La microhématurie est relativement fréquente dans la pratique clinique quotidienne. L’ étiologie sous-jacente est multiple et les conséquences peuvent être aussi bien bénignes que potentiellement mortelles. La confirmation d’ une microhématurie sur la base d’ un sédiment urinaire, une anamnèse ciblée et l’ examen clinique permettent d’ évaluer si un suivi néphrologique et/ou urologique plus approfondi est indiqué.
Microhematuria is relatively common in clinical practice. The underlying etiology is diverse and the consequences can be harmless as well as life-threatening. Confirmation of microhematuria with a urine sediment, a targeted history and clinical examination can be used to evaluate whether further nephrological and/or urological clarification is indicated.
Key Words: urine sediment; glomerular vs. non-glomerular; transient vs. persistent
Outre la recherche ciblée d’ une microhématurie en raison d’ une maladie présumée, il n’ est pas rare qu’ une hématurie microscopique soit diagnostiquée par hasard. La plupart du temps, le diagnostic initial est posé à la suite d’ un test urinaire par bandelette urinaire, qui possède une sensibilité élevée (équivalente à 1-2 érythrocytes/champ visuel) (1), mais une faible spécificité. Les faux négatifs sont donc rares. Cela a été décrit lors de la prise de doses importantes de vitamine C (2). Des faux positifs peuvent notamment survenir en cas d’ urine à pH élevé (>9), de détection de liquide séminal, d’ oxydant (nettoyage du périnée) ainsi qu’ en cas de myoglobinurie (rhabdomyolyse) et d’ hémoglobinurie (hémolyse).
En général, la microhématurie est définie par la présence de trois globules rouges ou plus par champ de vision (microscope à fort grossissement, 400 fois) dans un sédiment urinaire centrifugé.
En revanche, la macrohématurie est visible à l’ œil nu (environ 1 ml de sang/l d’ urine).
Prévalence
La prévalence réelle de la microhématurie est difficile à estimer sachant que les études montrent une grande variation entre 2-31 %. La prévalence dépend fortement de la population choisie, de la durée de l’ étude et de la fréquence des tests (3 ; 4).
