Das Bundesamt für Gesundheit (BAG) lässt das Krebsregistrierungsgesetz (KRG) extern evaluieren. Die Firma Infras Zürich hat dieses Mandat erhalten und führt die vierjährige Evaluation zwischen April 2020 und Mai 2024 durch. Im Zentrum stehen die Umsetzung des neuen Gesetzes und seine ersten Wirkungen. Die Ergebnisse dienen dem BAG als Grundlage für Optimierungsentscheide in Bezug auf die Umsetzung und auf einen allfälligen Revisionsbedarf des KRG und der KRV.
Im Rahmen der Evaluation sind unter anderem eine Online-Befragung und Interviews mit von krebsbetroffenen Menschen zu Themen wie Information und Widerspruch zur Registrierung der Daten vorgesehen. Bitte informieren Sie betroffene Patientinnen und Patienten über diesen Aufruf zur Online-Befragung um diese Evaluation zu unterstützen. Teilnehmen können alle Personen, die nach dem 1.1.2020 an Krebs oder einer Vorstufe davon erkrankt sind. Auch gesetzliche Vertreterinnen oder gesetzliche Vertreter eines Kindes unter 16 Jahren können an der Befragung teilnehmen.
Mit der Umfrage-Teilnahme können Krebspatientinnen und -patienten einen wertvollen Beitrag leisten, damit die Krebsregistrierung künftig im Sinne der Betroffenen verbessert wird.
Bei Fragen zur Evaluation steht Ihnen die BAG-interne Projektleiterin der Evaluation, Frau Christine Heuer von der Fachstelle Evaluation und Forschung, gerne zur Verfügung:
Tel. 058 462 63 55, E-Mail: christine.heuer@bag.admin.ch
Oncosuisse Netzwerkanlass zu Daten und Register: Kurzrückblick
Wo liegen die aktuellen Herausforderungen im Bereich Daten und Register in der Schweizer Onkologie? Welcher konkrete Handlungsbedarf ergibt sich in den Bereichen Datenerfassung, -Interoperabilität und -Linkage sowie im Datenschutz und der nachhaltigen Finanzierung von onkologischen Datensammlungen? Welche erarbeiteten Handlungsempfehlungen sind zu priorisieren?
Am Oncosuisse Netzwerk-Anlass vom 27.6.2022 in der Welle 7 in Bern wurden diese Fragen im Rahmen von acht separaten Workshops diskutiert und nach Antworten gesucht. Die über 100 Workshop-Teilnehmer:innen stammten primär aus den Bereichen Onkologie, Krebsregister, Datenwissenschaften, Pflege sowie aus nationalen und kantonalen Behörden (Bundesämter für Gesundheit, für Statistik sowie für Umwelt, Staatsekretariat für Wirtschaft, kantonale Gesundheitedirektionen sowie Kantonsärztliche Dienste und weitere). Nebst den Workshops wurden im Rahmen von Projektpräsentationen folgende Projekte vorgestellt:
Swiss Personalized Health Network SPHN, Thomas Geiger/Leiter Management Office
Die Projektpräsentationen sind auf der Oncosuisse Webseite abrufbar: (www.oncosuisse.ch/netzwerkanlass-4) und die Resultate der Workshops sind zur Zeit in der Nachbearbeitung. Ziel ist es, die in den Workshops entwickelten Handlungsempfehlungen im Rahmen des Masterplan 2030 von Oncosuisse zusammenzufassen und als Basis für einen zu schaffenden Schweizer Krebsplan zur Verfügung zu stellen. Wir werden Sie über die Fortschritte via Oncosuisse-Newsletter und hier im Heft informieren. Für Fragen wenden Sie sich an Michael Röthlisberger, Geschäftsführer Oncosuisse (info@oncosuisse.ch).
Des décennies de recherche ont désormais permis de mettre à la disposition des patients atteints d’ un cancer du poumon non à petites cellules présentant la mutation KRAS G12C un traitement ciblé efficace (1, 2). Dans le cadre de l’ étude pivot CodeBreaK 100, le sotorasib (Lumykras®), un inhibiteur hautement sélectif de KRAS G12C, a permis d’ obtenir une réponse rapide et durable chez les patients fortement prétraités (2). Le suivi à long terme de deux ans a également montré une efficacité et une tolérance prometteuses du traitement oral (3).
