Anamnese und Befunde
Wir berichten über vier Patienten, welche über den Zeitraum von 2014 bis 2021 mit rasch fortschreitenden kognitiven, neuropsychiatrischen und neurologischen Symptomen behandelt wurden. Der Fall von zwei Patienten wird im Folgenden exemplarisch geschildert.
Ein 67-jähriger Patient stellte sich mit progredienten Störungen des Kurzzeitgedächtnisses, Desorientiertheit, Tremor an verschiedenen Körperstellen sowie neu aufgetretener Urininkontinenz vor. Im Neurostatus fielen eine Ataxie, eine verminderte sensible Diskrimination (Spitz-Stumpf- sowie Warm-Kalt-Sensibilität und reduzierter Vibrationssinn an den unteren Extremitäten) sowie ein Rigor mit Zahnradphänomen aller Extremitäten auf. Des Weiteren fanden sich Myoklonien und okulomotorische Störungen mit sakkadierten Blickfolgebewegungen, ein blickgerichteter Nystagmus beidseits, eine Blickparese nach oben und deutlich verlangsamte vertikale Sakkaden.
Beim zweiten Patienten handelte es sich um einen 56-jährigen Mann, der über einen progredienten Verlust des Geruchssinns sowie einen Gewichtsverlust von 8 kg innerhalb von zwei Monaten klagte. Es fielen Konzentrationsstörungen mit Schwierigkeiten beim Folgen von Aufforderungen, eine Verschlechterung des Kurzzeitgedächtnisses sowie eine Schwäche der unteren Extremitäten auf. Im Weiteren fanden sich eine Desorientiertheit, eine Apraxie sowie eine inkomplette globale Aphasie mit insbesondere Wortfindungsstörungen und vermindertem Sprachfluss.
Differenzialdiagnostische Überlegungen
Aufgrund der Beteiligung von verschiedenen Hirnregionen bei vorherrschend kognitiven Beschwerden, aber auch zerebellären, extrapyramidalmotorischen und aphasischen Symptomen, ist die breite Differenzialdiagnostik der Enzephalopathie respektive des enzephalitischen Syndroms in Betracht zu ziehen. Dazu gehören neuroinvasive infektiöse Erkrankungen (wie Neuroborreliose, Neurolues und virale Erkrankungen wie HIV, Herpes- und FSME-Viren), autoimmun bedingte Enzephalitiden und Vaskulitiden, multifokale zerebrale Ischämien, beispielsweise im Rahmen einer Endokarditis, oder primäre und sekundäre ZNS-Neoplasien (Lymphome, ein metastasierender Tumor, seltener auch eine Meningeosis carcinomatosa oder paraneoplastische Enzephalopathien) (1).
Letztlich gibt es einige rasch progrediente neurodegenerative Erkrankungen, an die gedacht werden muss. Aufgrund der Multifokalität sollte eine Creutzfeldt-Jakob-Erkrankung erwogen werden, aber auch eine Alzheimer-Erkrankung oder eine frontotemporale Demenz können relativ rasch fortschreiten, mit aphasischen Störungen im Kontrast zu nur diskreten motorischen Beschwerden. Daneben gibt es Parkinson-Plus-Syndrome, zum Beispiel die supranukleäre Blickparese oder Lewy-Körperchen-Erkrankung, bei der typischerweise kognitive und extrapyramidalmotorische Störungen kombiniert auftreten. Auch eine Multisystematrophie ist möglich. Diese löst zwar oft weniger kognitive Symptome aus, kann aber sowohl zerebelläre als auch extrapyramidalmotorische Symptome mit Tremor und autonomen Störungen wie Urininkontinenz aufweisen. Auch eine kortikobasale Degeneration kann gelegentlich rasch fortschreiten mit kognitiven Einbussen und initial stark asymmetrischen extrapyramidalmotorischen Beschwerden mit ausgeprägtem Rigor, Myoklonien und Apraxie (1).