Quand un sédiment urinaire est-il utile ?
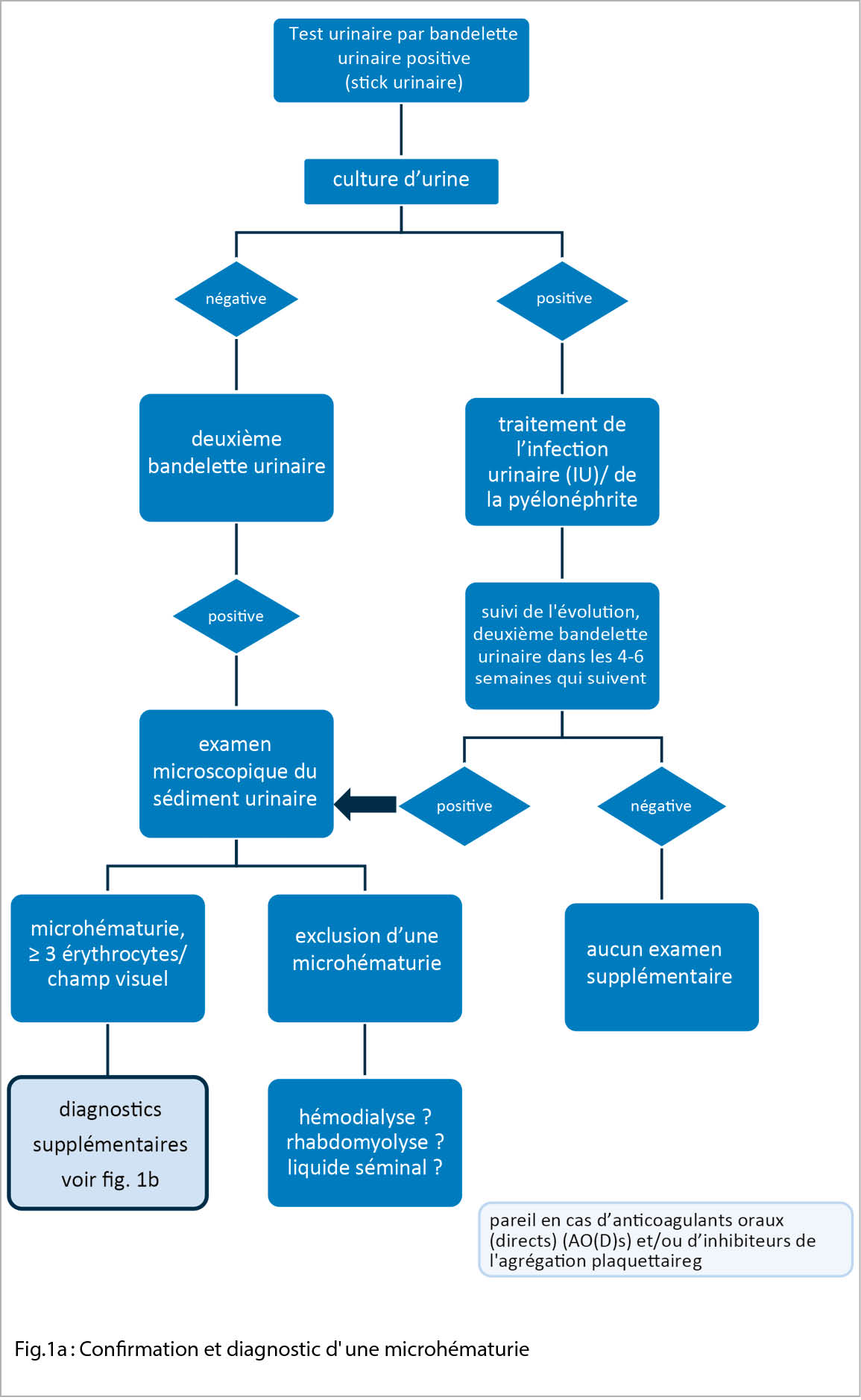
Le « gold standard » pour le diagnostic d’ une microhématurie est l’ examen microscopique du sédiment urinaire. Celui-ci est décisif pour confirmer une microhématurie réelle (fig.1a) et pour différencier entre une microhématurie glomérulaire et une microhématurie non-glomérulaire. En outre, le sédiment urinaire est recommandé chez presque tous les patients présentant une lésion rénale aiguë (IRA), le plus souvent lors de l’ examen d’ une maladie rénale chronique (MRC) ainsi qu’ en cas de protéinurie ou d’ albuminurie douteuse. Les exceptions peuvent être par exemple une infection urinaire/pyélonéphrite symptomatique ou une lithiase rénale ou urinaire confirmée.
Microhématurie glomérulaire versus microhématurie non-glomérulaire
La microhématurie non-glomérulaire se caractérise par la présence d’ érythrocytes isomorphes (forme biconcave uniforme). Elles peuvent être observées en rapport avec toutes les causes d’ hématurie. En cas de microhématurie glomérulaire, les érythrocytes montrent une morphologie modifiée. Il s’ agit d’ érythrocytes dysmorphiques et d’ acanthocytes. Les acanthocytes sont des érythrocytes annulaires avec des protubérances vésiculaires (“oreilles de Mickey”). La déformation est probablement due à des raisons méchaniques lors du passage à travers la membrane basale ainsi qu’ à un “stress” osmotique dans le néphron (5). Des cylindres érythrocytaires peuvent également apparaître.
Dans l’ urine, les acanthocytes, en particulier lorsqu’ ils sont détectés à ≥ 5 %, ont une grande spécificité pour un événement glomérulaire (6). En pratique clinique quotidienne, la mise en évidence d’ acanthocytes, même à un pourcentage inférieur, est suspecte de microhématurie glomérulaire. Le pourcentage d’ érythrocytes dysmorphiques requis dans le sédiment urinaire pour le diagnostic d’ une microhématurie glomérulaire n’ est pas uniforme. La plupart du temps, un pourcentage de > 30 % d’ érythrocytes dysmorphiques est exigé. Par rapport à l’ acanthocyturie, la sensibilité est nettement plus faible.
