Die ambulant erworbene Pneumonie (community-acquired pneumonia, CAP) bei Kindern und Jugendlichen ist eine häufige Diagnose und zugleich Herausforderung in Praxis und Spital. In den letzten Jahren gab es wesentliche Fortschritte im Verständnis der Diagnose und Therapie der CAP, welche das Management beeinflussen und im Fokus dieser Übersicht stehen.
Community-acquired pneumonia (CAP) in children and adolescents is both a common diagnosis and a challenge in practice and hospital settings. In recent years, there have been significant advances in the understanding of the diagnosis and treatment of CAP, which influence management and are the focus of this review.
Key Words: Community-acquired pneumonia (CAP)
Die ambulant erworbene Pneumonie (community-acquired pneumonia, CAP) ist eine akute Infektion des Lungenparenchyms, welche ausserhalb vom Spital oder anderen Gesundheitseinrichtungen erworben wird. Sie ist eine der häufigsten Hospitalisationsgründe bei Kindern in Industrieländern (1) und immer noch die häufigste Todesursache von Kindern in Entwicklungsländern (2, 3). Die klinische Diagnose der CAP ist schwierig, weil die Symptome mit dem Alter variieren und bei Kleinkindern unspezifisch sein können. Zudem ist die Erregerdiagnose der CAP eine Herausforderung.
Neben der Verringerung der diagnostischen Unsicherheit gilt es, die Exposition der an CAP erkrankten Kinder gegenüber Antibiotika zu reduzieren. Aktuell sind dazu Richtlinien für Kinder und Jugendliche von verschiedenen Gesellschaften in der Schweiz und Europa unter Mitwirkung der Pädiatrischen Infektiologiegruppe Schweiz (PIGS, https://pigs.ch/) in Überarbeitung, welche u.a. Empfehlungen dieser Übersicht beinhalten werden (4).
Epidemiologie
Vor Einführung der Konjugatimpfstoffe gegen Haemophilus influenzae Typ b (Hib) und Streptococcus pneumoniae (pneumococcal conjugate vaccine, PCV) wurden bei Kindern mit CAP hauptsächlich Hib und Pneumokokken nachgewiesen (5). Die Inzidenz und das Erregerspektrum der CAP haben sich durch die Einführung der Hib- und PCV-Impfprogramme wesentlich verändert. In neueren, gross angelegten Studien wurden umfangreiche mikrobiologische Tests durchgeführt, um die Ätiologie bei Kindern und Jugendlichen mit einer radiologisch bestätigten CAP zu untersuchen (1-3). Ein viraler und/oder bakterieller Erreger wurde in 81–99% dieser Kinder in den oberen Atemwegen nachgewiesen. Viren machten dabei die Mehrheit der Erreger aus (1-3, 6), insbesondere bei Kleinkindern (>90%) (2).
Vor der COVID-19-Pandemie war der häufigste nachgewiesene Erreger bei hospitalisierten Kindern mit CAP in den USA das Respiratorische Synzytial-Virus (RSV), vor anderen respiratorischen Viren wie humane Rhinoviren, humane Metapneumoviren, Adenoviren, Parainfluenzaviren und Influenza A und B. Der häufigste bakterielle Erreger war Mycoplasma pneumoniae. Die Ätiologie der CAP variiert mit dem Alter (Tabelle 1), was auch diese Studie zeigte: Der Anteil an RSV war bei Kindern unter 5 Jahren signifikant höher als bei älteren Kindern (37% vs. 8%), umgekehrt war bei Kindern ab 5 Jahren der Anteil von M. pneumoniae höher als bei jüngeren Kindern (19% vs. 3%) (1).

Im März 2020 ist es durch die Einführung von nicht-pharmazeutischen Massnahmen zur Eindämmung der COVID-19-Pandemie (u.a. Maskenpflicht) zu einem massiven Rückgang der Fallzahlen mit teilweise komplettem Verschwinden nahezu aller Atemwegsinfektionen gekommen, was sich in einer signifikanten Abnahme von Hospitalisierungen durch die CAP widerspiegelte (7, 8). Die Zirkulation von SARS-CoV-2 hatte hingegen keinen Einfluss auf die Inzidenz der CAP, da sich COVID-19 bei immunkompetenten Kindern und Jugendlichen nicht als CAP manifestiert (9). Nach den Lockerungen der Massnahmen im Jahre 2021 zirkulierten die meisten Erreger von Atemwegsinfektionen wieder (10). Anstatt der üblicherweise jährlich im Winter auftretenden RSV-Welle kam es zu einem aussersaisonalen Anstieg der RSV-Aktivität im Sommer 2021 und nach dem Wegfallen aller Massnahmen zu einer ungewöhnlich frühen und starken RSV-Saison im Herbst/Winter 2022 mit absoluten Rekordzahlen an Hospitalisationen von RSV-infizierten Kindern (11). Eine Zirkulation von M. pneumoniae konnte auch nach dem Wegfallen aller Massnahmen noch nicht wieder beobachtet werden (10). Zurzeit wird die Aktivität von M. pneumoniae mit einer weltweiten, prospektiven Überwachung monitorisiert (www.escmid.org (12)).