Aufgrund der breiten Differenzialdiagnose sind eine detaillierte zerebrale Bildgebung mittels MRI des Schädels mit Kontrastmittel und Angiographie indiziert, eventuell zusätzlich mit Dark-blood-Sequenzen bei Vaskulitis-verdacht. Die zeitnahe Labor- und Liquorabklärung ist essenziell. Im Serum werden Antikörper auf ausgewählte infektiöse Erreger und im Liquor infektiöse Erreger mittels PCR oder Liquor-/Serum-Index analysiert. Zudem werden autoimmune und paraneoplastische Antikörper gesucht. Im Weiteren sollte bei Lymphom- oder Tumorverdacht eine Zytologie abgenommen werden wie auch Demenzmarker bei Alzheimer-Verdacht und Bestimmung des Proteins 14-3-3 und «Real-time quaking induced conversion» (RT-QuIC) bei möglicher Prionenerkrankung. Auch die Durchführung eines Elektroenzephalogramms (EEG) ist bei starker Desorientierung, Wesensänderung und Myoklonien indiziert, um Hinweise für eine Allgemeinveränderung oder epilepsietypische Potenziale zu erhalten (1).
Weitere Abklärungsschritte und Verlauf
Das MRI des Schädels des ersten Patienten war unauffällig. Die Basisanalytik in der Liquoruntersuchung fiel unauffällig bei Zellzahl und Protein in der Norm aus. In Liquorkultur, -PCR und