En cas d’ une microhématurie glomérulaire isolée, la fonction rénale est normale, sans qu’ une protéinurie ou hypertension artérielle soient détectées et sans qu’ une maladie systémique soit suspectée.
Microhématurie transitoire ou persistante
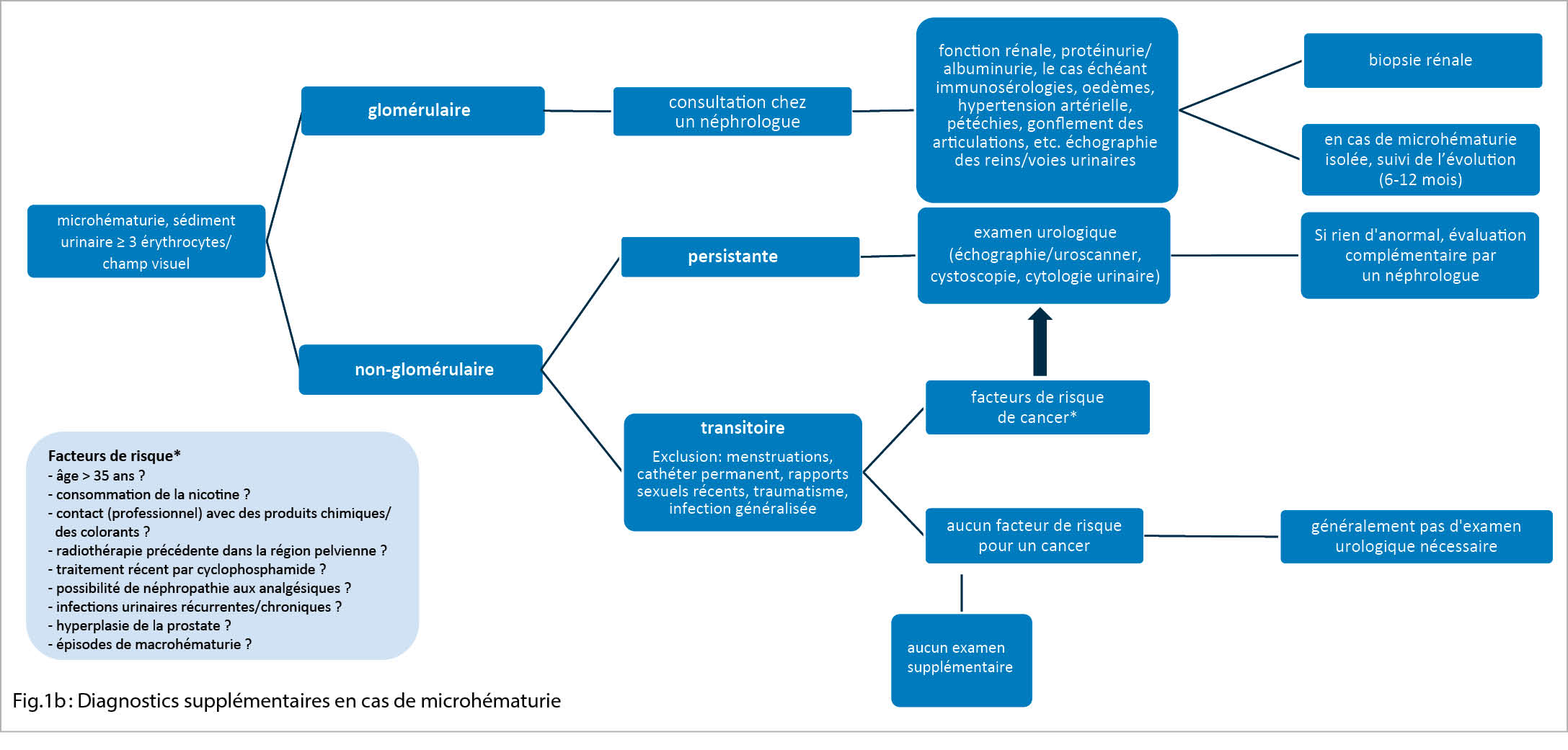
La microhématurie peut être persistante ou transitoire (fig. 1b).
