
In den meisten Studien zum asymptomatischen Myelom wird als primärer Endpunkt die Progression in ein symptomatisches Myelom gewählt. Doch stellen sich die folgenden Fragen: kann damit auch das Überleben verbessert werden und führt diese Strategie nicht zu einer Überbehandlung von Patienten, die auch ohne Therapie keine Progression erleiden? In den bis anhin durchgeführten und publizierten Studien wurden nur Patienten mit hohem oder zum Teil mit intermediärem Risiko eingeschlossen. Der folgende Beitrag fasst die derzeitigen Kenntnisse zusammen.
In most studies of asymptomatic myeloma, progression to symptomatic myeloma is chosen as the primary endpoint. However, the following questions arise: can this also improve survival and does this strategy not lead to overtreatment of patients who do not experience progression even without therapy? In the studies conducted and published to date, only patients with high or, in some cases, intermediate risk were included. The following article summarizes the current knowledge.
Key Words: asymptomatic myeloma, overtreatment, survival
Definition
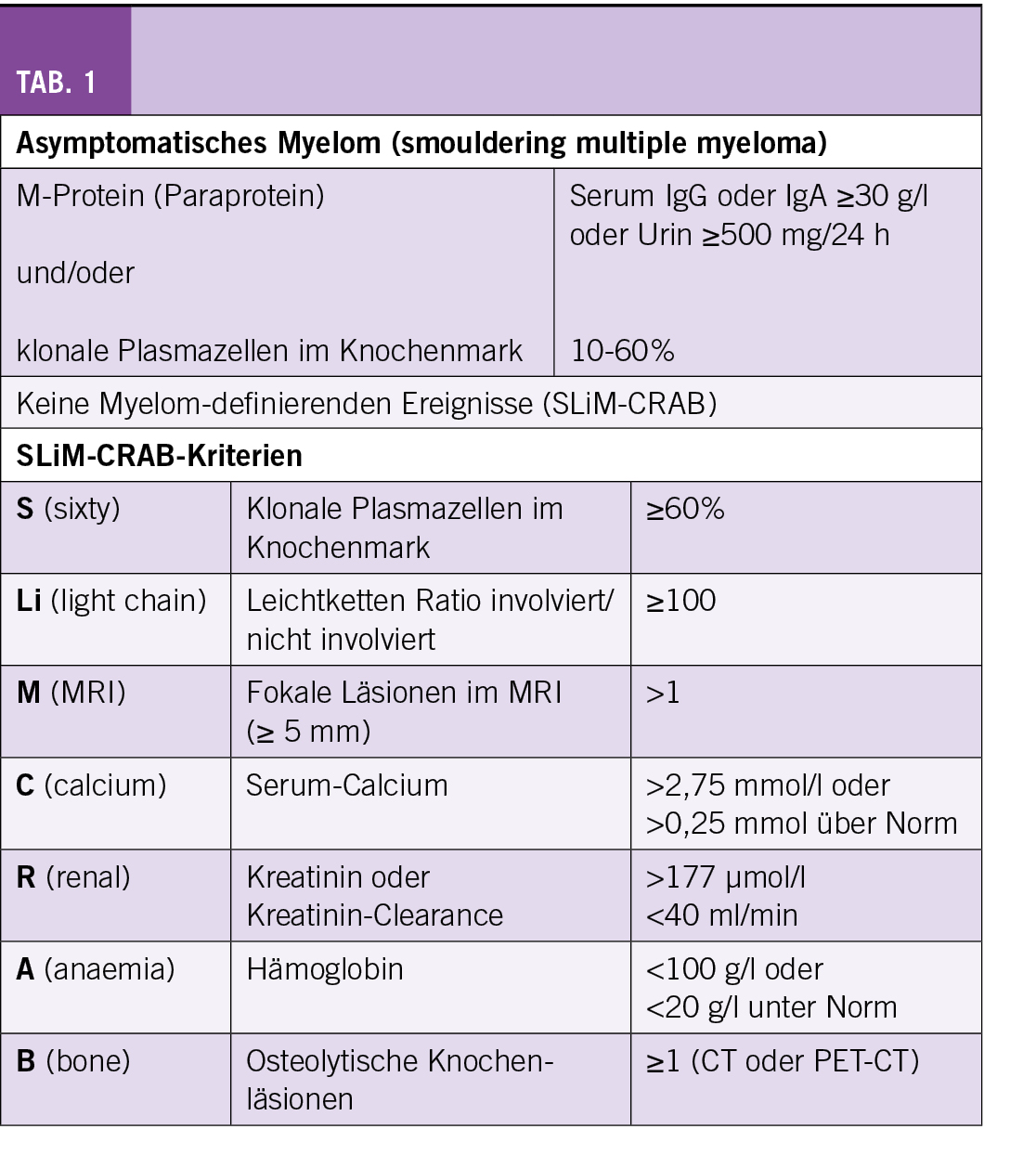
Das asymptomatische Myelom, auch smoldering multiple myeloma (SMM) genannt, wurde durch die International Myeloma Working Group 2014 neu definiert. Während vorher ein M-Protein im Serum ≥ 30 g/l und/oder eine Knochenmarksinfiltration von ≥ 10% klonaler Plasmazellen sowie das Fehlen von Endorganschäden und Symptomen als Kriterien galten (1), wurden die Diagnosekriterien 2014 folgendermassen angepasst (2):
- Monoklonales Serumprotein (IgG or IgA) ≥ 30 g/l oder monoklonales Protein im Urin ≥ 500 mg/24 h und/oder Knochenmarksinfiltration durch Plasmazellen 10-60%
- Abwesenheit von Myelom-definierenden Ereignissen und Amyloidose
Zu den Myelom-definierenden Ereignissen gehören die sogenannten SLiM-CRAB-Kriterien (Tab. 1). Neu gelten mehr als eine fokale Läsion im MRI als ein Myelom-definierendes Ereignis. Das bedeutet, dass im Falle eines unauffälligen low-dose Ganzkörper CTs ein Ganzkörper MRI durchgeführt werden sollte. Alternativ kann auch ein FDG-PET/CT veranlasst werden. Mit der Erweiterung der Diagnosekriterien wurde eine Untergruppe von ultra-high risk SMM als aktive multiple Myelome definiert (ca. 15%).