serologisch fanden sich keine Hinweise auf eine infektiöse Ursache. Es zeigte sich jedoch eine positive RT-QuIC für Prionen sowie ein positives Protein 14-3-3, passend zur Verdachtsdiagnose einer sporadischen Creutzfeldt-Jakob-Krankheit («sporadic Creutzfeldt Jakob disease», sCJD). Im EEG fanden sich eine Verlangsamung der Grundaktivität und triphasische Potenziale (Abb. 1).
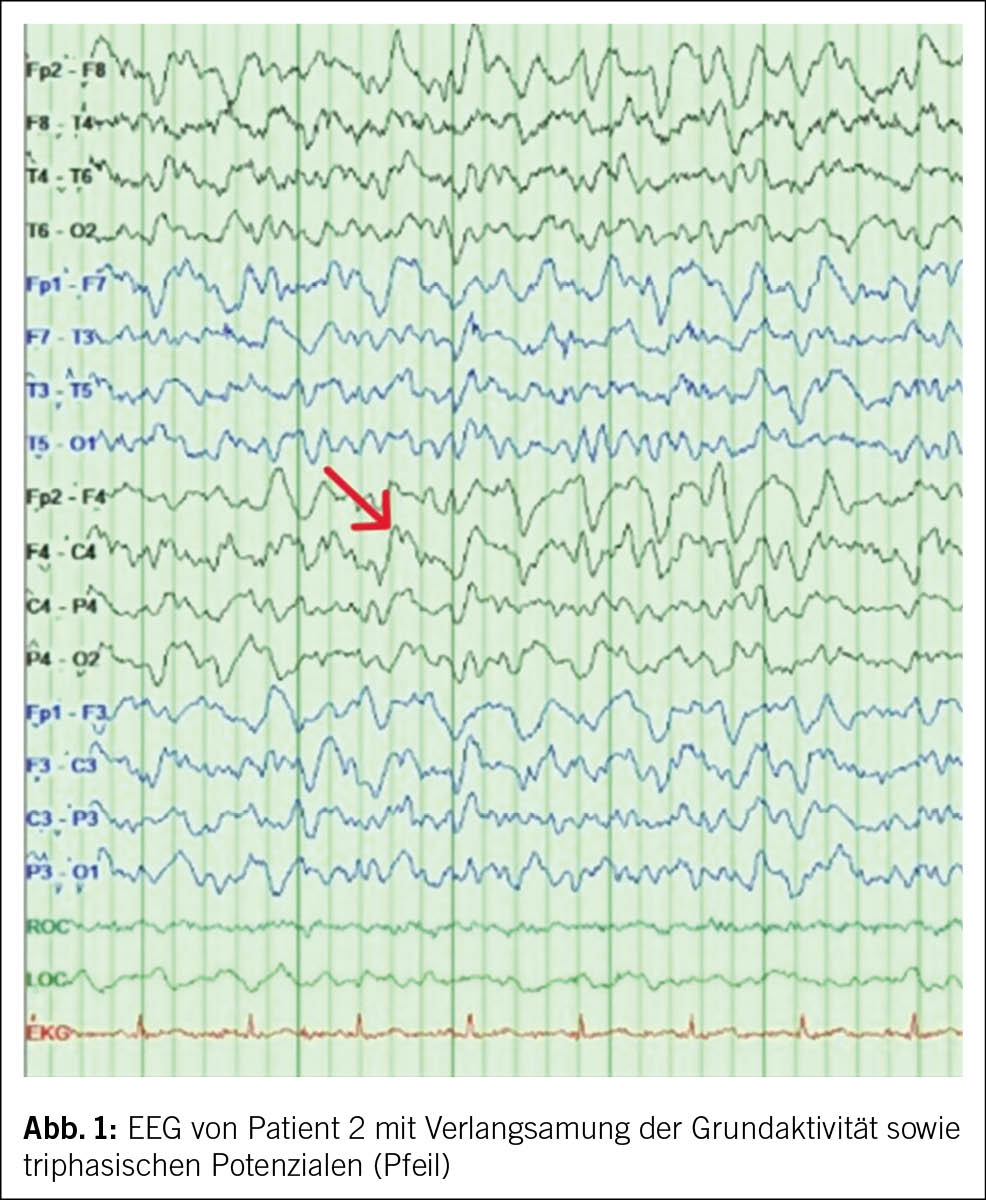
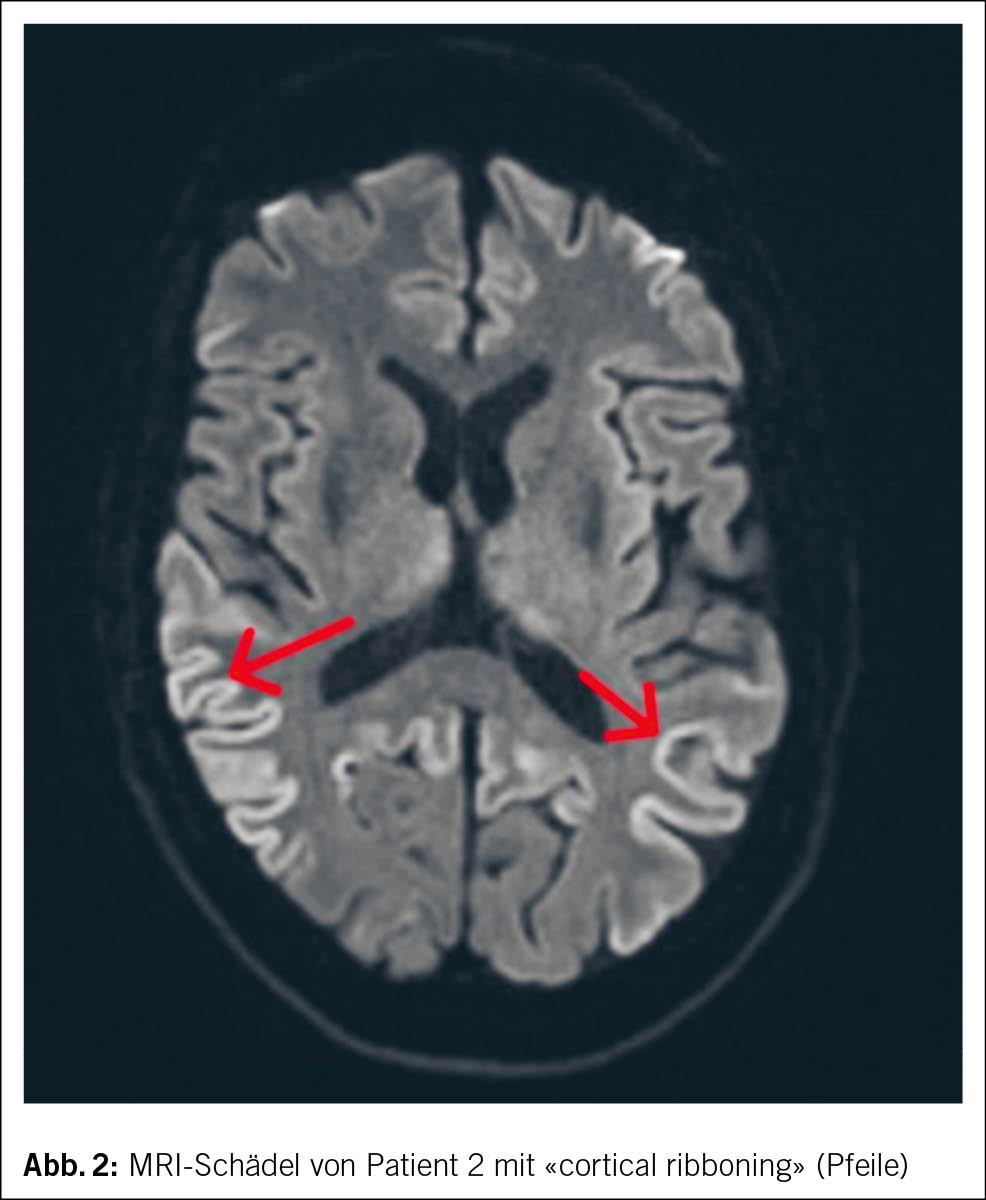
Im MRI des Schädels des zweiten Patienten zeigten sich streng kortikale, hyperintense Signalveränderungen ohne Einbezug der subkortikalen weissen Substanz, ein sogenanntes «cortical ribboning» (Abb. 2). Im Liquor wurden ebenfalls eine positive RT-QuIC für Prionen und ein positives Protein 14-3-3 festgestellt. Hinweise für eine infektiöse oder entzündliche Ursache fanden sich nicht.
Das klinische Erscheinungsbild war bei beiden Patienten rasch progredient und ohne Möglichkeit zur spezifischen Therapie. Beide Patienten verstarben im kurzzeitigen Verlauf innerhalb eines Jahres nach Diagnosestellung.
Diagnose
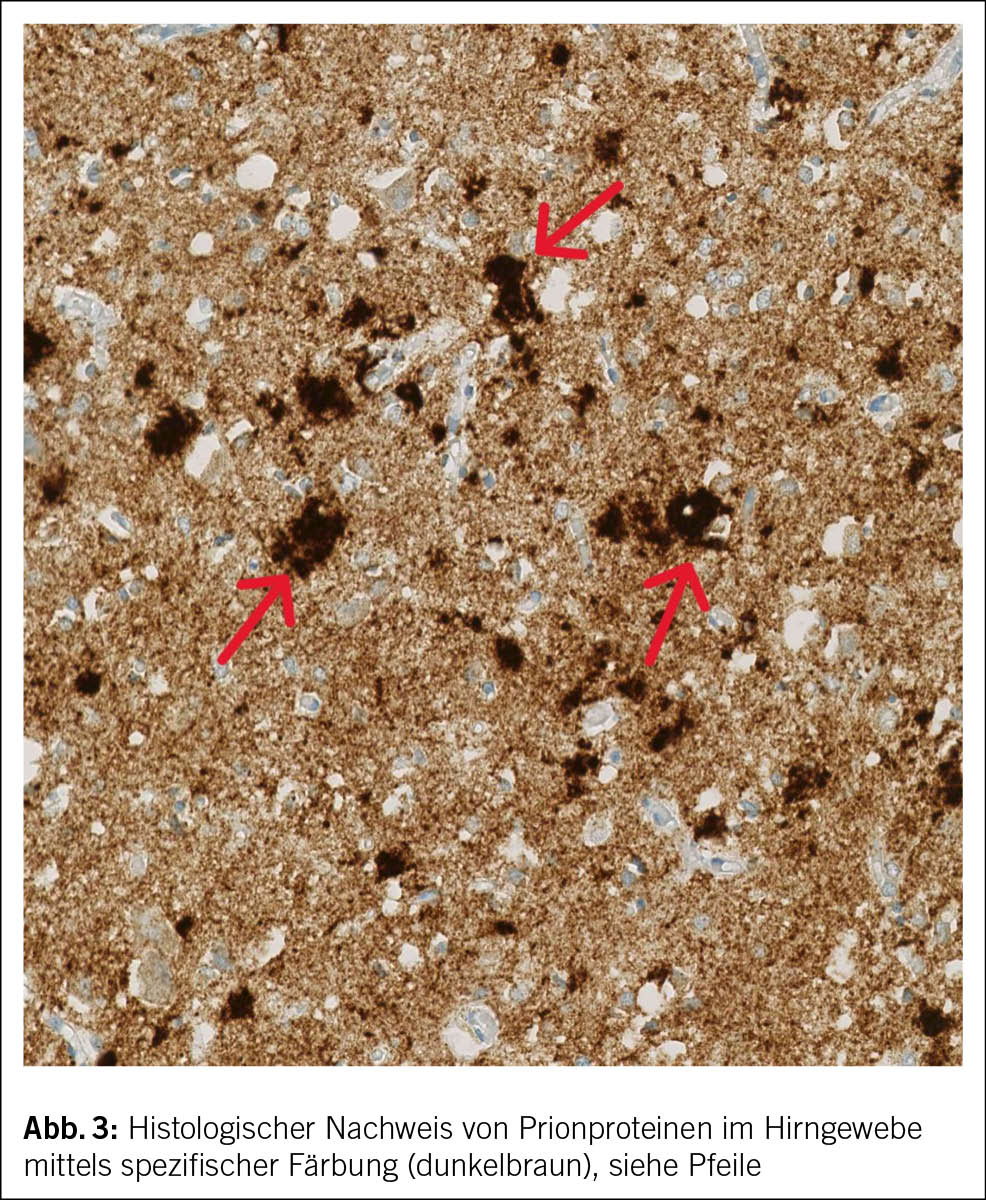