Une microhématurie persistante, asymptomatique et isolée doit être investiguée, car elle est également associée à un risque global plus élevé de maladie rénale nécessitant une dialyse (7). L’ exercice physique (exercise-induced hematuria), les rapports sexuels, les infections urinaires/prostatites, l’ endométriose, les traumatismes, la néphrolithiase ou la fièvre peuvent causer une microhématurie transitoire. Les patients de sexe féminin doivent également être interrogés sur des causes gynécologiques possibles (menstruation, grossesse, atrophie génitale, etc.). En l’ absence d’ autres symptômes/anomalies, il n’ est pas nécessaire de procéder immédiatement à un diagnostic complémentaire. L’ analyse urinaire doit être répétée dans les semaines suivantes afin de déterminer s’ il s’ agit d’ une microhématurie transitoire ou persistante. L’ étiologie de la microhématurie transitoire n’ est parfois pas clairement identifiable.
En présence de facteurs de risque de malignité (fig. 1b), il convient également d’ examiner une microhématurie transitoire.
Diagnostic et démarche d’ investigation
Un dépistage général de la microhématurie n’ est pas recommandé. Si le test de la bandelette urinaire révèle une microhématurie (fig. 1a), après avoir exclu une infection des voies urinaires, l’ examen doit être répété dans un délai de 4 à 6 semaines environ (micro-hématurie transitoire?). Si une microhématurie (asymptomatique) est à nouveau détectée, l’ étape suivante consiste à effectuer un sédiment urinaire et à déterminer la protéinurie (physiologiquement jusqu’ à 150mg/jour) et l’ albuminurie. De plus, une anamnèse ciblée peut aider à différencier les causes :
- Clinique : (nouvelle) hypertension artérielle ? Urine mousseuse (protéinurie) ? Douleurs sur les flancs (néphrolithiase) ?
- Antécédents familiaux : microhématurie ? Surdité, troubles visuels (syndrome d’ Alport) ? Polykystose rénale autosomique dominante (PKRAD) ?
- Facteurs de risque de malignité ?
- Activité sportive récente ? Menstruations ?
Selon la cause présumée ou la présentation clinique / les facteurs de risque, la démarche d’ investigation est différente (fig. 1b). S’ il existe une microhématurie non-glomérulaire sans indices d’ une maladie rénale à la base (fonction rénale limitée, hypertension artérielle, œdèmes, protéinurie), d’ une néphrolithiase ou d’ une infection, un examen urologique est indiqué après exclusion d’ une microhématurie transitoire dont l’ étiologie serait connue. Une tumeur maligne doit être exclue, en particulier en présence de facteurs de risque tels que la consommation de tabac, un âge > 35 ans, des épisodes de macrohématurie, un contact avec des produits chimiques (par ex. amines aromatiques), une radiothérapie dans la région pelvienne ou un traitement par alkylants (cyclophosphamide). En plus d’ une tomodensitométrie, un diagnostic plus approfondi est réalisé à l’ aide de la cytologie urinaire et de la cystoscopie. Si ces examens ne révèlent pas de pathologie, il faut passer à une étape ultérieure et voir un néphrologue.