Mikrobiologie
Die rechtzeitige und zuverlässige Erregerdiagnose der CAP ist entscheidend für die Einleitung einer gezielten und wirksamen Antibiotika-Therapie. Der «Goldstandard» für die mikrobiologische Diagnose der CAP ist der Nachweis von Erregern direkt am Ort der Infektion, d.h. in der Lunge, durch invasive Methoden wie bronchoalveoläre Lavage, Pleurapunktion oder Lungenbiopsie. Sputum und Trachealsekret sind Proben der unteren Atemwege, welche oft durch Erreger aus den oberen Atemwegen kontaminiert sind. Zudem kann ein Kind im Unterschied zum Jugendlichen und Erwachsenen Sputum nur selten auf Abruf aushusten. Im klinischen Alltag behilft man sich daher mit Proben, die leichter zu gewinnen, aber deshalb vom Ort der Infektion entfernt sind: Sekrete der oberen Atemwege (PCR, Antigentest), Blut (Serologie, Blutkulturen) und Urin (Antigentest). Jedoch hat keine einzelne Methode, die auf solchen nicht-pulmonalen Proben beruht, gleichzeitig eine hohe Sensitivität und Spezifität für die Erregerdiagnose der CAP bei Kindern und Jugendlichen (13).
Sekrete der oberen Atemwege (Rachenabstrich, Nasopharyngealabstrich oder -sekret): Beispielsweise wurden in einer Studie bei jeweils mehr als der Hälfte der Fälle bei Kindern mit einer CAP (59%) und bei gesunden Kindern (54%) mittels Multiplex-PCR ≥4 Erreger in den oberen Atemwegen nachgewiesen (3). Diese Tatsache erschwert die Zuordnung eines Erregers in den oberen Atemwegen zur CAP. Der Nachweis mehrerer potentieller Erreger in den oberen Atemwegen von Kindern und Jugendlichen mit einer CAP bedeutet insgesamt lediglich eine Kolonisation, eine Persistenz nach früherer Infektion oder eine obere Atemwegsinfektion, muss aber nicht ursächlich für die CAP sein (13). Nur RSV detektiert man seltener bei gesunden als bei kranken Kindern (2, 3).
Serologie: Eine Kolonisation kann auch eine systemische Antikörperreaktion hervorrufen (IgM und IgG) und somit den Aussagewert der Serologie im Blut für die Erregerdiagnose der CAP wesentlich einschränken (13). Zudem ist eine Verlaufs-Serologie zum Nachweis einer Serokonversion und/oder eines signifikanten Anstiegs des Messwertes im Abstand von ≥2 Wochen für das akute klinische Management der CAP nicht hilfreich.
Blutkulturen: Seit Einführung der Hib- und PCV-Impfprogramme sind Blutkulturen nur noch in <1% der Fälle von nicht-schwerer CAP positiv (14). Falls tatsächlich positiv, sind sie jedoch hochspezifisch für die ätiologische Diagnose einer CAP.
Urin: Antigentests für Pneumokokken im Urin sind bei kolonisierten Kindern positiv und weisen daher nur eine geringe Spezifität auf (3). Eine Kolonisation mit Pneumokokken findet man in bis zu 77% bei gesunden Kindern (13).
Diese Aspekte widerspiegeln die Herausforderungen der Erregerdiagnose bei der CAP. Deshalb werden mikrobiologische Tests generell nur für Kinder und Jugendliche mit einer CAP empfohlen, welche hospitalisiert werden müssen (14, 15).
Vielversprechende innovative diagnostische Ansätze für die Zukunft sind neue Biomarker (16-18), eine multidimensionale Analyse der Abwehrreaktion (19, 20), die molekulare Analyse der Ausatmungsluft (21), und neue analytische Ansätze (3). Bei der CAP durch M. pneumoniae konnten wir zeigen, dass der Nachweis von Erreger-spezifischen Antikörper-sezernierenden Zellen (ASC, sog. Plasmablasten) mittels Enzyme-linked ImmunoSpot (ELISpot) Test ein effizienter diagnostischer Indikator einer Infektion mit M. pneumoniae ist – im Unterschied zu einer Kolonisation, bei der keine solchen B-Zellen im Blut vorhanden sind (22, 23). Die Methode wird nun validiert und auf andere Erreger der CAP ausgeweitet.
Klinik und Diagnose
Die CAP wird in erster Linie klinisch diagnostiziert. Sie muss bei Fieber und Tachypnoe (nach Fiebersenkung) in Betracht gezogen werden (15). Die Definition von Fieber (und Messmethode) ist altersabhängig wie folgt festgelegt: <3 Monate, ≥38.0°C (rektal); 3–12 Monate, ≥38.5°C (rektal); und ab 1 Jahr, ≥38.5°C (aurikulär) (24).
Die Tachypnoe scheint das wichtigste klinische Zeichen zu sein, da sie mit Hypoxämie, pneumonischen Infiltraten im Thoraxröntgenbild und insgesamt mit dem Schweregrad der CAP korreliert (15). Sie ist je nach Alter entsprechend definiert: <2 Monate, >60 Atemzüge/min; 2–12 Monate, >50/min; 1–5 Jahre, >40/min; und >5 Jahre, >20/min (14). Die Atemfrequenz sollte wenn möglich über eine ganze Minute bei ruhigem Kind gezählt werden. Fieber allein kann die Atemfrequenz um 10 Atemzüge pro Minute und Grad Celsius Körpertemperatur erhöhen (25). Neben der Tachypnoe sind weitere Zeichen der Atemnot thorakale Einziehungen (subkostal, interkostal oder jugulär), exspiratorisches Stöhnen («grunting»), Nasenflügeln und Apnoen (im Säuglingsalter) (14). Auch die Bronchitis und Bronchiolitis gehen oft mit Fieber und Tachypnoe einher. Die klinische Unterscheidung ist daher entsprechend schwierig. Kinder und Jugendliche mit Pfeifen und/oder Giemen («wheeze») in der Lungenauskultation oder mit Symptomen der oberen Atemwege (z.B. Rhinitis, Pharyngitis) haben jedoch selten eine CAP (15).
Allgemeine Symptome der CAP sind sehr vielseitig: reduzierter Allgemeinzustand (auch nach Fiebersenkung), Schüttelfrost, Husten, Thorax- und Bauchschmerzen, sowie eine Vigilanzminderung können auf eine CAP hindeuten. Die Symptome variieren mit dem Alter und können bei Kleinkindern unspezifisch sein.