Bei charakteristischer Klinik mit rasch progredientem Verlauf und den typischen Befunden im MRI des Schädels sowie in Liquor und EEG lag bei beiden Patienten das Bild einer sCJD vor. Bei Patient 2 wurde die Diagnose post mortem in der Histopathologie des Hirngewebes gesichert (Abb. 3).
Kommentar
Die sCJD gehört zur Gruppe der subakuten spongiformen Enzephalopathien, welche durch abnorm gefaltete Prionen verursacht wird (2). Im Gehirn und in den hinteren Augenabschnitten ist die Konzentration der Prionen am höchsten, was den Symptomenkomplex aus rasch progredienter Demenz, extrapyramidalen Störungen, Myoklonien sowie visuellen und Kleinhirnstörungen verursacht (2, 3). Aufgrund der heterogenen und unspezifischen Symptomatik, insbesondere bei Krankheitsbeginn, gestaltet sich die klinische Diagnosestellung schwierig (2). Um unnötige Diagnostik zu vermeiden und möglichst Klarheit in den differenzialdiagnostischen Herausforderungen zu schaffen, ist es wichtig, die Erkrankung als mögliche Differenzialdiagnose bei rasch progredienten neurodegenerativen Erkrankungen einzubeziehen. Die rasche Dynamik der neurokognitiven sowie motorischen Symptome gerade bei Krankheitsbeginn führt zum klinischen Verdachtsmoment, die Fremdanamnese durch nahe Angehörige ist oft hilfreich. Diese Fallserie hat den Zweck, das Krankheitsbild bekannter und auf den Symptomkomplex aufmerksam zu machen.
