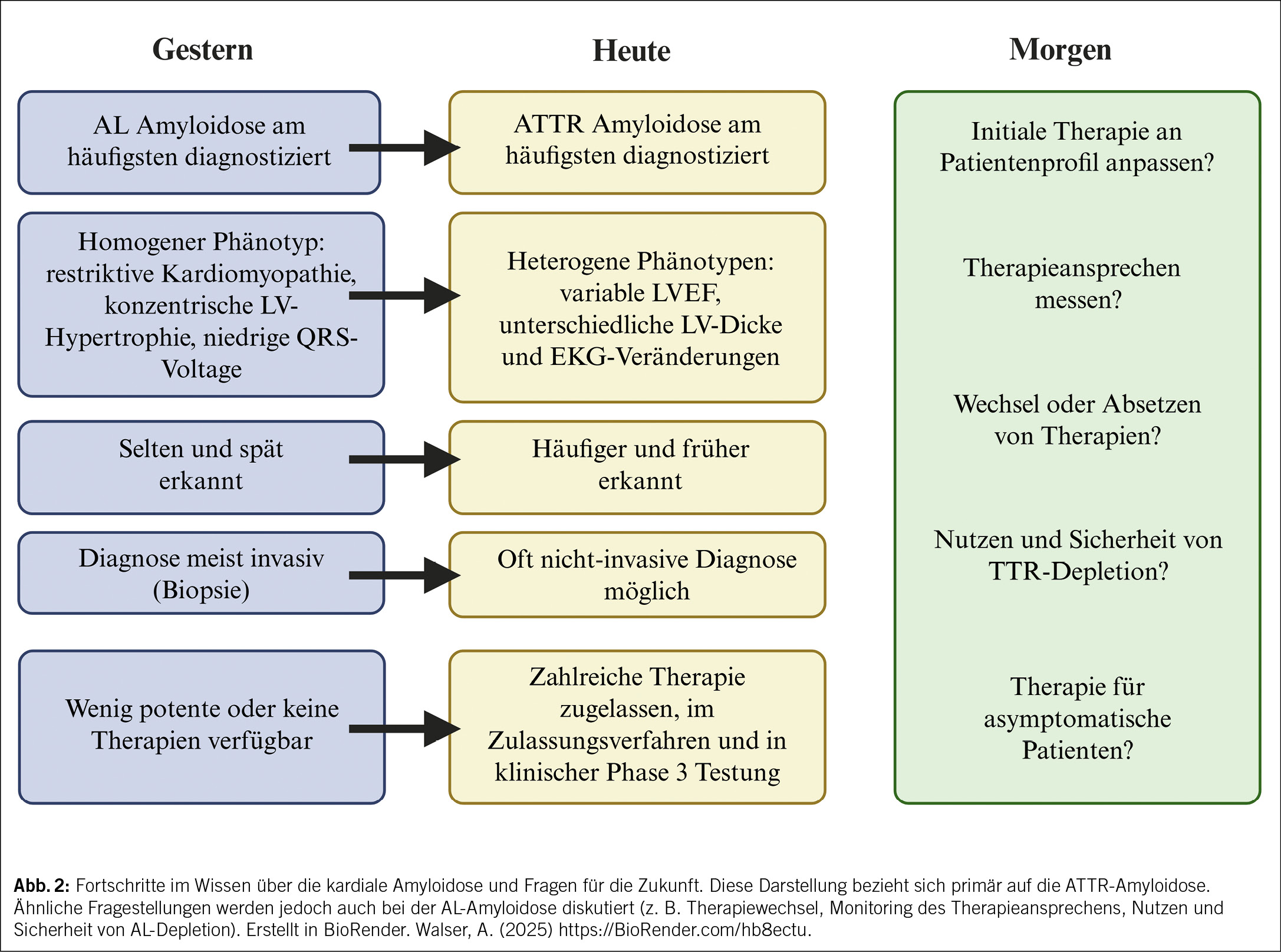
Herausforderungen für die Zukunft
Trotz enormer Fortschritte in Diagnostik und Therapie bestehen weiterhin Herausforderungen. Die derzeit für die ATTRwt-CM zugelassenen Behandlungen können die Krankheit nicht rückgängig machen, und bei etwa 30–40 % der Patienten kommt es trotz Therapie weiterhin zu einem kardiovaskulären Ereignis oder sogar zum Tod. Vor diesem Hintergrund und der gleichzeitig wachsenden Anzahl an Therapieoptionen stellen sich zunehmend neue Fragen. Welche initiale Therapie ist für welchen Patienten am besten geeignet? Wie lässt sich das Ansprechen auf die Therapie zuverlässig beurteilen? Wann und unter welchen Bedingungen sollte von einem Wirkstoff auf einen anderen gewechselt werden? Und in welchen Situationen ist es sinnvoll, eine Therapie abzusetzen? Um diese Fragen beantworten zu können, sind grössere, multizentrische Studien mit langen Beobachtungszeiträumen notwendig. Eine wichtige Rolle spielen dabei auch Register, wie sie beispielsweise vom Schweizerischen Amyloidose-Netzwerk mit dem Schweizer Amyloidose Register (SAR) aufgebaut und kontinuierlich erweitert werden, da sie die Generierung grosser Mengen an Real-World-Daten ermöglichen (16). Diese Studien und Register müssen funktionelle Tests, Biomarker, patientenberichtete Outcomes sowie bildgebende Verfahren beinhalten, um Schwellenwerte für das Fortschreiten der Krankheit wie auch das Therapieansprechen festzulegen. Schlussendlich wird ein multimodaler Ansatz, der klinische Parameter, Biomarker und Bildgebung kombiniert, entscheidend sein, um personalisierte Therapiestrategien zu entwickeln und die Versorgung von Patienten mit kardialer Amyloidose weiter zu verbessern (17).
Amely Walser 1
Prof. Dr. med. Andreas J. Flammer 2
