
La fibrose pulmonaire idiopathique (FPI) est la plus fréquente des pneumopathies interstitielles idiopathiques. Elle survient principalement à un âge avancé, et son évolution demeure fatale. Elle doit être évoquée dans toutes les situations associant toux sèche, dyspnée et râles crépitants. La découverte d’ agents anti-fibrotiques, la pirfenidone et le nintedanib, a modifié de manière significative le pronostic de cette affection en ralentissant son évolution. Ces progrès thérapeutiques ainsi que la possible indication à une transplantation pulmonaire requièrent donc une rigueur diagnostic afin de les initier sans délai. Une approche palliative précoce est également essentielle à la prise en charge de la FPI.
La FPI appartient au groupe des pneumopathies interstitielles, groupe hétérogène de plus de 150 affections, et en représente le 55 % (1). Elle consiste en un remaniement de l’ épithélium alvéolaire entraînant une atteinte bibasale du parenchyme, associée à une dilatation des bronchioles et à une fibrose progressive interstitielle, qui a pour conséquence une diminution de la capacité vitale forcée (CVF), une altération des échanges gazeux, et une hypoxie progressive (2).
Son incidence augmente avec l’ âge, et se manifeste le plus souvent au-delà de 60 ans avec une prédominance masculine. Le tabagisme (70 % des patients sont des fumeurs), l’ exposition aux poussières et aux virus, ainsi que les facteurs génétiques représentent de potentiels facteurs de risque (3). Elle est souvent associée à des comorbidités : hypertension pulmonaire, reflux gastro-œsophagien, BPCO, diabète et coronaropathie (1).
L’ évolution est difficilement prévisible et varie entre une progression lente (décès survenant 3 à 5 ans après le diagnostic avant l’ introduction d’ anti-fibrotiques efficaces), ou rapide à la faveur d’ une exacerbation aiguë (2).
Diagnostic
La démarche diagnostique a fait l’ objet d’ une mise à jour publiée par l’ ATS/ERS/JRS/ALAT en 2018 (4). Le diagnostic doit être évoqué par la triade toux sèche, dyspnée, et râles inspiratoires crépitants. La spirométrie peut être initialement normale et évolue vers un syndrome restrictif. Il doit écarter les autres causes de pneumopathies interstitielles idiopathiques. Sa confirmation se fonde sur l’ imagerie CT en haute résolution (image en nid d’ abeilles) et sur la biopsie chirurgicale. Cette dernière, non dénuée de risque chez les patients âgés souffrant de comorbidités, peut être omise en présence d’ une imagerie suffisamment évocatrice (2). Un diagnostic précoce est un élément déterminant du pronostic.
Traitement
Le traitement, pharmacologique et non pharmacologique, vise à ralentir le déclin de la CVF, à maintenir une oxygénation satisfaisante, à réduire les symptômes et les exacerbations, et à minimiser les effets secondaires des anti-fibrotiques (1).
Avant 2014, seuls des traitements empiriques tels que l’ association prednisone-azathioprine, inefficace et entraînant une augmentation de la morbi-mortalité, ou l’ acétylcystéine, également inefficace, étaient proposés.
L’ arrivée sur le marché d’ anti-fibrotiques ayant fait la preuve de leur sécurité et efficacité au terme d’ études de phase 3 (5 – 6), a radicalement changé le pronostic de la FPI, et entraîné de nouvelles recommandations thérapeutiques. La prise en charge par les assureurs maladie de ces traitements requiert une démarche diagnostique conforme aux recommandations internationales (4).
Les anti-fibrotiques
La pirfenidone (Esbriet®)
Il s’ agit d’ un anti-fibrotique oral dont le mécanisme d’ action est peu clair. Il inhibe la prolifération des fibroblastes et la synthèse du collagène en régulant l’ activité du facteur de croissance transformant β et du TNFα.
L’ étude ASCEND (5) a montré, lorsque l’ on compare la pirfenidone à un placebo, une diminution de 54 % du déclin de la CVF après 1 an de traitement (122 vs 262 ml, p < 0,001) ainsi qu’ une survie à 5 ans significativement améliorée (55,9 % vs 31,5 %, p < 0.02).