Hinweisend für eine CAP sind zudem ein verstärktes oder auch abgeschwächtes Atemgeräusch sowie feinblasige Rasselgeräusche bei Inspiration. Eine Klopfschalldämpfung spricht für ein lobäres Infiltrat und/oder einen Pleuraerguss (14, 15).
Zahlreiche Studien haben gezeigt, dass man anhand von klinischen, laborchemischen und radiologischen Kriterien bei Kindern und Jugendlichen mit einer nicht-schweren CAP nicht zuverlässig zwischen einer bakteriellen und viralen Ätiologie unterscheiden kann (14, 15). Daher sollten sich Therapieentscheidungen vielmehr am klinischen Bild und den zu erwartenden Erregern anhand des Alters und der Epidemiologie orientieren.
Management
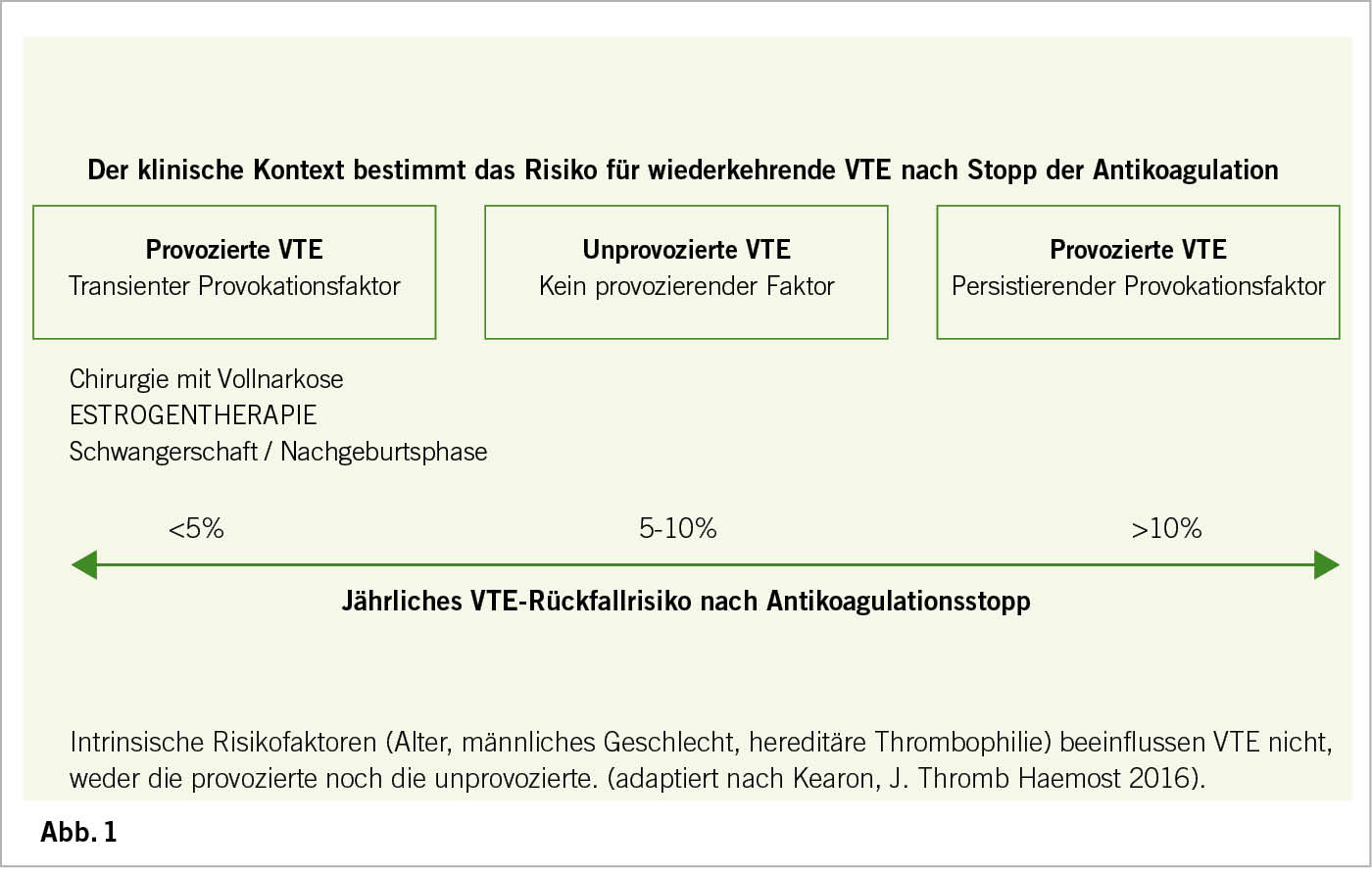
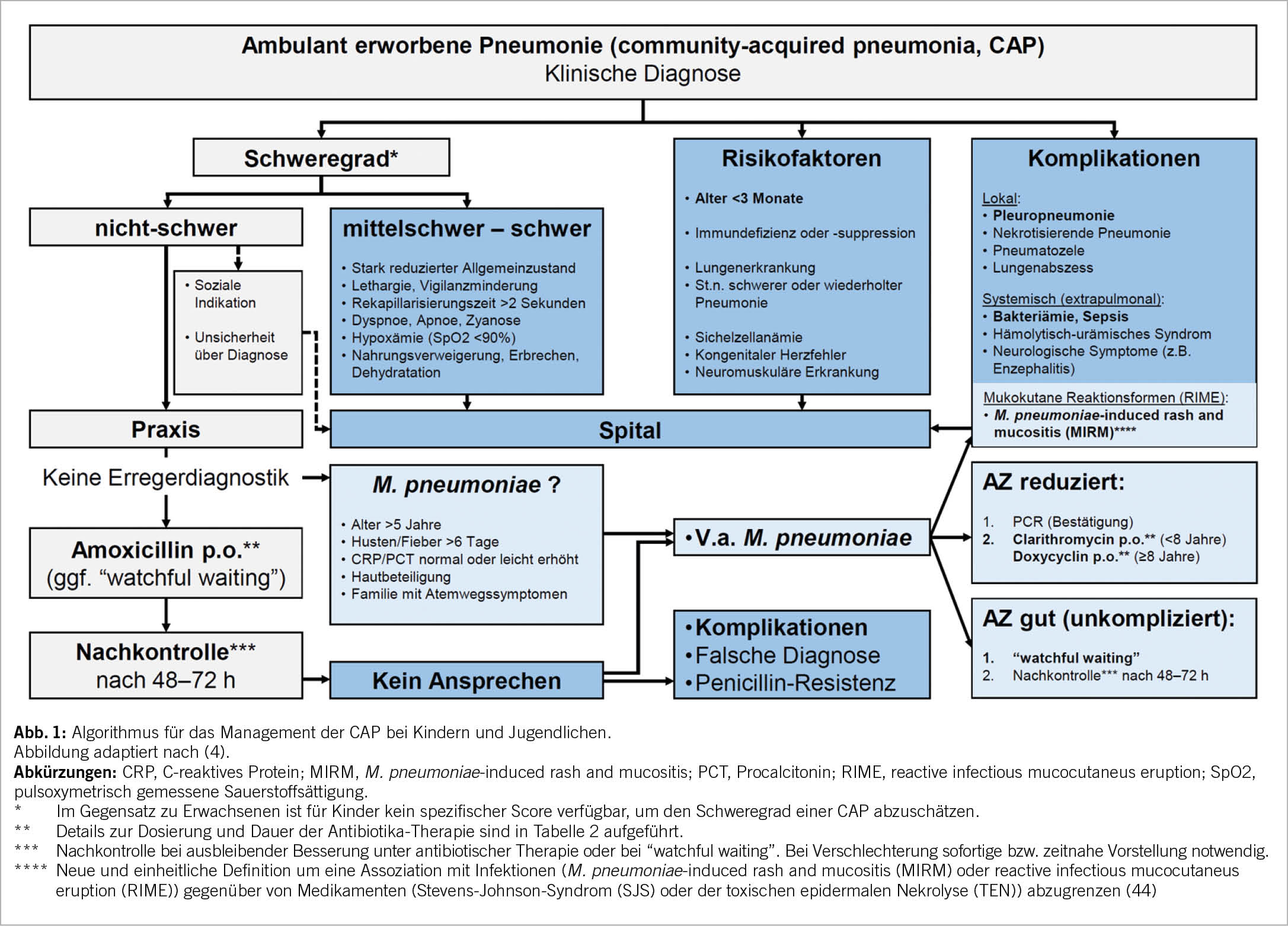
Ein Algorithmus für das Management der CAP bei Kindern und Jugendlichen ist in Abbildung 1 dargestellt. Die CAP ist ein Hauptgrund für die Verschreibung von Antibiotika bei Kindern. Sie werden bei Kindern mit einer CAP häufig «nur für den Fall» verschrieben, aus Angst vor einer raschen klinischen Verschlechterung, einer zukünftigen Hospitalisation oder vor Komplikationen durch eine bakterielle Infektion (26). Tatsächlich nimmt die Verschreibung von Antibiotika mit der diagnostischen Unsicherheit in Praxis und Spital zu (27, 28). Die Verringerung der diagnostischen Unsicherheit durch die Identifizierung derjenigen Kinder und Jugendlichen mit CAP, welche ein sehr geringes Risiko für eine bakterielle Infektion und daraus resultierende Komplikationen haben, könnte die unangemessene Verschreibung und Anwendung von Antibiotika erheblich reduzieren. Deshalb sollte nach der klinischen Diagnose der CAP bei jedem Kind und Jugendlichen eine Risikobeurteilung stattfinden. Eine Zuweisung ins Spital wird bei einer mittelschweren bis schweren CAP, bei Vorliegen von Risikofaktoren oder Hinweisen auf Komplikationen empfohlen. Die meisten Kinder und Jugendlichen mit einer CAP können ambulant betreut werden.