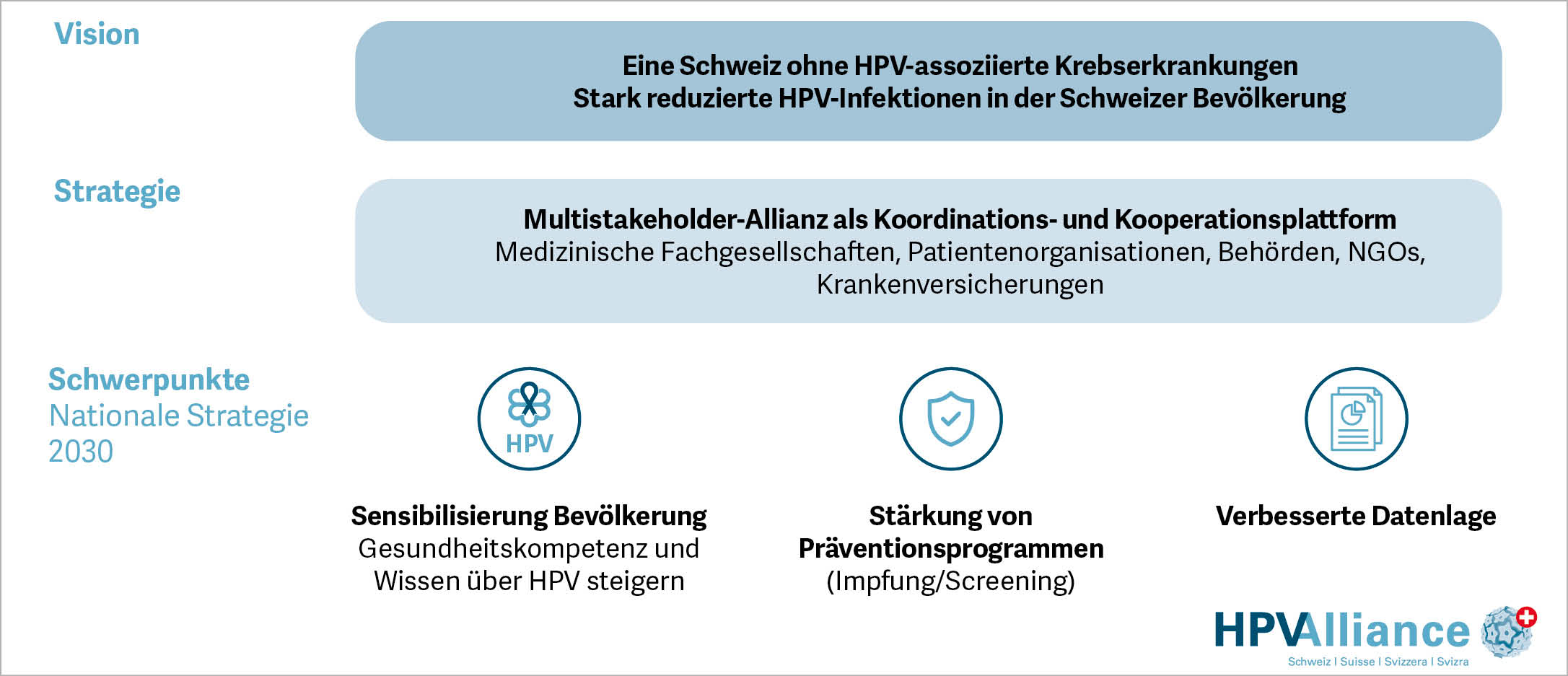
Seit Mitte Juni gibt es den Verein «HPV Alliance Schweiz». Die sechs Gründungsmitglieder wollen die Bevölkerung stärker für das Thema HPV sensibilisieren, Präventionsmassnahmen fördern und weitere Datengrundlagen schaffen. Ihr Vorhaben deckt sich mit dem Ziel der Weltgesundheitsorganisation, die erreichen will, dass niemand mehr an HPV-bedingtem Krebs erkrankt (1).