En pratique, la posologie est progressive : 3 x 1 gélule de 267 mg/j la première semaine, 3 x 2 gélules/j la 2ème semaine, puis 3 x 3 gélules/j ou 1 gélule de 801 mg/j dès la 3ème semaine.
Le nintedanib (Ofev®)
Bien que le mécanisme d’ action soit incomplètement élucidé, le nintedanib présente des propriétés anti-inflammatoires et anti-fibrotiques, en interférant avec la migration, la prolifération, la différenciation des fibroblastes, et la synthèse du collagène. C’ est un inhibiteur intracellulaire de plusieurs tyrosine-kinases impliquées dans le processus fibrotique.
Comparé à un placebo, le nintedanib réduit le déclin de la CVF de 52 % (115 vs 240 ml, p < 0,001) après 52 semaines de traitement chez 1000 patients. Il réduit également la fréquence des exacerbations (5,3 vs 8,2 / 100 patients-années) (6). En pratique, la posologie est de 2 x 150 mg/j, susceptible d’ être réduite à 2 x 100 mg/j lors d’ intolérance.
Des méta-analyses montrent que la pirfenidone et le nintedanib ont un effet similaire sur le déclin de la CVF. Ni l’ un ni l’ autre n’ ont cependant d’ effet significatif sur l’ amélioration des symptômes cliniques.
Les traitements combinés
L’ étude INJOURNEY (7) a évalué la sécurité et la tolérance du nintedanib + pirfenidone vs nintedanib seul chez 105 patients sur une période 12 semaines. Le déclin de la CVF s’ est avéré moindre dans le groupe combiné (-13,3 ml vs -40,9 ml). Nausées et vomissements ont néanmoins été observés plus fréquemment dans le groupe combiné. Bien que cette étude soit prometteuse, une étude de plus longue durée sur un plus grand collectif, évaluant son efficacité, est néanmoins nécessaire avant de recommander un tel traitement combiné.
D’ autres études sont en cours pour déterminer l’ utilité d’ associations basées sur les comorbidités, anti-fibrotiques + sildénafil dans l’ hypertension pulmonaire par exemple.
Les effets secondaires des anti-fibrotiques
Les effets secondaires les plus fréquents liés aux anti-fibrotiques touchent le système gastro-intestinal (2).
Sous nintedanib, les diarrhées ont été reportées chez 61,5 % des patients. La majorité ont cependant pu poursuivre leur traitement après une réduction de la posologie associée à des anti-diarrhéiques. Nausées, vomissements, inappétence, douleurs abdominales, perturbations des tests hépatiques, perte de poids et hypertension ont également été observés (1). Un risque hémorragique augmenté a encore été reporté en raison de l’ inhibition du récepteur du facteur de croissance de l’ endothélium vasculaire (VEGF), ce qui exige une pesée du risque-bénéfice chez les patients à risque hémorragique. De même des cas de thromboses artérielles ont été décrits, requérant la prudence chez les patients présentant des risques cardio-vasculaires élevés.
Sous pirfenidone, l’ effet secondaire le plus fréquemment reporté est la nausée (35,5 % des patients). Cet effet secondaire est géré par la réduction de dose, la prise du traitement avec les repas, voire son interruption. Une photosensibilisation et un rash cutané ont également été décrits imposant aux patients de minimiser leur exposition au soleil (1).
Le nintedanib et la pirfenidone peuvent entraîner une perturbation des tests hépatiques, ALAT et ASAT, généralement réversible après réduction de dose ou arrêt. Le nintedanib est à proscrire lors d’ atteinte hépatique préexistante (Child B, C) et la posologie réduite à 2 x 100 mg/j pour une atteinte Child A.
Traitements non-pharmacologiques
Si les anti-fibrotiques occupent une place essentielle dans le traitement de la FPI, d’ autres approches font également partie de leur prise en charge.