Da die Mehrheit der CAP bei Kindern durch virale Erreger bedingt ist, muss nicht jeder Patient mit einer nicht-schweren CAP ohne Risikofaktoren antibiotisch behandelt werden (26). Im Gegenteil: Es ist durchaus vertretbar, in einer solchen Situation abzuwarten und zu beobachten («watchful waiting»). Voraussetzungen dafür sind die ausführliche Aufklärung der Eltern über Warnzeichen, welche auf eine mittelschwere bis schwere CAP hindeuten und somit zu einer sofortigen bzw. zeitnahen Wiedervorstellung führen müssen («safety netting»), sowie die Gewährleistung einer klinischen Kontrolle nach 48 bis 72 Stunden.
Antibiotika
Eine Übersicht über Antibiotika-Wahl, -Dosierung und -Dauer ist in Tabelle 2 zusammengestellt.
Wahl: Für die Behandlung der CAP bei Kindern und Jugendlichen ist gemäss aktueller Richtlinien immer noch Amoxicillin die erste Wahl (14, 15, 29), da es gegen die meisten bakteriellen Erreger der CAP (v.a. Pneumokokken) wirksam, gut verträglich und preiswert ist. Die Verabreichung sollte immer peroral erfolgen, wenn die orale Medikamenteneinnahme und enterale Resorption gewährleistet ist. Cephalosporine und Makrolide sollen wegen der Resistenzentwicklung von Bakterien generell nicht für die primäre Behandlung der CAP bei Kindern und Jugendlichen eingesetzt werden. Nur bei einer Penicillin-Allergie oder bei Bestätigung einer CAP durch M. pneumoniae (siehe unten) oder Chlamydia pneumoniae können Makrolide oder Tetrazykline (ab 8 Jahren) eingesetzt werden.