Gemäss Schätzungen des Bundesamtes für Gesundheit (BAG) infizieren sich in der Schweiz 70–80% der sexuell aktiven Menschen mindestens einmal im Leben mit humanen Papillomaviren (HPV); etwa 5’000 von ihnen erhalten jährlich die Diagnose einer HPV-bedingten Krebsvorstufe. Mit jährlich rund 250 neuen Fällen bei Frauen im Alter zwischen 20 und 49 Jahren ist der Gebärmutterhalskrebs, die häufigste HPV-bedingte Krebserkrankung, die fünfthäufigste Krebserkrankung der Frauen in der Schweiz (2).
Handlungsbedarf für die Schweiz
In der Bevölkerung in der Schweiz ist der Begriff HPV weniger bekannt als in Europa. So wussten in einer Umfrage nur 48% der in der Schweiz Befragten, was HPV ist, im Vergleich zum europäischen Durchschnitt von 63% (3). Generell werden in der Schweiz Präventionsmassnahmen zur Verminderung von HPV-assoziierten Krebserkrankungen zu wenig genutzt:
Es existieren keine einheitlichen Impf- oder populationsbasierten Krebsführerkennungsprogramme.
Die Durchimpfungsraten bei Jugendlichen/jungen Erwachsenen sind mit 59% (Mädchen) und 17% (Jungen) weit unter dem definierten Ziel des Bundesamtes für Gesundheit (BAG) von 80% (4).
Die Umsetzung der Empfehlungen der Schweizerischen Gesellschaft für Gynäkologie und Geburtshilfe (SGGG) zur Vorsorge von Gebärmutterhalskrebs sind unzureichend (5); u.a. infolge fehlender nationaler Richtlinien.
Es fehlt eine übergeordnete und koordinierte Zusammenarbeit auf nationaler Ebene.
«HPV betrifft alle Menschen. Deshalb müssen die Massnahmen und Informationen breiter verankert werden – damit jede Person geschützt und informiert über ihre Sexualität bestimmen kann», sagt Barbara Berger, Präsidentin HPV Alliance Schweiz & Geschäftsleiterin Sexuelle Gesundheit Schweiz. Für ihre künftige Arbeit haben die Mitglieder der HPV Alliance Schweiz folgende Schwerpunkte erarbeitet: Sie wollen langfristig HPV-assoziierte Krebserkrankungen eliminieren, die HPV-Gesundheitskompetenz stärken, evidenzbasierte Präventionsmassnahmen fördern, die Datengrundlage verbessern und eine bisher fehlende nationale Koordination sicherstellen.
1. World Health Organization (WHO), 17. November 2020. Global strategy to
accelerate the elimination of cervical cancer as a public health problem.
Verfügbar unter: https://www.who.int/publications/i/item/9789240014107, zuletzt eingesehen: Juni 2022.
2. Bundesamt für Gesundheit (BAG). Empfehlungen zur Impfung gegen humane Papillomaviren (HPV), Februar 2008. Verfügbar unter:
https://www.bag.admin.ch/bag/de/home/krankheiten/krankheiten-im-ueberblick/hpv.html, zuletzt eingesehen: Juli 2022.
3. Mücke C. Humanes Papillomavirus: Schweiz zeigt im europäischen Vergleich grosse Lücken in Wissen und Bewusstsein. Ars Medici 2019(9):332-335.
4. Bundesamt für Gesundheit BAG, Eidgenössische Kommission für Impffragen (EKIF). HPV-Impfung: ergänzende Impfempfehlung für Jungen und Männer im Alter von 11 bis 26 Jahre. Bulletin, 2015(10): 141-149.