C’ est le cas de l’ oxygénothérapie qui est clairement indiquée chez les patients hypoxémiques au repos (8).
De même, la réhabilitation pulmonaire doit être envisagée pour améliorer la tolérance à l’ effort et la qualité de vie des patients. Elle permet également d’ apporter conseils et soutien psychologique aux patients et à leurs proches (8).
La transplantation pulmonaire (uni-pulmonaire, bi-pulmonaire, cœur-poumons) représente une option thérapeutique pour une minorité de patients en raison des fréquentes comorbidités et de l’ âge avancé des patients. Elle doit néanmoins faire l’ objet d’ une évaluation au stade précoce de la maladie, avant même la détérioration spirométrique, afin de maximaliser les chances d’ éligibilité chez les patients de moins de 65 ans (9). En Suisse, la fibrose pulmonaire représente le quart des transplantations, bi-pulmonaires le plus souvent.
La progression de la maladie et la fréquence des exacerbations sont par ailleurs significativement réduites par l’ arrêt du tabac et la vaccination (grippe et pneumocoques).
Le traitement des comorbidités et des exacerbations
Les comorbidités, hypertension pulmonaire, reflux gastro-œsophagien (RGO), BPCO, diabète et coronaropathie, sont responsables de 30-40 % des décès de la FPI, et sont associées à un mauvais pronostic. Leur traitement fait donc partie intégrante de la prise en charge de la FPI, et permet d’ améliorer l’ espérance de vie de la FPI.
Ainsi lors de comorbidités cardio-vasculaires, les inhibiteurs de la thrombine, tel que le dabigatran, seront préférés aux coumariniques qui peuvent péjorer le pronostic de la FPI. Les statines ayant une action anti-inflammatoire ont également un effet protecteur dans l’ évolution de la FPI.
Lors de RGO, malgré le faible niveau d’ évidence et le risque majoré d’ infection, les inhibiteurs de la pompe à protons sont recommandés dans la FPI.
Les exacerbations aiguës peuvent survenir n’ importe quand et sont associées à une mortalité de 50 %. Si les corticoïdes ne font plus partie des recommandations du traitement chronique de la FPI, leur place reste avérée, souvent en association avec des antibiotiques, lors d’ exacerbations aiguës malgré l’ absence d’ études contrôlées. Par ailleurs les autres immunosuppresseurs (tacrolimus, cyclophosphamide) sont également une option envisagée dans certaines recommandations cliniques (8).
Perspectives futures
Afin d’ améliorer le diagnostic et le traitement de la FPI, les recherches actuelles portent sur le développement de bio-marqueurs. Le diagnostic pourrait bénéficier de marqueurs sanguins des lésions épithéliales et de la dégradation de la matrice (métalloprotéinase MMP7, chitinase-like protéine) afin d’ éviter des biopsies à risque. De même des marqueurs pronostiques sont étudiés, telle que la C réactive protéine. Des marqueurs génétiques sont aussi étudiés dans le cadre de la médecine prédictive. Ces marqueurs n’ ont cependant pas encore d’ application clinique (9).
Plusieurs évidences mettant en avant le rôle d’ une altération du microbiome dans la progression de la maladie, l’ utilisation d’ antibiotiques, tels que le co-trimoxazole, susceptibles de réduire la charge bactérienne des voies aériennes, est en cours d’ étude (9).
Soins palliatifs
Bien que la qualité de vie et l’ espérance de survie soient souvent inférieures à de nombreux cancers, plusieurs études montrent que le recours à une approche palliative est souvent oublié dans la FPI (10-12). Or, l’ intolérance à l’ effort, la dyspnée progressive, les hospitalisations, et les exacerbations péjorent la qualité de vie des patients souffrant de FPI.