In unserem Spital wurde im Zeitraum von 2014 bis 2021 bei vier männlichen Patienten im Alter von 56–85 Jahren mit rasch progredienter neurokognitiver Symptomatik eine sCJD diagnostiziert, bei drei Patienten wurde die Diagnose post mortem autoptisch gesichert. Das Einzugsgebiet des Spitals zählt etwa 150 000 Bewohner. In der Literatur wird eine Krankheitsinzidenz von 1–2:1 000 000 in der Durchschnittsbevölkerung angegeben (4), sodass die Krankheit eventuell unterdiagnostiziert wird. In der Schweiz liegt die Inzidenz bei 0.4–2.63/1 000 000 Einwohner/-innen, wobei es Anfang des 21. Jahrhunderts zu einem vorübergehend signifikanten Anstieg der Fallzahlen kam. Dies wurde im Rahmen der Etablierung neuer diagnostischer Methoden mit infolgedessen vermehrter Aufmerksamkeit für die Erkrankung interpretiert (5 [WG1.1]).
Allen Patienten in unserer Fallserie gemeinsam war eine rasche Progredienz der Symptomatik in Kombination mit einer Demenz und neurologischen Auffälligkeiten wie pyramidalen und extrapyramidalen Zeichen sowie visuellen und zerebellären Symptomen oder Myoklonien (Tab. 1).