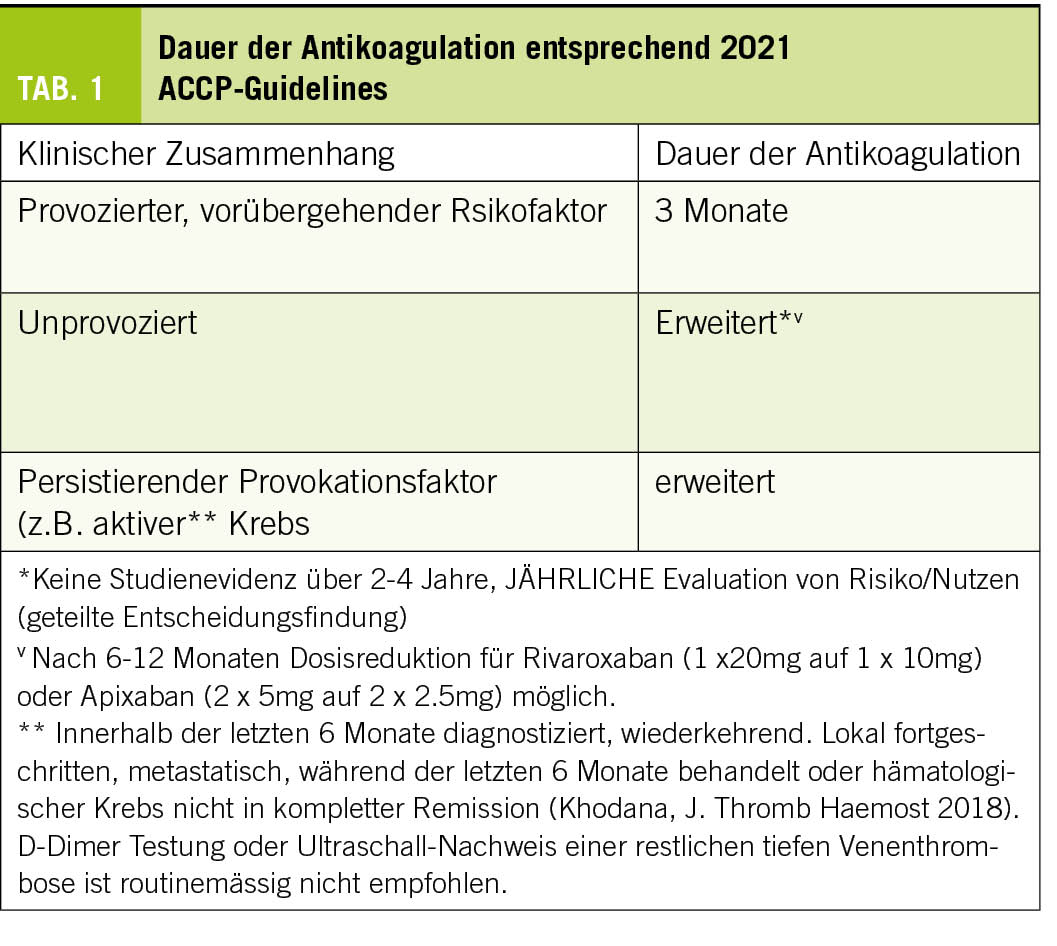
Dosierung und Dauer: Die erschwerte Erregerdiagnose ist auch ein Problem bei Studien, welche den Nutzen einer Antibiotika-Therapie bei der CAP evaluieren. Aufgrund der schwierigen klinischen Unterscheidung zwischen einer bakteriellen und viralen CAP wird der Effekt von Antibiotika oft auch bei vermutlich viraler CAP beurteilt und dadurch möglicherweise bezogen auf eine bakterielle CAP unterschätzt bzw. verzerrt (Pollyanna-Prinzip) (26). Zwei neue randomisiert-kontrollierte Studien haben die Dauer einer oralen Therapie mit Amoxicillin bei Kindern mit CAP analysiert:
Die SAFER-Studie (USA, 2 Zentren, 281 Kinder) konnte bei radiologisch bestätigter CAP die Empfehlungen aktueller Richtlinien bekräftigen, dass die 5- einer 10-tägigen Behandlung nicht unterlegen ist (30). Jedoch wurden auch bei der SAFER-Studie bei ca. zwei Dritteln der Patienten mittels PCR im Nasopharyngealabstrich Viren nachgewiesen (v.a. RSV).
In der CAP-IT-Studie (UK, 29 Zentren, 824 Kinder) konnte sogar gezeigt werden, dass eine 3- bzw. 7-tägige Therapie einen vergleichbaren Behandlungserfolg hat (31). Zudem war für beide Therapiedauern eine niedrige Amoxicillin-Dosis (30–50 mg/kg/Tag) einer hohen Dosierung (70–90 mg/kg/Tag) nicht unterlegen. Jedoch wurde in dieser Studie die CAP ausschliesslich klinisch diagnostiziert (keine Röntgenuntersuchungen und keine mikrobiologische Diagnostik). Da v.a. sehr junge Kinder eingeschlossen wurden, ist es auch bei dieser Studie sehr wahrscheinlich, dass zahlreiche Kinder mit einer viralen CAP eingeschlossen wurden und der Effekt der Antibiotika möglicherweise verzerrt, also gegenüber der Behandlung einer bakteriellen CAP unterschätzt wurde. Die Diagnosestellung in der CAP-IT-Studie entspricht aber den aktuellen Empfehlungen und der Realität in der Praxis. Die Resultate können deshalb in der Praxis auf Kinder mit einer nicht-schweren CAP übertragen werden.