Si les besoins d’ une approche palliative augmentent avec la progression de la maladie, il est essentiel d’ y recourir dès la confirmation du diagnostic afin de minimiser l’ angoisse engendrée par l’ incertitude du pronostic. Un accompagnement individualisé permet de prendre en compte les aspects psycho-sociaux des patients et de leurs proches, ainsi que leurs besoins, tout en leur apportant informations et soutien du diagnostic au décès. L’ accompagnement dans la rédaction de directives anticipées permet de s’ assurer que les traitements entrepris sont conformes aux souhaits du patient et évite des traitements futiles, tels qu’ une intubation ou la mise en place d’ une circulation extracorporelle non désirées.
Des outils tels que le NECPAL (13) peuvent aider les médecins à évaluer de manière qualitative et quantitative les patients susceptibles de bénéficier d’ une approche palliative et du moment le plus approprié d’ intervention.
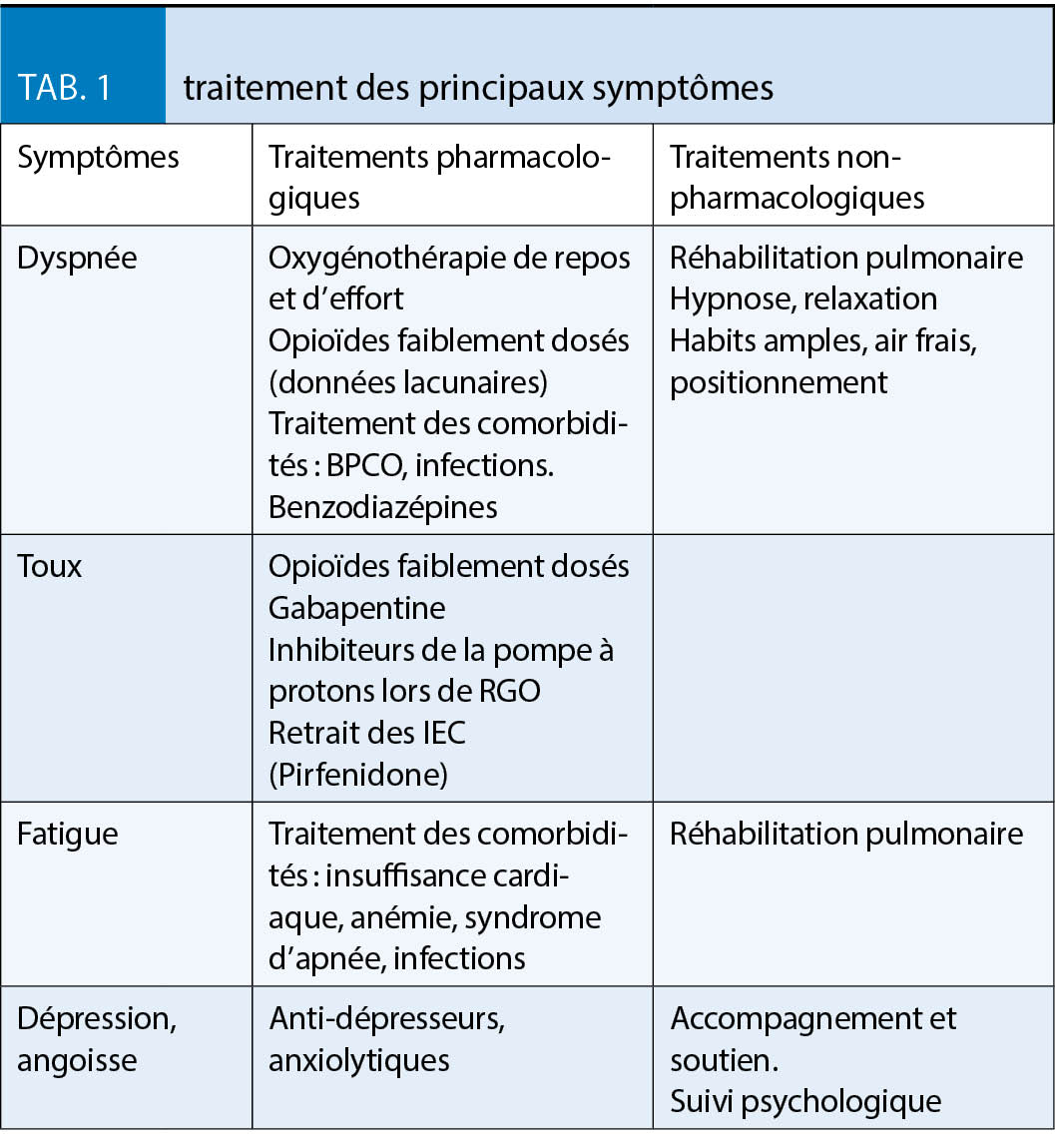
La gestion des symptômes, dyspnée, toux, fatigue, dépression et anxiété, qui ne doit pas être négligée dans la prise en charge de la FPI, est résumée dans le tableau 1 (10). La plupart des traitements symptomatiques proposés le sont cependant avec un faible niveau d’ évidence.