Es gibt 4 Hauptformen der Creutzfeldt-Jakob-Erkrankung: sporadisch, genetisch, iatrogen und erworben; die sporadische Form stellt mit 80–95 % die häufigste Form dar (4, 6). Das mediane Erkrankungsalter liegt bei 67 Jahren (4, 6), wobei es auch deutlich jüngere betroffene Personen gibt (6). Die Geschlechtsverteilung variiert je nach Region (7, 8). Bis heute gibt es keine kurative Therapie, die meisten Betroffenen sterben innerhalb eines Jahres nach Diagnosestellung (4, 6, 9). Jüngere Patienten scheinen eher einen längeren Krankheitsverlauf zu haben (6). Die sporadische Form der Creutzfeldt-Jakob-Erkrankung wird durch abnorm gefaltete, nicht lösliche Prionen (PrPsc) (10) verursacht, welche teilweise bereits Jahrzehnte vor den ersten Symptomen nachweisbar sind (4). Die Fehlfaltung der Prionen beginnt spontan (6) und führt zu Ablagerungen als Amyloid im Gewebe (10), was wiederum zu einer spongiformen Degeneration von Hirngewebe, neuronalem Untergang und astrozytärer Gliose führt (9, 11). Die Ursache der Erkrankung ist bisher nicht bekannt, es gibt jedoch Hinweise auf eine mögliche genetische Komponente (12). Das normale Prionprotein (PrPc) wird durch Chromosom 20 kodiert. Bisher ist keine klare Funktion des physiologischen Prion-proteins bekannt, es könnte jedoch eine Rolle im Kupferstoffwechsel und in antioxidativen Systemen spielen (10). Interessanterweise wird das Krankheitsbild in asiatischen Ländern kaum beschrieben, wobei dies eventuell auch durch die Seltenheit der Erkrankung und somit diagnostische Herausforderung sowie fehlende Dokumentation und Informationsweitergabe bedingt sein könnte (6).
Im Folgenden werden die wichtigsten diagnostischen Abklärungen und Analysen beschrieben, die bei der klinischen Verdachtsdiagnose einer sCJD angewendet werden. Die Diagnosekriterien sind in Tab. 2 aufgeführt (siehe auch S1-Leitlinie zur Creutzfeldt-Jakob-Krankheit der DGN [7]).

MRI-Schädel
Bei den meisten Patienten sind der zerebrale Kortex und die Basalganglien involviert. Typisch ist ein sogenanntes «cortical ribboning» (2, 6), also kortikale Signalalterationen in den Sequenzen der «fluid attenuated inversion recovery» (FLAIR) sowie in den diffusionsgewichteten Aufnahmen («diffusion weighted imaging», DWI) (13).
Liquoruntersuchung
In der Basisanalytik im Liquor kann die Proteinmenge leicht erhöht sein. Zudem kann eine Pleozytose mit normaler Glukose im Liquor vorliegen (6).
Die Liquoruntersuchung dient insbesondere dazu, Infektionen und Entzündungen des zentralen Nervensystems oder andere Differenzialdiagnosen auszuschliessen. Es gibt typische Befunde, welche auf eine sCJD hinweisen, dazu zählen das Protein 14-3-3 und die RT-QuIC (6).
Die Sensitivität und Spezifität des Proteins 14-3-3 sind sehr variabel. In einer Metaanalyse wurde über eine Sensitivität von ca. 92 % bei einer Spezifität von ca. 80 % (14) berichtet, wobei in frühen Krankheitsstadien die Sensitivität geringer zu sein scheint (15).
Die Spezifität des RT-QuIC liegt bei nahezu 100 % bei einer Sensitivität von 70 % (6, 16) bis 90 % (17, 18, 19) bei Tests der ersten Generation. Solche der zweiten Generation zeigen eine etwas höhere Sensitivität von 92 % (20) bis 97 % (21).
Weiter kann eine starke Erhöhung des totalen Tau im Liquor die Diagnose untermauern (15, 22), aktuell ist dies jedoch nicht in den Diagnosekriterien für sCJD integriert (13). Wichtige Differenzialdiagnosen für eine Erhöhung des Tau-Proteins im Liquor sind die Alzheimer-Demenz sowie entzündliche und neoplastische ZNS-Erkrankungen (3).