Zusammenfassend wird aktuell eine kürzest mögliche Behandlungsdauer empfohlen, in der Regel für 5 Tage für nicht-schwere CAP (29). Falls das Kind bereits vorher gesund ist, kann wohl eine Dauer von 3 Tagen ausreichen (31).
Nachkontrolle
Im Falle einer klinischen Verschlechterung muss eine sofortige bzw. zeitnahe Wiedervorstellung erfolgen. Eine Nachkontrolle der nicht-schweren CAP nach 48 bis 72 Stunden ist in folgenden Situationen notwendig:
- ausbleibende Besserung bei «watchful waiting»
- fehlendes Ansprechen auf antibiotische Therapie (Amoxicillin)
Gründe für ein fehlendes Ansprechen trotz adäquater Therapie können vielseitig sein:
Falsche Diagnose: Die klinische Diagnose einer CAP muss bei ausbleibender Besserung mit oder ohne antibiotische Therapie re-evaluiert werden. Zu diesem Zeitpunkt kann auch eine erweiterte Diagnostik mittels Thoraxröntgenbild indiziert sein.
Penicillin-Resistenz: Resistenzen von Pneumokokken gegenüber Penicillinen sind in der Schweiz noch eher selten (2%), gegenüber Makroliden jedoch höher (6%) (www.anresis.ch). Am höchsten sind die Resistenzraten von Pneumokokken in der Region Genf (Penicilline, 10%; Makrolide, 8%). Die Antibiotikaresistenzen von Pneumokokken haben nach Einführung von PCV7 bzw. PCV13 abgenommen, weil weniger empfindliche Serotypen in der Impfung eingeschlossen wurden (32).
Komplikationen: Mögliche Komplikationen der CAP bei Kindern und Jugendlichen sind in Abbildung 1 zusammengestellt. In ca. 1% der Fälle kommt es bei der CAP zur Entwicklung einer Pleuropneumonie (33). In den meisten Fällen von Pleuropneumonien konnten wir in Zürich (2001–2015) Pneumokokken in Pleurapunktat und/oder Blutkultur nachweisen (79%), gefolgt von Streptococcus pyogenes (11%) und Staphylococcus aureus (6%) (34). Weitere lokale Komplikationen der CAP sind sehr selten (<1%) und beinhalten eine nekrotisierende Pneumonie und eine Pneumatozele (S. pyogenes, S. aureus), sowie einen Lungenabszess (S. aureus, Anaerobier). Systemische Komplikationen sind die Bakteriämie und Sepsis (Pneumokokken) und sehr selten auch das hämolytisch-urämische Syndrom (Pneumokokken).
Mycoplasma pneumoniae: Kein Ansprechen auf Amoxicillin ist auch ein zuverlässiger diagnostischer Hinweis auf eine Infektion mit M. pneumoniae (35). Zusätzliche klinische und laborchemische Kriterien können bei der Diagnose einer CAP durch M. pneumoniae helfen: Alter >5 Jahre, prolongierte Symptomdauer (>6 Tage), Familie auch mit Symptomen eines Atemweginfektes, sowie CRP- und Procalcitonin-Werte, welche normal oder nur leicht erhöht sind (35). Zudem hinweisend sind extrapulmonale Manifestationen, welche in bis zu einem Drittel der Fälle bei der CAP durch M. pneumoniae auftreten und nahezu jedes Organ betreffen können (v.a. Haut und Nervensystem) (36, 37). Dabei ist eine schwere Form der Hautmanifestation mit vowiegend Beteiligung der Schleimhäute die M. pneumoniae-induced rash and mucositis (MIRM) (37, 38).
Therapieoptionen für die CAP durch M. pneumoniae sind Makrolide oder Tetrazykline (ab 8 Jahren) (Tabelle 2). Es ist jedoch weiterhin unklar, ob diese Antibiotika tatsächlich wirksam sind (39, 40). Trotzdem werden Makrolide weltweit sehr grosszügig eingesetzt, was zu alarmierenden Resistenzraten von M. pneumoniae geführt hat (Asien: >90%, Schweiz: 2–9%) (36, 41). Die CAP durch M. pneumoniae ist in der Regel mild und selbst-limitierend (23, 35). Diese Beobachtung und neue Forschungsergebnisse stützen die Hypothese einer immunvermittelten Pathogenese dieser Infektion, das heisst, die CAP wird nicht durch M. pneumoniae direkt, sondern durch die einsetzende Immunantwort hervorgerufen (möglicherweise durch eine spezifische T-Zell-Immunantwort) (36, 42). Deshalb kann auch bei diesen Patienten mit einer Antibiotika-Therapie zugewartet und das Kind beobachtet werden. Falls eine Antibiotika-Therapie in Betracht gezogen wird, sollte in dieser speziellen Situation zuvor ein Erregernachweis mittels PCR angestrebt werden (Abb. 1). Die Wirksamkeit von Makroliden bei der CAP durch M. pneumoniae bei Kindern und Jugendlichen werden wir in einer grossen, multizentrischen Studie untersuchen, welche durch den Schweizerischen Nationalfonds finanziert ist (www.mythic-study.ch).
Teile dieser Arbeit sind auch in PAEDIATRICA 2023;34(1):32-41 erschienen.