Conclusion
On estime qu’ en Suisse 2000 personnes souffrent de FPI, une maladie mortelle, dont la médiane de survie est de 3-5ans. De nombreux patients ne bénéficient pas d’ un diagnostic et d’ un traitement initié précocement.
L’ évolution imprévisible de la FPI relève d’ une évaluation initiale interdisciplinaire et holistique dans des centres spécialisés.
L’ instauration d’ un traitement anti-fibrotique, un bilan pré-greffe chez les moins de 65 ans, le traitement des comorbidités, les mesures préventives ainsi qu’ une approche palliative précoce, représentent actuellement la meilleure attitude susceptible d’ améliorer le pronostic et la qualité de vie des patients souffrant de fibrose pulmonaire idiopathique.
Hôpital de Lavaux, service de médecine et réadaptation
Colombaires 31
1096 Cully
gerard.pralong@hopitaldelavaux.ch
L’auteur n’a déclaré aucun conflit d’intérêts en relation avec cet article.
1. Glassberg MK. Overview of idiopathic pulmonary fibrosis, evidence-based guidelines, and recent developments in the treatment landscape. Am J Manag Care 2019 ; S195-S203
2. Pleasants R and Tighe RM. Management of idiopathic pulmonary fibrosis. Annals of pharmacotherapy 2019 ; 1-11
3. Hostettler K. Fibrose pulmonaire idiopathique. Swiss medical forum 2017 ; 17(50) :1115-1123
4. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2018 ; 198 : e44-e68
5. King TE, Bradford WZ, Castro-Bernardini S, et al. ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014 ; 370 : 2083-2092
6. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014 ; 370 : 2071-2082
7. Vancheri C, Kreuter M, Richeldi L, et al. Nintedanib with add-on pirfenidone in idiopathic pulmonary fibrosis. Results of the INJOURNEY trial. Am J Respir Crit Care Med 2018 ; 197 : 356-363
8. Homma S, Bando M, Azuma A, et al. Japanese guideline for the treatment of idiopathic pulmonary fibrosis. Respiratory investigation 2018 ; 56 : 268-291
9. Somogyi V, Chaudhuri N, Torrisi SE, et al. The therapy of idiopathic pulmonary fibrosis : what is next ? Eur Respir Rev 2019; 28: 190021
10. Kreuter M, Bendstrup E, Russell AM, et al. Palliative care in interstitial lung disease: living well.Lancet Respi Med 2017; 5 : 968-80.
11. Faverio P, et al. Early referral to palliative care services in patients with IPF : a tool to take a step forward. BMJ Supportive & Palliative Care 2019 ; Aug 29.
12. Ferrara G, Luppi F, Birring SS, et al. Best supportive care for idiopathis pulmonary fibrosis : current gaps and future directions. Eur Respir Rev 2018 ; 27 : 170076
13. Gómez-Batiste X, Martínez-Muñoz M, Blay C, et al. Identifying patients with chronic conditions in need of palliative care in the general population : development of the NECPAL tool and preliminary prevalence rates in Catalonia. BMJ Support Palliat Care 213 ; 3 : 300-8

la gazette médicale
- Vol. 8
- Ausgabe 7
- Dezember 2019