EEG
Im EEG sind periodische «Sharp wave»-Komplexe («periodic sharp wave complexes», PSWC) mit einer Frequenz von ca. 1–1.5 Hz typisch für sCJD (6). PSWC können jedoch auch bei anderen Erkrankungen auftreten (2). In sehr frühen Krankheitsstadien kann das EEG noch normal sein, das Fehlen von solchen Komplexen schliesst eine sCJD somit nicht aus (2, 6, 23).
Biopsie
Da eine definitive Diagnose nur histopathologisch gestellt werden kann, ist die Hirnautopsie bzw. -biopsie weiterhin der Goldstandard zur Diagnose einer sCJD (6, 24). Die Hirnbiopsie erfolgt aufgrund der Invasivität und oft auch fehlender Verfügbarkeit der Diagnostik (24) in der Regel erst post mortem. Es besteht das Risiko eines falsch negativen Resultats, da die fehlgefalteten Prionen nicht in allen Hirnregionen zu finden sind (15).
Bei potenziellem Kontakt mit Gewebe bei Verdacht auf sCJD muss ein mögliches Übertragungsrisiko auf das beteiligte Gesundheitspersonal berücksichtigt werden. Dies gilt insbesondere beim Umgang mit Probematerial aus dem zentralen Nervensystem, da hier die Prionenkonzentration am höchsten ist (2). Gerade [WG2.1] bei einer Autopsie mit Verdacht auf eine sCJD sind strenge Sicherheitsvorkehrungen zu treffen. Dazu gehören beispielsweise ein Speziallabor (Biosicherheitslabor, BSL3) sowie Ganzkörperschutzanzüge für das Personal (25).
Janine Badertscher, Gaby Schoch, Robert Escher, Gabriel Waldegg
Klinik für Allgemeine Innere Medizin, Spital Emmental, Burgdorf
Abkürzungen
DWI Diffusion weighted imaging
EEG Elektroenzephalogramm
FLAIR fluid attenuated inversion recovery
FSME Frühsommer-Meningoenzephalitis
HIV Humanes Immundefizienz-Virus
MRI Magnetic resonance imaging
PCR Polymerase chain reaction
PSWC Periodic sharp wave complexes
RT-QuIC Real-time quaking induced conversion
sCJD sporadic Creutzfeldt-Jakob-Disease
Historie
Manuskript eingegangen: 28.10.2025
Angenommen nach Revision: 11.02.2026
Assistenzärztin
Spital Emmental Burgdorf
Oberburgstrasse 54
3400 Burgdorf
janine.badertscher@gmail.com
Die Autorin hat keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Hermann P, Zerr I. Rapidly progressive dementias — aetiologies, diagnosis and management. Vol. 18, Nature Reviews Neurology. Nature Research; 2022. p. 363–76.
2. Salehi P, Clark M, Pinzon J, Patil A. Sporadic Creutzfeldt-Jakob disease. American Journal of Emergency Medicine. 2022 Feb 1;52:267.e1-267.e3.
3. Lehmann S, Paquet C, Malaplate-Armand C, Magnin E, Schraen S, Quillard-Muraine M, et al. Diagnosis associated with Tau higher than 1200 pg/mL: Insights from the clinical and laboratory practice. Clinica Chimica Acta. 2019 Aug 1;495:451–6.
4. Uttley L, Carroll C, Wong R, Hilton DA, Stevenson M. Review Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation [Internet]. 2020. Available from: www.thelancet.com/infection
5. Stoeck K, Hess K, Amsler L, Eckert T, Zimmermann D, Aguzzi A, et al. Heightened incidence of sporadic Creutzfeldt-Jakob disease is associated with a shift in clinicopathological profiles. J Neurol. 2008 Oct;255(10):1464–72.
6. Khan S, Khan S. Sporadic Creutzfeldt-Jakob disease: Diagnosing typical and atypical presentations under limited circumstances. Dement Geriatr Cogn Disord. 2021 Jun 1;50(1):36–42.