Das SMM stellt eine Vorstufe des Multiplen Myeloms dar. Durch verschiedene Mutationen (KRAS, NRAS, FAM46C), strukturelle Ereignisse (t(MYC), del(1p)/gain(1q)), biallele Ereignisse oder die APOBEC Signatur kann eine Progression in ein multiples Myelom auftreten (3).
Epidemiologie
Die Prävalenz des SMM bei Personen > 40 Jahren liegt bei circa 0,5%, die der MGUS (monoklonale Gammopathie unklarer Signifikanz) bei 2-3% (4). Das Risiko für eine Progression in ein multiples Myelom wird beim SMM in den ersten fünf Jahren auf 10% pro Jahr geschätzt, in den zweiten fünf Jahren beträgt es 3% pro Jahr und anschliessend 1% pro Jahr. Im Gegensatz dazu zeigt sich beim MGUS ein linearer Anstieg mit einer Progression in ein multiples Myelom von 1% pro Jahr (5).
Risikostratifizierung
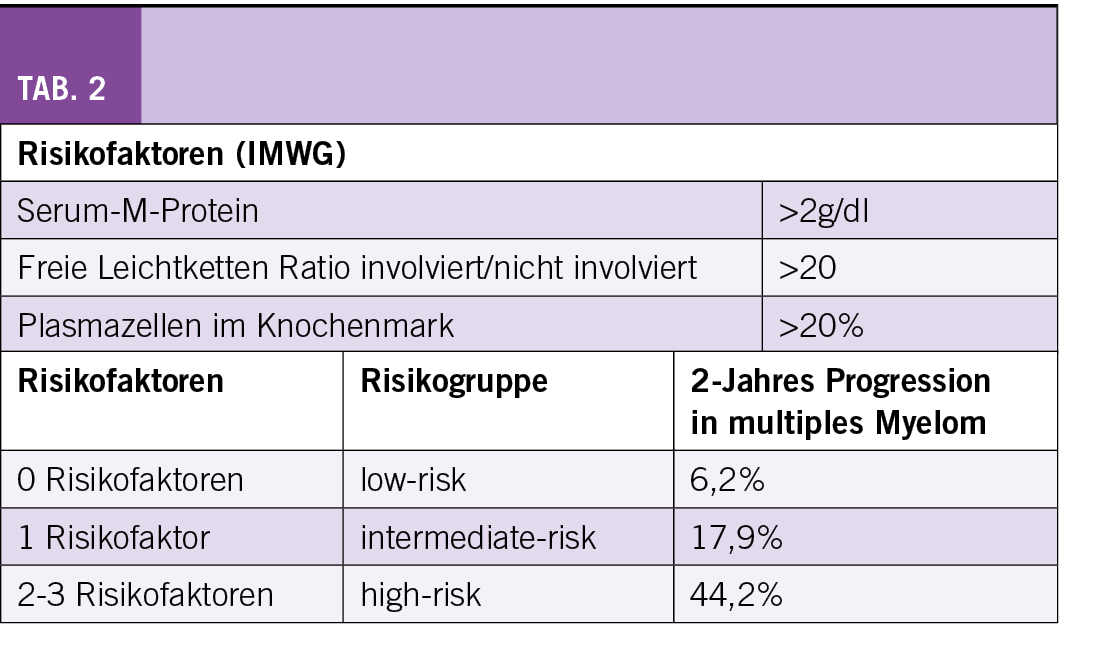
Risikofaktoren für eine Progression des SMM in ein multiples Myelom sind gemäss einer Untersuchung der Mayo-Klinik ein M-Gradient von ≥ 30 g/l, ≥ 10% klonale Plasmazellen im Knochenmark und eine Ratio der freien Leichtketten < 0,125 oder > 0,8. Die spanische Studiengruppe definierte als Risikofaktoren ≥ 95% abnormale Plasmazellen und eine Immunoparese, d.h. eine Reduktion der nicht involvierten Immunglobulinen (6). Zur Risikostratifizierung des SMM werden durch die IMWG drei Risikofaktoren definiert: M-Protein > 2 g/dl, FLC Ratio > 20, Knochenmarksinfiltration > 20% (Tab. 2) (7). Ohne Risikofaktor (low-risk) liegt das Risiko für eine Progression nach 2 Jahren bei 6,2%, mit einem Risikofaktor (intermediate-risk) bei 17,9% und bei 2-3 Risikofaktoren (high-risk) bei 44,2%. Wenn zusätzlich die Zytogenetik berücksichtigt wird und als zusätzlicher Risikofaktor eine t(4;14), t(14;16), +1q und/oder eine del13q/Monosomie miteinbezogen werden, erfolgt eine Aufteilung in vier Risikogruppen mit einer Progressionsrate nach 2 Jahren von 6,0% (low-risk) bis 63,1% (high-risk).