Dr. med. Rahel Schwotzer 3
PD Dr. med. Dominik C. Benz 1,2,4
1 Klinik für Nuklearmedizin, Universitätsspital Zürich, Rämistrasse 100, 8091 Zürich
2 Klinik für Kardiologie, Universitätsspital Zürich, Rämistrasse 100, CH-8091 Zürich
3 Klinik für Medizinische Onkologie und Hämatologie, Universitätsspital Zürich, Rämistrasse 100, 8091 Zürich
4 Advanced Clinician Scientist Programm, Universitäre Medizin Zürich (UMZH)
Copyright
Aerzteverlag medinfo AG
Klinik für Nuklearmedizin
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
- Klinik für Nuklearmedizin, Universitätsspital Zürich,
Rämistrasse 100, 8091 Zürich
- Klinik für Kardiologie, Universitätsspital Zürich,
Rämistrasse 100, CH-8091 Zürich
- Advanced Clinician Scientist Programm,
Universitäre Medizin Zürich (UMZH)
dominik.benz@usz.ch
Stipendien: Dr. Benz gibt an, ein Karrierestipendium des Advanced Clinician Scientist Programms der Universitären Medizin Zürich (UMZH) zu erhalten.
Amely Walser berichtet über keine Interessenkonflikte.
Dr. Flammer berichtet über Forschungsförderung von AstraZeneca und Novartis sowie Honorare von Alnylam, AstraZeneca, Bayer, Boehringer Ingelheim, Novartis, Pfizer, Roche und Vifor. Er ist nationaler Koordinator der DepleTTR-CM-Studie (Alexion/AstraZeneca).
Dr. Schwotzer berichtet über Forschungsförderung der Mach-Gaensslen-Stiftung; Förderung des Swiss Amyloidosis Registry durch Alnylam, AstraZeneca, SOBI und Pfizer; sowie Beratungshonorare von Alnylam, AstraZeneca und Johnson & Johnson.
Dr. Benz berichtet über Forschungsförderung der Olga Mayenfisch-Stiftung und der Immanuel-und-Ilse-Straub-Stiftung; investigator-initiierte Forschungsförderung von AstraZeneca und Life Molecular Imaging; sowie Beratungshonorare von Pfizer, Alnylam, Bayer Healthcare und AstraZeneca.