7. Hermann P, Treig J, Unkel S, Goebel S, Bunck T, Jünemann M, et al. Sporadic Creutzfeldt-Jakob disease among physicians, Germany, 1993–2018. Emerg Infect Dis. 2020 Aug 1;26(8):1710–9.
8. Feng S, Zhao X, Zhou X, Ye X, Yu X, Jiang W, et al. Epidemiological and Clinical Characteristics of Sporadic Creutzfeldt–Jakob Disease: A Retrospective Study in Eastern China. Front Neurol. 2021 Oct 6;12.
9. Kojima G, Tatsuno BK, Inaba M, Velligas S, Masaki K, Liow KK. Creutzfeldt-Jakob Disease: A Case Report and Differential Diagnoses. Vol. 72. 2013.
10. Knight RSG. PRION DISEASES. J Neurol Neurosurg Psychiatry [Internet]. 2004 Mar 1;75(90001):36i–42. Available from: https://jnnp.bmj.com/lookup/doi/10.1136/jnnp.2004.036137
11. Fabiano Marin L, Carvalho Felício A, Bernardi Bichuetti D, Adolfo Celso dos Santos W, Raquel Rodrigues Borges L, Parissi Buainain R, et al. CliniCal findingS in Creutzfeldt-Jakob diSeaSe mimiCking dementia with lewy bodieS. Vol. 66, Arq Neuropsiquiatr. 2008.
12. Windl O, Dempster M, Collinge J. Genetic basis of Creutzfeldt-Jakob disease in the United Kingdom a systematic analysis of predisposing mutations and allelic variation in the PRNP gene. 1996;
13. Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009 Oct;132(10):2659–68.
14. Muayqil T, Gronseth G, Camicioli R. Evidence-based guideline: Diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease Report of the Guideline Development Subcommittee of the American Academy of Neurology [Internet]. 2012. Available from: www.neurology.org
15. Hermann P, Appleby B, Brandel JP, Caughey B, Collins S, Geschwind MD, et al. Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. Vol. 20, The Lancet Neurology. Lancet Publishing Group; 2021. p. 235–46.
16. Groveman BR, Orrú CD, Hughson AG, Bongianni M, Fiorini M, Imperiale D, et al. Extended and direct evaluation of RT-QuIC assays for Creutzfeldt-Jakob disease diagnosis. Ann Clin Transl Neurol. 2017 Feb 1;4(2):139–44.
17. McGuire LI, Peden AH, Orrú CD, Wilham JM, Appleford NE, Mallinson G, et al. Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 2012 Aug;72(2):278–85.
18. Hermann P, Laux M, Glatzel M, Matschke J, Knipper T, Goebel S, et al. Validation and utilization of amended diagnostic criteria in Creutzfeldt-Jakob disease surveillance. Neurology. 2018 Jul 24;91(4):e331–8.
19. Rudge P, Hyare H, Green A, Collinge J, Mead S. Imaging and CSF analyses effectively distinguish CJD from its mimics. J Neurol Neurosurg Psychiatry. 2018 May 1;89(5):461–6.
20. Foutz A, Appleby BS, Hamlin C, Liu X, Yang S, Cohen Y, et al. Diagnostic and prognostic value of human prion detection in cerebrospinal fluid. Ann Neurol. 2017 Jan 1;81(1):79–92.
21. Franceschini A, Baiardi S, Hughson AG, McKenzie N, Moda F, Rossi M, et al. High diagnostic value of second generation CSF RT-QuIC across the wide spectrum of CJD prions. Sci Rep. 2017 Dec 1;7(1).
22. Otto M, Wiltfang J, Cepek L, Neumann M, Mollenhauer B, Steinacker P, et al. Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease [Internet]. 2002. Available from: https://www.neurology.org
23. Geschwind MD. Prion Diseases. Vol. 21, CONTINUUM Lifelong Learning in Neurology. Lippincott Williams and Wilkins; 2015. p. 1612–38.
24. Ahmad S. Neurodegenerative Diseases. 2012.
25. Reimann Regina. CJD autopsy guidelines (external) Änderungshinweis: fachlich unverändert. 2024.
26. Leitfaden zur Meldepflicht übertragbarer Krankheiten und Erreger 2025 [Internet]. [cited 2025 Oct 25]. Available from: https://www.bag.admin.ch/de/meldepflichtige-infektionskrankheiten