5. Schweizerische Gesellschaft für Gynäkologie und Geburtshilfe (SGGG). Expertenbrief Nr. 50, 1. März 2018. Empfehlungen für die Gebärmutterhalskrebsvorsorge. Verfügbar unter: https://www.sggg.ch/fileadmin/user_upload/Formulardaten/akt_50_D_Gebaermutterhals-krebsvorsorge_01.03.18.pdf
Auf Einladung von Oncosuisse kamen am 8. November 2019 vierzig Expertinnen und Experten aus diversen Bereichen zusammen, um Massnahmen zu entwickeln, die den Zugang zu Krebsmedikamenten sicherstellen sollen. Im Rahmen dieser Oncosuisse Initiative haben in der Folge drei weitere Workshops sowie diverse Sitzungen stattgefunden, in denen die Massnahmen in sieben Projekten konkretisiert wurden. Mit der Unterstützung eines Netzwerkes von rund 100 Fachpersonen werden einige dieser Massnahmen in einer nächsten Phase in die Praxis umgesetzt.
Oncosuisse organisiert die Workshops und Sitzungen und übernimmt die Koordination der Projekte. Für die Entwicklung und Umsetzung der Projekte selbst sind die involvierten Expertinnen und Experten sowie die beteiligten Organisationen zuständig. Das Jahr 2022 steht im Zeichen der Umsetzung dieser verschiedenen Projekte.
Das sind die laufenden Projekte
Massnahme 1 «Erweitertes Antragsrecht»
Massnahme 2 «Wissenstransfer gewährleisten»
Massnahme 3 «Expertengremium für schwierige Fälle»
Massnahme 4 «Kosten & Nutzen»
Massnahme 5 «Internationale Zusammenarbeit»
Massnahme 6 «Empfehlungsliste»
Massnahme 7 «Zugang verkürzen»
Vorstellung der Massnahme 6 «Empfehlungsliste»
Beim Oncosuisse-Workshop am 8. November 2019 hat eine Gruppe von Akteuren des Gesundheitswesens auf das Verfahren im Zusammenhang mit einer bestimmten Art von Vergütungsanträgen als Hindernis für einen raschen Zugang zu Krebsbehandlungen hingewiesen. Diese Anträge beziehen sich auf «Off-Label»-Anwendungen, die fast immer von den Krankenkassen vergütet werden. Dabei geht es um Altmedikamente, die nicht mehr durch ein Patent geschützt sind, für die der therapeutische Nutzen der «Off-Label»-Behandlung jedoch wissenschaftlich belegt ist (Art. 71a KVV). Da das Patent für diese Medikamente abgelaufen ist, erwartet man hier auch keine Indikationserweiterungen mehr, und daher müssen für diese Anwendungen auch weiterhin Vergütungsanträge bei den Krankenversicherern eingereicht werden. Damit verursachen sie nach wie vor einen vermeidbaren Mehraufwand von Onkologinnen und Onkologen, Krankenversicherern und deren Vertrauensärztinnen und -ärzten.
Zielsetzung des Projekts
Diese Massnahme zielt darauf ab, Onkologinnen und Onkologen, Krankenversicherer sowie deren Vertrauensärztinnen und -ärzte von der zusätzlichen Arbeitsbelastung zu entlasten, die durch das Schreiben und Analysieren von Vergütungsanträgen für die oben beschriebenen Anwendungen entsteht. Das ultimative Ziel ist es, Krebspatientinnen und -patienten einen raschen Zugang zu diesen Behandlungen zu ermöglichen.
Projektstatus und Zeitplan 2022
In Absprache mit den Mitgliedern von curafutura und SWICA wurde eine Liste von Medikamenten und Indikationen erstellt und eine gemeinsame Erklärung unterzeichnet. Mehrere Spitäler haben bereits ihr Interesse bekundet an der Pilotphase dieses Projekts teilzunehmen. Der Antragsprozess erfordert keine besonderen Anpassungen der Schnittstellen in den Spitälern. Alle Ärzte oder Institutionen, die an dieser Projektteilnahme interessiert sind, können sich direkt beim Projektleiter Dimitri Kohler, Krebsliga Schweiz, melden (dimitri.kohler@krebsliga.ch).