Si une microhématurie glomérulaire persistante est mise en évidence, il est judicieux de procéder à une consultation plus approfondie par un néphrologue. Si le diagnostic d’ une microhématurie glomérulaire isolée et asymptomatique est posé, il faut penser -surtout chez les jeunes patients – à une néphropathie à IgA (parfois accompagnée d’ épisodes de macrohématurie, surtout en relation avec des infections respiratoires ou gastro-intestinales) ou à une maladie associée au collagène de type IV (8). Dans ce dernier cas, il s’ agit d’ une mutation génétique dans le collagène de type IV, qui entraîne un spectre de néphropathies différentes. Cela va d’ une microhématurie souvent isolée (à l’ époque syndrome de la membrane basale mince) à une néphropathie nécessitant une dialyse dans le cadre d’ un syndrome d’ Alport (accompagné de manifestations extrarénales telles que surdité/troubles de la vision) (9).
En cas de microhématurie glomérulaire isolée, il est recommandé de procéder à des contrôles réguliers de l’ évolution tous les 6 à 12 mois. Il s’ agit notamment de déterminer la protéinurie/l’ albuminurie, la créatinine sérique et de contrôler la pression artérielle. Si les résultats sont stables, le pronostic à long terme est très bon et une biopsie rénale n’ est pas nécessaire (8). Une biopsie rénale est considérée ou bien indiquée si l’ un ou plusieurs de ces paramètres changent (8).
La détérioration rapide de la fonction rénale, en particulier en présence d’ une microhématurie glomérulaire supplémentaire et/ou l’ apparition d’ autres anomalies cliniques (p. ex. une hypertension artérielle nouvellement diagnostiquée) nécessitent une consultation d’ urgence auprès d’ un néphrologue.
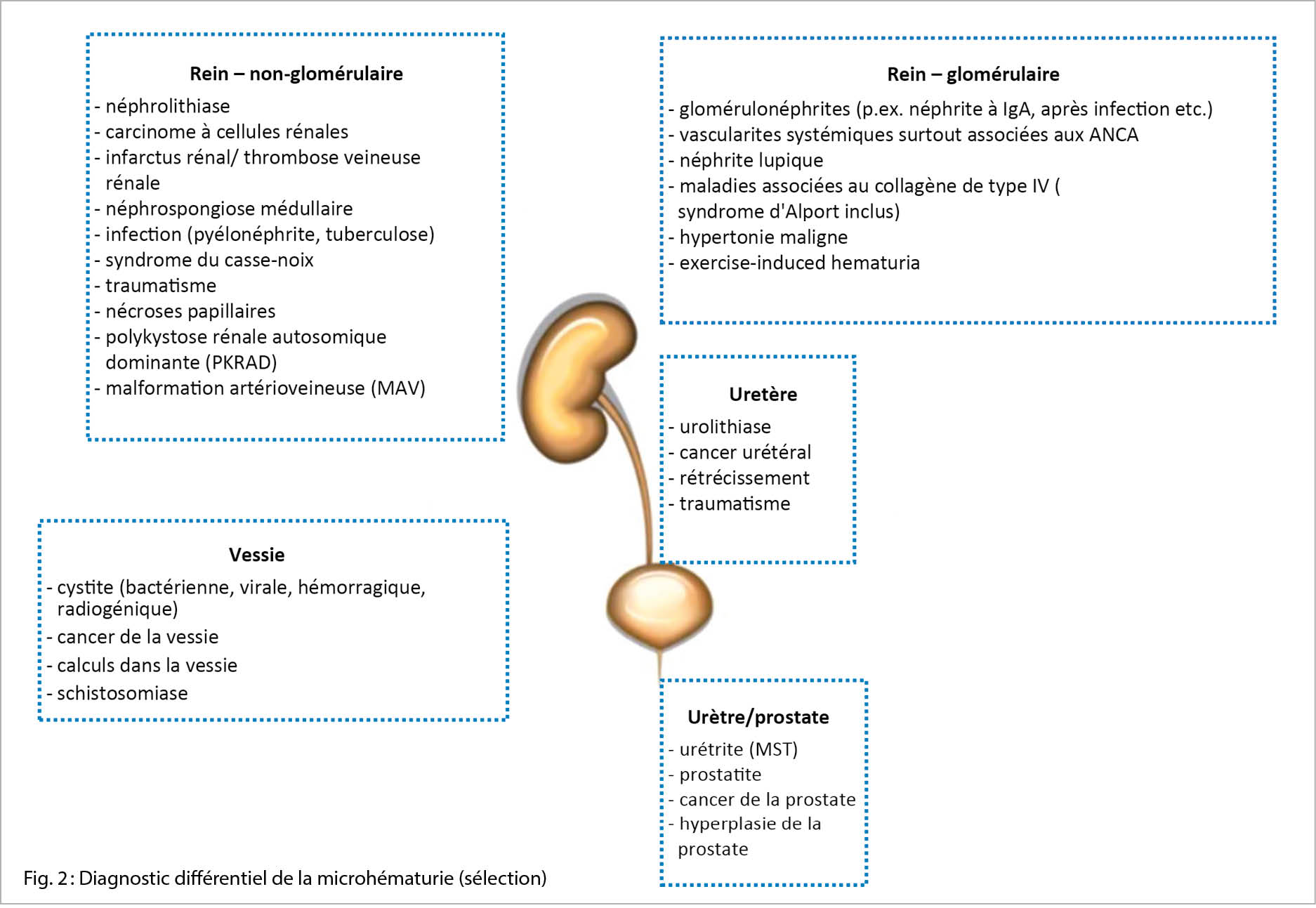
Diagnostics différentiels