Copyright bei Aerzteverlag medinfo AG
Oberarzt mbF Infektiologie und Spitalhygiene
Forschungsgruppenleiter Infektiologie
Abteilung Infektiologie und Spitalhygiene
Universitäts-Kinderspital Zürich
Steinwiesstrasse 75
8032 Zürich
Der Autor hat keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Jain S, Williams DJ, Arnold SR, Ampofo K, Bramley AM, Reed C, et al. Community-acquired pneumonia requiring hospitalization among U.S. children. N Engl J Med. 2015;372(9):835-45.
2. Zar HJ, Barnett W, Stadler A, Gardner-Lubbe S, Myer L and Nicol MP. Aetiology of childhood pneumonia in a well vaccinated South African birth cohort: a nested case-control study of the Drakenstein Child Health Study. Lancet Respir Med. 2016;4(6):463-72.
3. Pneumonia Etiology Research for Child Health (PERCH) study group. Causes of severe pneumonia requiring hospital admission in children without HIV infection from Africa and Asia: the PERCH multi-country case-control study. Lancet. 2019;394(10200):757-79.
4. Meyer Sauteur PM. Diagnose und Therapie der Pneumonie bei Kindern und Jugendlichen. PAEDIATRICA. 2023;34(1):32-41.
5. Feikin DR, Hammitt LL, Murdoch DR, O’Brien KL and Scott JAG. The enduring challenge of determining pneumonia etiology in children: considerations for future research priorities. Clin Infect Dis. 2017;64(suppl_3):S188-S96.
6. Pratt MTG, Abdalla T, Richmond PC, Moore HC, Snelling TL, Blyth CC, et al. Prevalence of respiratory viruses in community-acquired pneumonia in children: a systematic review and meta-analysis. Lancet Child Adolesc Health. 2022;6(8):555-70.
7. Meyer Sauteur PM, Beeton ML, Uldum SA, Bossuyt N, Vermeulen M, Loens K, et al. Mycoplasma pneumoniae detections before and during the COVID-19 pandemic: results of a global survey, 2017 to 2021. Euro Surveill. 2022;27(19):pii=2100746.
8. Kohns Vasconcelos M, Meyer Sauteur PM, Keitel K, Santoro R, Heininger U, van den Anker J, et al. Strikingly decreased community-acquired pneumonia admissions in children despite open schools and day-care facilities in Switzerland. Pediatr Infect Dis J. 2021;40(4):e171-e2.
9. Ponmani C and Roland D. Fifteen-minute consultation: Does this child have COVID-19 (and does it matter)? Arch Dis Child Educ Pract Ed. 2021;106(5):278-83.
10. Meyer Sauteur PM, Chalker VJ, Berger C, Nir-Paz R, Beeton ML, ESGMAC and the ESGMAC–MyCOVID study group. Mycoplasma pneumoniae beyond the COVID-19 pandemic: where is it? Lancet Microbe. 2022;3(12):e897.
11. von Hammerstein AL, Aebi C, Barbey F, Berger C, Buettcher M, Casaulta C, et al. Interseasonal RSV infections in Switzerland – rapid establishment of a clinician-led national reporting system (RSV EpiCH). Swiss Med Wkly. 2021;151:w30057.
12. ESGMAC MAPS study. Available from: https://www.escmid.org/research-
projects/study-groups/study-groups-g-n/mycoplasma-and-chlamydia/
esgmac-maps-study (accessed June 5, 2023).
13. Meyer Sauteur PM. Challenges and progress toward determining pneumonia etiology. Clin Infect Dis. 2020;71(3):514-6.
14. Bradley JS, Byington CL, Shah SS, Alverson B, Carter ER, Harrison C, et al. The management of community-acquired pneumonia in infants and children older than 3 months of age: clinical practice guidelines by the Pediatric Infectious Diseases Society and the Infectious Diseases Society of America. Clin Infect Dis. 2011;53(7):e25-76.
15. Harris M, Clark J, Coote N, Fletcher P, Harnden A, McKean M, et al. British Thoracic Society guidelines for the management of community acquired pneumonia in children: update 2011. Thorax. 2011;66 Suppl 2:ii1-23.
16. Rhedin S, Elfving K and Berggren A. Novel biomarkers differentiating viral from bacterial infection in febrile children: Future perspectives for management in clinical praxis. Children (Basel). 2021;8(11):1070.
17. Rhedin S, Eklundh A, Ryd-Rinder M, Peltola V, Waris M, Gantelius J, et al. Myxovirus resistance protein A for discriminating between viral and bacterial lower respiratory tract infections in children – The TREND study. Clin Microbiol Infect. 2022;28(9):1251-7.
18. Papan C, Argentiero A, Porwoll M, Hakim U, Farinelli E, Testa I, et al. A host signature based on TRAIL, IP-10, and CRP for reducing antibiotic overuse in children by differentiating bacterial from viral infections: a prospective, multicentre cohort study. Clin Microbiol Infect. 2022;28(5):723-30.
19. Walter JM, Ren Z, Yacoub T, Reyfman PA, Shah RD, Abdala-Valencia H, et al. Multidimensional assessment of the host response in mechanically ventilated patients with suspected pneumonia. Am J Respir Crit Care Med. 2019;199(10):1225-37.
20. Walter JM, Helmin KA, Abdala-Valencia H, Wunderink RG and Singer BD. Multidimensional assessment of alveolar T cells in critically ill patients. JCI Insight. 2018;3(17):e123287.
21. van Oort PM, Brinkman P, Slingers G, Koppen G, Maas A, Roelofs JJ, et al. Exhaled breath metabolomics reveals a pathogen-specific response in a rat pneumonia model for two human pathogenic bacteria: a proof-of-concept study. Am J Physiol Lung Cell Mol Physiol. 2019;316(5):L751-L6.
22. Meyer Sauteur PM, Seiler M, Truck J, Unger WWJ, Paioni P, Relly C, et al. Diagnosis of Mycoplasma pneumoniae pneumonia with measurement of specific antibody-secreting cells. Am J Respir Crit Care Med. 2019;200(8):1066-9.