Alle Details zu den weiteren Massnahmen sind unter folgendem Link zu finden: www.oncosuisse.ch/gesundheitspolitik/oncosuisse-initiative/
Weitere Informationen zu den Massnahmen erhalten Sie vom Koordinator der Oncosuisse Initiative, Dimitri Kohler, d.kohler@oncosuisse.ch
Im Folgenden werden krebspolitisch relevante Entscheide aus der Sommersession 2022 vorgestellt. Zu beiden Themen werden Stellungnahmen von Oncosuisse-Mitgliedern eingereicht.
Änderungen der KVV und KLV: Arzneimittelmassnahmen (2021/74) Geschäftstyp: Geschäft des Bundesrates Stand der Beratung: Botschaft Nächster Schritt: Kommission Erstrat
Mit dieser Revision sollen einerseits Massnahmen zur Kostendämpfung im Bereich der obligatorischen Krankenpflegeversicherung (OKP) umgesetzt werden. Andererseits sind Anpassungen geplant, die der Prozessoptimierung sowie der Erhöhung der Transparenz und der Schaffung von mehr Klarheit und Rechtssicherheit dienen sollen. Gleichzeitig sind Anpassungen im Bereich der Gebühren für die Verwaltungsverfahren vorgesehen. Schliesslich sollen auch die Bestimmungen über die Vergütung im Einzelfall angepasst werden. Die Vernehmlassung läuft bis zum 30. September 2022. Der Bundesrat will die Verordnungsänderungen im ersten Halbjahr 2023 in Kraft setzen.
Zurzeit erarbeiten die Oncosuisse Krebsorganisationen eine gemeinsame Stellungnahme. In kaum einem Fachgebiet werden Therapien so häufig ausserhalb ihrer zugelassenen Indikation eingesetzt wie in der Onkologie: Rund ein Drittel aller erwachsenen Krebsbetroffenen und fast alle Kinder mit Krebs werden im sogenannten Off-Label-Use behandelt. Der Zugang für Patientinnen und Patienten zu diesen mehrheitlich lebensnotwendigen Therapien ist abhängig davon, ob die obligatorische Grundversicherung die Kosten vergütet. Gemäss Art. 71a bis 71d KVV müssen die Krankenkassen entsprechende Kostengutsprachegesuche im Einzelfall beurteilen.
Der Schlussbericht des Bundesamtes für Gesundheit zur Evaluation der Vergütung im Einzelfall zeigt die hohe Bedeutung der Artikel 71a–71d KVV für den raschen Zugang zu lebenswichtigen Medikamenten und bestätigt gleichzeitig, was zahlreiche Fachleute und die Krebsliga seit Jahren bemängeln: Die heutige Situation ist in der Praxis unbefriedigend. Es besteht eine stossende Ungleichbehandlung der Versicherten bei der Kostenübernahme von Off-Label-Medikamenten und der administrative Aufwand für alle beteiligten Akteure ist unverhältnismässig gross.
Mit der rasanten medizinischen Entwicklung und der modernen Präzisionsmedizin werden Off-Label-Behandlungen weiter stark zunehmen, damit ist die Zukunftsfähigkeit der als Ausnahmeregelung konzipierten Verordnungsbestimmungen fraglich. Oncosuisse begrüsst dementsprechend, dass der Handlungsbedarf zur Verbesserung der Einzelfallvergütung auch seitens des Bundes erkannt ist. Die vorgeschlagenen Massnahmen gehen in die richtige Richtung, enthalten leider zu wenige verbindliche Massnahmen, um die heute bestehende Ungleichbehandlung substanziell zu vermindern.
Bessere Betreuung und Behandlung von Menschen am Lebensende (18.3384) Geschäftstyp: Postulat Stand der Beratung: Antrag Abschreibung Urheber/in: SGK-S
Im Postulatsbericht 18.3384 «Bessere Betreuung und Behandlung von Menschen am Lebensende» hat der Bundesrat Handlungsbedarf aufgezeigt. Um die Rahmenbedingungen und Qualitätsstandards der Gesundheitlichen Vorausplanung zu verbessern, haben das BAG, Sektion Nationale Gesundheitspolitik, und die SAMW im Auftrag des Bundesrates eine nationale Arbeitsgruppe «Gesundheitliche Vorausplanung» eingesetzt. Diese hat ein Modell zur Förderung und Umsetzung der Gesundheitlichen Vorausplanung in der Schweiz erarbeitet. Dieses Modell wird nun einer breiten Vernehmlassung unterzogen.