Bustoros et al. konnten mittels next generation sequencing (NGS) aufzeigen, dass die meisten genetischen Alterationen bei Diagnose des SMM bereits stattgefunden haben (8). Als unabhängige Risikofaktoren für eine Progression zeigten sich Alterationen im MAPK-Signalweg (KRAS, NRAS), in den DNA-Reparatur-Signalwegen (del 17p; TP53 und ATM) und Aberrationen in MYC (Translokationen oder copy-number variations).
Behandlungsstrategien
Soll das SMM behandelt werden und was ist das Ziel der Therapie? In den meisten Studien wird als primärer Endpunkt die Progression in ein symptomatisches Myelom gewählt. Doch kann damit auch das Überleben verbessert werden? Und führt diese Strategie nicht zu einer Überbehandlung von Patienten, die auch ohne Therapie keine Progression erleiden?
In den bis anhin durchgeführten und publizierten Studien wurden nur high-risk und zum Teil intermediate-risk Patienten eingeschlossen. Eine der ersten Untersuchungen wurde 2013 von der spanischen Studiengruppe unter María-Victoria Mateos publiziert (9, 10). In dieser Phase 3 Studie wurden 119 Patienten mit einem, nach spanischen Kriterien, high-risk SMM eingeschlossen und randomisiert mit Lenalidomid/Dexamethason behandelt oder beobachtet. Der primäre Endpunkt, die Zeit bis zur symptomatischen Erkrankung, konnte signifikant verlängert werden (not reached versus 21 Monate, Hazard Ratio 0,24). Zudem zeigte sich auch eine signifikante Verbesserung des Gesamtüberlebens (Hazard Ratio 0,43). Nach einem Follow up von 10 Jahren zeigt sich ein anhaltender Nutzen bezüglich der Progression und des Gesamtüberlebens (Hazard Ratio für OS 0,54) (11). Das Gesamtüberleben ab der Progression in ein Myelom war in beiden Gruppen vergleichbar. Eine frühe Exposition mit Lenalidomid führte somit nicht zu einer resistenteren Krankheit im Rezidiv.
In einer weiteren Phase 3 Studie von S. Lonial et al. wurden Patienten mit einem intermediate oder high-risk SMM mit Lenalidomid mono behandelt oder beobachtet (12). Auch hier zeigte sich eine Verbesserung des progressionsfreien Überlebens nach 3 Jahren von 66% auf 91% (HR 0,28). Das Gesamtüberleben war jedoch nicht unterschiedlich. Zu beachten ist, dass die Risikoklassifizierung in den beiden Studien unterschiedlich war.