23. Meyer Sauteur PM, Truck J, van Rossum AMC and Berger C. Circulating antibody-secreting cell response during Mycoplasma pneumoniae childhood pneumonia. J Infect Dis. 2020;222(1):136-47.
24. Seiler M, Geiser A and Berger C. Das Kind mit Fieber: Wie messen? Wie handeln? Wie beraten? PAEDIATRICA. 2019;30(2):5-8.
25. Stuckey-Schrock K, Hayes BL and George CM. Community-acquired pneumonia in children. Am Fam Physician. 2012;86(7):661-7.
26. Meyer Sauteur PM. A limited role for microbiological testing for childhood lower respiratory tract infections in primary care: managing diagnostic uncertainty by withholding antibiotics and watchful waiting. Clin Microbiol Infect. 2022;28(9):1189-92.
27. Hagedoorn NN, Borensztajn DM, Nijman R, Balode A, von Both U, Carrol ED, et al. Variation in antibiotic prescription rates in febrile children presenting to emergency departments across Europe (MOFICHE): A multicentre observational study. PLoS Med. 2020;17(8):e1003208.
28. Pandolfo AM, Horne R, Jani Y, Reader TW, Bidad N, Brealey D, et al. Understanding decisions about antibiotic prescribing in ICU: an application of the Necessity Concerns Framework. BMJ Qual Saf. 2022;31(3):199-210.
29. National institute for Health and Care Excellence (NICE). Pneumonia (community-acquired): antimicrobial prescribing (NICE guideline NG138). 2019; https://www.nice.org.uk/guidance/ng138/resources/pneumonia-communityacquired-antimicrobial-prescribing-pdf-66141726069445.
30. Pernica JM, Harman S, Kam AJ, Carciumaru R, Vanniyasingam T, Crawford T, et al. Short-course antimicrobial therapy for pediatric community-acquired pneumonia: the SAFER randomized clinical trial. JAMA Pediatr. 2021;175(5):475-82.
31. Bielicki JA, Stohr W, Barratt S, Dunn D, Naufal N, Roland D, et al. Effect of amoxicillin dose and treatment duration on the need for antibiotic re-treatment in children with community-acquired pneumonia: the CAP-IT randomized clinical trial. JAMA. 2021;326(17):1713-24.
32. Hauser C, Kronenberg A, Allemann A, Muhlemann K and Hilty M. Serotype/serogroup-specific antibiotic non-susceptibility of invasive and non-invasive Streptococcus pneumoniae, Switzerland, 2004 to 2014. Euro Surveill. 2016;21(21).
33. Dykes JKB, Lawton A, Burchett S and Gupta A. Fifteen-minute consultation: A structured approach to children with parapneumonic effusion and empyema thoracis. Arch Dis Child Educ Pract Ed. 2023;108(2):86-90.
34. Meyer Sauteur PM, Burkhard A, Moehrlen U, Relly C, Kellenberger C, Ruoss K, et al. Pleural tap-guided antimicrobial treatment for pneumonia with parapneumonic effusion or pleural empyema in children: a single-center cohort study. J Clin Med. 2019;8(5):698.
35. Meyer Sauteur PM, Krautter S, Ambroggio L, Seiler M, Paioni P, Relly C, et al. Improved diagnostics help to identify clinical features and biomarkers that predict Mycoplasma pneumoniae community-acquired pneumonia in children. Clin Infect Dis. 2020;71(7):1645-54.
36. Meyer Sauteur PM, Unger WWJ, Nadal D, Berger C, Vink C and van Rossum AMC. Infection with and carriage of Mycoplasma pneumoniae in children. Front Microbiol. 2016;7:329.
37. Meyer Sauteur PM, Theiler M, Buettcher M, Seiler M, Weibel L and Berger C. Frequency and clinical presentation of mucocutaneous disease due to Mycoplasma pneumoniae infection in children with community-acquired pneumonia. JAMA Dermatol. 2020;156(2):144-50.
38. Canavan TN, Mathes EF, Frieden I and Shinkai K. Mycoplasma pneumoniae-induced rash and mucositis as a syndrome distinct from Stevens-Johnson syndrome and erythema multiforme: a systematic review. J Am Acad Dermatol. 2015;72(2):239-45.
39. Gardiner SJ, Gavranich JB and Chang AB. Antibiotics for community-acquired lower respiratory tract infections secondary to Mycoplasma pneumoniae in children. Cochrane Database Syst Rev. 2015;1:CD004875.
40. Biondi E, McCulloh R, Alverson B, Klein A and Dixon A. Treatment of mycoplasma pneumonia: a systematic review. Pediatrics. 2014;133(6):1081-90.
41. Wagner K, Imkamp F, Pires VP and Keller PM. Evaluation of Lightmix Mycoplasma macrolide assay for detection of macrolide-resistant Mycoplasma pneumoniae in pneumonia patients. Clin Microbiol Infect. 2019;25(3):383.e5-e7.
42. Panisova E, Unger WWJ, Berger C and Meyer Sauteur PM. Mycoplasma pneumoniae-specific IFN-gamma-producing CD4(+) effector-memory T cells correlate with pulmonary disease. Am J Respir Cell Mol Biol. 2021;64(1):143-6.
43. Haq IJ, Battersby AC, Eastham K and McKean M. Community acquired pneumonia in children. BMJ. 2017;356:j686.
44. Ramien M and Goldman JL. Pediatric SJS-TEN: Where are we now? F1000Res. 2020;9:F1000 Faculty Rev-982.



 Der Frühjahrskongress der SGAIM stand unter dem Motto «Together for better care».
Der Frühjahrskongress der SGAIM stand unter dem Motto «Together for better care».