Oncosuisse Mitgliederorganisationen haben hierzu eine Stellungnahme eingereicht. Oncosuisse begrüsst es, dass der Bundesrat beauftragt ist, die Definition von Palliative Care-Leistungen und deren Tarifierung und Vergütung über die ganze Versorgungskette einschliesslich Schnittstellen verbindlich zu regeln. Der Bericht des Bundesrates «Bessere Betreuung und Behandlung von Menschen am Lebensende» zeigt, dass in der Schweiz nicht alle Menschen Zugang zu bedarfsgerechten Palliative Care Angeboten haben und entsprechende Angebote nicht ausreichend in die Gesundheitsversorgung integriert sind. Mit den heutigen Strukturen und Leistungsangeboten wird es künftig nicht möglich sein, die zunehmende Anzahl sterbender Menschen und ihre Angehörigen angemessen zu behandeln und zu betreuen. Weiter ist die Finanzierung von Palliative Care nur ungenügend geklärt. Es bestehen heute massive Finanzierungslücken im Rahmen der Langzeitpflege sowie der Akutbehandlung im stationären und im ambulanten Bereich. Ebenso begrüssenswert ist die Erarbeitung des Modells zur gesundheitlichen Vorausplanung durch die Arbeitsgruppe unter dem Lead vom BAG und der SAMW, die im Rahmen der Umsetzung zum Postulatsbericht eingesetzt wurde.
Pflegerische Beratungen in der onkologischen Rehabilitation sind zentral für eine wirksame Selbstmanagement-Förderung. Wenige dieser Beratungen wurden bisher im Beizug von Theorie und Forschungsergebnissen entwickelt. Dieser Artikel berichtet über die forschungsbasierte Entwicklung einer Pflegeberatung und deren Testung in drei Rehabilitationszentren.
Ziele in der Pflegeberatung
In der Rehabilitation hat das Erreichen von anspruchsvollen Zielen das Potential, die Lebensqualität von Krebsbetroffenen zu verbessern (Levack et al., 2015). Ebenso ist in Selbstmanagement-Theorien das Setzen und Erreichen von persönlichen Zielen ein fester Bestandteil (Lorig & Holman, 2003). Eine kontinuierliche Beratung dient dabei als Methode zur Förderung von Wissen, Motivation und Kompetenzen (Kessler, 2018). Pflegefachpersonen übernehmen in der onkologischen Rehabilitation eine Schlüsselrolle in der Unterstützung von Betroffenen in deren Selbstmanagement (Mayrhofer, Mrak, Kobleder, & Kohler, 2021). Während onkologische Pflegeberatungen als Tätigkeit einer Advanced Practice Nurse im Schweizer Akutsetting bereits forschungsbasiert entwickelt und implementiert sind (Kobleder, Mayer, & Senn, 2020; Serena et al., 2015), befindet sich deren Entwicklung in der onkologischen Rehabilitation noch in ihren Anfängen. Einzelne Rehabilitationszentren bieten bereits onkologische Pflegesprechstunden an, jedoch wurden diese nicht systematisch entwickelt. In diesem Projekt wurde daher ein Gesprächsleitfaden für eine Pflegeberatung in drei Rehabilitationszentren forschungsbasiert entwickelt und dessen Machbarkeit getestet. Während ein Zentrum noch keine pflegerischen Beratungen anbot, war ein Zentrum im Aufbau, und in einem weiteren Zentrum war das Beratungsgespräch bereits ein fester Bestandteil. Wir sehen den Gesprächsleitfaden als ein forschungsbasiertes Grundgerüst, welches mit individuellen Inhalten je nach Rehabilitationszentrum gefüllt werden kann.