Le diagnostic différentiel de la microhématurie est large et va de l’ anodin à des maladies potentiellement mortelles. Les causes les plus fréquentes sont les infections des voies urinaires, la néphro-/urolithiase et – avec l’ âge- les tumeurs malignes de l’ appareil urogénital. En outre, toute forme de glomérulonéphrite peut provoquer une microhématurie. Les diagnostics différentiels de la microhématurie sont énumérés ci-dessous (fig. 2) (liste incomplète, certaines étiologies peuvent se manifester dans différentes localisations).
Cet article est une traduction de « der informierte arzt » 12_2022
Copyright Aerzteverlag medinfo AG
Médecin cadre en néphrologie et médecine interne
Hôpital Zollikerberg
Trichtenhauserstrasse 20
8125 Zollikerberg
simone.rieder@spitalzollikerberg.ch
Médecin-chef en néphrologie
Hôpital Zollikerberg
Trichtenhauserstrasse 20
8125 Zollikerberg
joerg.bleisch@spitalzollikerberg.ch
Les auteurs déclarent aucun conflit d’ intérêt en rapport avec cet article.
1. Cohen RA et al. Clinical practice. Microscopic hematuria. N Engl J Med. 2003;348(23):2330.
2. Brigden ML et al. High incidence of significant urinary ascorbic acid concentrations in a west coast population-implications for routine urinalysis., G Clin Chem. 1992;38(3):426.
3. Hole B et al. Investigating asymptomatic invisible haematuria. BMJ 2014;349:g6768.
4. Davis R et al. Diagnosis, evaluation and follow-up of asymptomatic microhematuria (AMH) in adults: AUA guideline. J Urol 2012; 188: 2473–81.
5. U. Kuhlmann Nephrologie Pathophysiologie-Klinik-Nierenersatzverfahren,
6. Auflage
6. Köhler H et al. Acanthocyturia-a characteristic marker for glomerular bleeding. Kidney Int. 1991;40(1):115.
7. Vivante A et al. Persistent asymptomatic isolated microscopic hematuria in
Israeli adolescents and young adults and risk for end-stage renal disease. JAMA. 2011;306(7):729.
8. CL Hall et al. Clinical value of renal biopsy in patients with asymptomatic
microscopic hematuria with and without low-grade proteinuria. Clin Nephrol. 2004;62(4):267.
9. Kashtan CE et al. Alport syndrome: a unified classification of genetic disorders of collagen IV α345: a position paper of the Alport Syndrome Classification Working Group Kidney Int. 2018;93(5):1045–51.