1. Maurer MS, Witteles RM, Garcia-Pavia P, Sheikh FH, Morbach C, Rodriguez Duque D, et al. Impact of Heart Failure Severity on Vutrisiran Efficacy in Transthyretin Amyloidosis With Cardiomyopathy. J Am Coll Cardiol. 2025;85(20):1927-39.
2. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016;133(24):2404-12.
3. Porcari A, Razvi Y, Cappelli F, Nitsche C, Serenelli M, Longhi S, et al. Clinical Phenotype and Prognosis of Asymptomatic Patients With Transthyretin Cardiac Amyloid Infiltration. JAMA Cardiol. 2025;10(5):437-45.
4. Smiley DA, Einstein AJ, O’Gorman KJ, Santana D, Teruya S, Chan N, et al. Early Detection of Transthyretin Cardiac Amyloidosis Using (124)I-Evuzamitide Positron Emission Tomography/Computed Tomography. JACC Cardiovasc Imaging. 2025;18(7):799-811.
5. Patel RK, Ioannou A, Sheikh A, Razvi Y, Mansell J, Martinez-Naharro A, et al. Transthyretin amyloid cardiomyopathy: natural history and treatment response assessed by cardiovascular magnetic resonance. Eur Heart J. 2025.
6. Benz DC, Clerc OF, Cuddy SAM, Singh V, Kijewski MF, Bianchi G, et al. Changes in Myocardial Light Chain Amyloid Burden After Plasma Cell Therapy. JACC Cardiovasc Imaging. 2025.
7. Kamel MA, Abbas MT, Kanaan CN, Awad KA, Baba Ali N, Scalia IG, et al. How Artificial Intelligence Can Enhance the Diagnosis of Cardiac Amyloidosis: A Review of Recent Advances and Challenges. J Cardiovasc Dev Dis. 2024;11(4).
8. Oikonomou EK, Sangha V, Vasisht Shankar S, Coppi A, Krumholz HM, Nasir K, et al. Artificial intelligence-enabled electrocardiography and echocardiography to track preclinical progression of transthyretin amyloid cardiomyopathy. Eur Heart J. 2025.
9. Pascoe MA, Kolodziej A, Birks EJ, Vaidya G. Using electronic medical records to identify patients at risk for underlying cardiac amyloidosis. J Cardiol. 2025;85(1):43-4.
10. Grogan M, Lopez-Jimenez F, Guthrie S, Kumar N, Langevin R, Lousada I, et al. Value of Artificial Intelligence for Enhancing Suspicion of Cardiac Amyloidosis Using Electrocardiography and Echocardiography: A Narrative Review. J Am Heart Assoc. 2025;14(8):e036533.
11. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med. 2018;379(11):1007-16.
12. Gillmore JD, Judge DP, Cappelli F, Fontana M, Garcia-Pavia P, Gibbs S, et al. Efficacy and Safety of Acoramidis in Transthyretin Amyloid Cardiomyopathy. N Engl J Med. 2024;390(2):132-42.
13. Fontana M, Berk JL, Gillmore JD, Witteles RM, Grogan M, Drachman B, et al. Vutrisiran in Patients with Transthyretin Amyloidosis with Cardiomyopathy. N Engl J Med. 2024.
14. Bianchi G, Zhang Y, Comenzo RL. AL Amyloidosis: Current Chemotherapy and Immune Therapy Treatment Strategies: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2021;3(4):467-87.
15. Kastritis E, Palladini G, Minnema MC, Wechalekar AD, Jaccard A, Lee HC, et al. Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N Engl J Med. 2021;385(1):46-58.
16. Brouwers S, Heimgartner R, Laptseva N, Aguzzi A, Ehl NF, Fehr T, et al. Historic characteristics and mortality of patients in the Swiss Amyloidosis Registry. Swiss Med Wkly. 2024;154:3485.
17. Gonzalez-Lopez E, Maurer MS, Garcia-Pavia P. Transthyretin amyloid cardiomyopathy: a paradigm for advancing precision medicine. Eur Heart J. 2025;46(11):999-1013.


info@herz+gefäss
- Vol. 15
- Ausgabe 5
- Dezember 2025