Vorgehensweise im Projekt
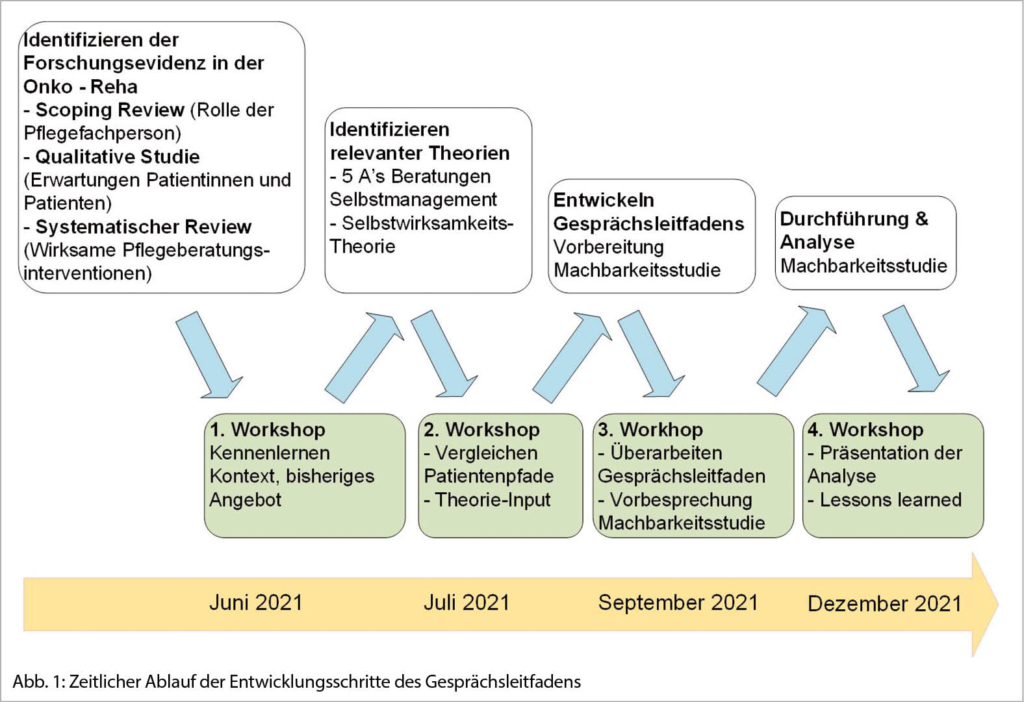
Anhand des Medical Research Council Frameworks (MRC) (Skivington et al., 2021) wurde der Gesprächsleitfaden mit den involvierten Pflegefachpersonen der drei Rehabilitationszentren entwickelt und verfeinert. Die regelmässigen Workshops mit den Rehabilitationszentren (vgl. Abb. 1) garantierten uns, dass Kontextfaktoren in der Erarbeitung des Gesprächsleitfadens berücksichtigt wurden. Eine Pflegeexpertin aus einem Rehabilitationszentrum absolvierte zusätzlich ein Forschungspraktikum im vorliegenden Projekt und konnte somit Erkenntnisse aus der Forschung und Praxis ideal miteinander verbinden. Während des Erarbeitungsprozesses orientierten wir uns am Leitfaden für die Entwicklung und Implementierung einer onkologischen Pflegesprechstunde der Onkologiepflege Schweiz (Sivanathan & Kaufmann-Molnár, 2021).