2021 publizierte D. Kazandjian et al. aus der Gruppe von Ola Landgren eine Phase 2 Studie, in der die Behandlung des SMM mit Carfilzomib, Lenalidomid und Dexamethason, gefolgt von einer Erhaltungstherapie mit Lenalidomid untersucht wurde (13). Mit dieser Therapie konnte eine Rate an MRD-negativen kompletten Remissionen von 70,4% erreicht werden. Das progressionsfreie Überleben nach 8 Jahren lag bei 91,2%.
In verschiedenen aktiven Studien wird die Behandlung des SMM untersucht, unter anderem mit Kombinationen mit Carfilzomib, Daratumumab, Isatuximab, Elotuzumab, Pembrolizumab und Vakzinen.
Aktuelle Empfehlungen zur Therapie
Die NCCN-Guidelines empfehlen aktuell in der low risk Situation den Einschluss in klinische Studien oder die Beobachtung und in der high risk Situation den Einschluss in klinische Studien, die Beobachtung oder eine Therapie mit Lenalidomid.
Gemäss Empfehlung der EHA-ESMO steht die «Watch-and-wait» Strategie bei den meisten Patienten mit einem SMM weiterhin an erster Stelle. Zudem gilt es zu berücksichtigen, dass in der Schweiz für das SMM aktuell keine zugelassenen Substanzen zur Verfügung stehen. High-risk Patienten sollten, sofern verfügbar, im Rahmen von Phase 3 Studien behandelt werden.
Follow up
Unabhängig vom Vorgehen (Observation oder Therapie) ist eine engmaschige Kontrolle empfohlen. Diese beinhaltet die dreimonatliche Kontrolle des Blutbildes, der Serumeiweisselektrophorese und der freien Leichtketten im Serum sowie die Nierenfunktion und des Calciumwertes. Die Indikation für eine Knochenmarkspunktion ist vom Verlauf abhängig. Zudem wird eine jährliche Bildgebung mit einem Ganzkörper MRI, einem Ganzköper-CT oder einem FDG-PET-CT empfohlen.
Copyright bei Aerzteverlag medinfo AG
Chefarzt Onkologie STGAG
Kantonsspital Münsterlingen
Spitalcampus 1
8596 Münsterlingen
Der Autor hat keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. International Myeloma Working Group Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br. J. Haematol 2003;1245:749-757
2. Vincent Rajkumar S et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014;15:e538-548
3. Langenbucher, A., et al. An extended APOBEC3A mutation signature in cancer. Nat Commun 12, 1602 (2021). https://doi.org/10.1038/s41467-021-21891-0Nat. Commun 2021;12
4. Thorsteindottir S et al ASH 20121; Abstr 151
5. Kyle RA et al Clinical Course and Prognosis of Smoldering (Asymptomatic) Multiple Myeloma.. NEJM 2007;356:2583-1590
6. Landgren O Shall we treat smoldering multiple myeloma in the near future? Hematology Am Soc Hematol Educ Program. 2017 Dec 8;2017(1):194-204. doi: 10.1182/asheducation-2017.1.194
7. Mateos, MV et al. International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J. 10, 102 (2020). https://doi.org/10.1038/s41408-020-00366-3Blood Cancer Jouirnal 2020; 10
8. Bustoros M et al. Genomic Profiling of Smoldering Multiple Myeloma Identifies Patients at a High Risk of Disease Progression. J Clin Oncol 2020;38:2380-2389
9. Mateos MV et al. Lenalidomide plus Dexamethasone for High-Risk Smoldering Multiple Myeloma. NEJM 2013; 369:438-447
10. Mateos MV et al. Lenalidomide plus dexamethasone versus observation in patients with high-risk smouldering multiple myeloma (QuiRedex): long-term follow-up of a randomised, controlled, phase 3 trialL.ancet Oncol 2016;17: 1127-1136
11. HemaSpere 2020; EP 950
12. Lonial S et al Randomized Trial of Lenalidomide Versus Observation in Smoldering Multiple Myeloma. J Clin Oncol 2019;38:1126-1137
13. Kazandji D et al. an Carfilzomib, Lenalidomide, and Dexamethasone Followed by Lenalidomide Maintenance for Prevention of Symptomatic Multiple Myeloma in Patients With High-risk Smoldering Myeloma: A Phase 2 Nonrandomized Controlled Trial. JAMA Oncol 2021;7:1678-1685
14. Callander NS et al. NCCN Guidelines® Insights: Multiple Myeloma, Version 3.2022. J Natl Compr Canc Netw 2022 Jan;20(1):8-19. doi: 10.6004/jnccn.2022.0002.