Aufbau des Gesprächsleitfadens
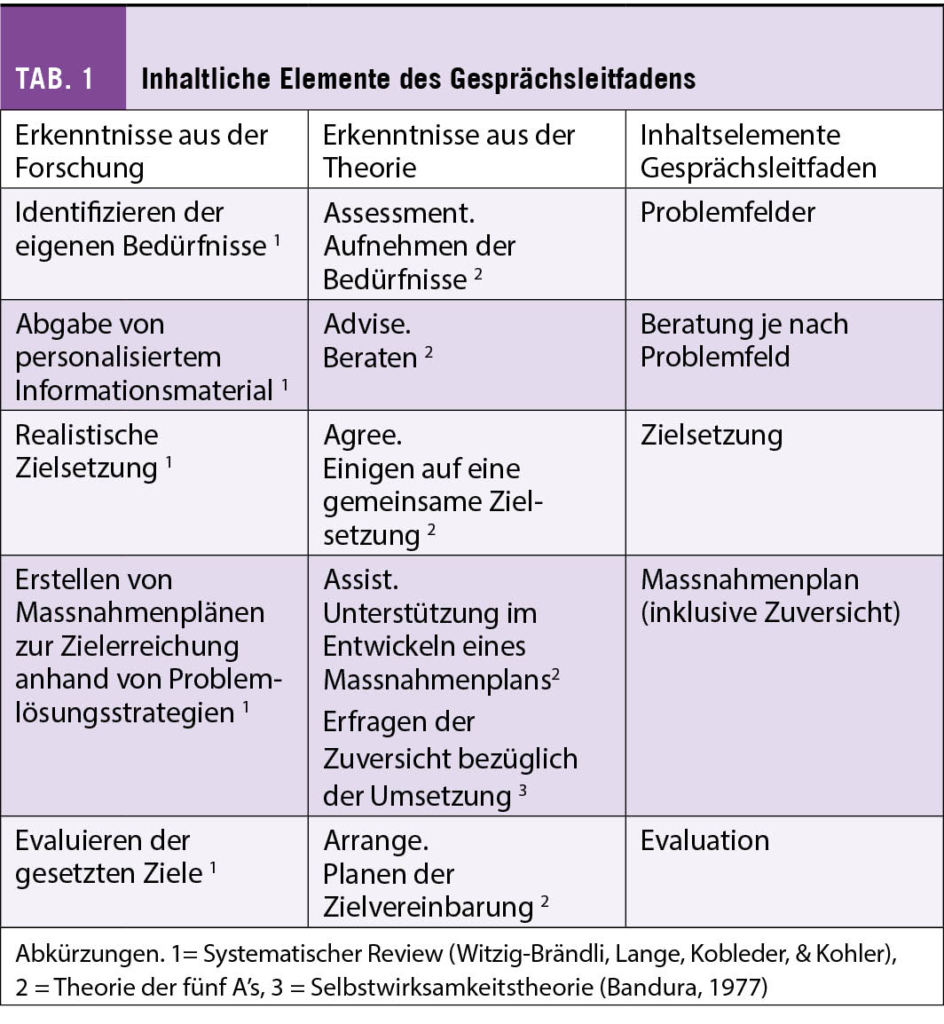
Der Gesprächsleitfaden wurde anhand der Resultate des systematischen Reviews und den dazu gezogenen Theorien erstellt (vgl. Tab. 1). Die Methode der fünf A’s unterstützen Krebsbetroffene in der Entwicklung von Massnahmenpläne und Zielsetzungen mit der Absicht der Selbstmanagement-Förderung (Glasgow, Davis, Funnell, & Beck, 2003). Ebenso empfehlen die «Global Partners on Self-Management in Cancer» diesen Beratungsansatz für eine nachhaltige Verhaltensänderungen im Selbstmanagement (Howell et al., 2020).
Die Problemfelder werden im Gesprächsleitfaden mit dem Selbsteinschätzung-Instrument, dem Distress Thermometer erhoben. Nationale (Sivanathan & Kaufmann-Molnár, 2021), wie auch internationale Richtlinien (NCCN, 2022) empfehlen das Distress Thermometer als Screeninginstrument. Nachdem das ausgefüllte Distress Thermometer mit der krebsbetroffenen Person besprochen ist, findet eine pflegerische Beratung bezogen auf die Problemfelder statt. Gemeinsam mit der krebsbetroffenen Person werden anspruchsvolle Ziele nach der SMART Methode (Doran, 1981) definiert.
Um die gesetzten Ziele zu erreichen, ist die Umsetzung von konkreten Massnahmen im Rehabilitationsalltag von zentraler Bedeutung, Sobald die krebsbetroffene Person zusammen mit der Pflegefachperson eine Massnahme erstellt hat, wird die Zuversicht der Umsetzung der Massnahme im Alltag mit einer numerischen Skala (0 – 10) gemessen. Eine hohe Zuversicht ist ein wichtiger Selbstwirksamkeitsindikator und trägt zur Zielerreichung bei (Bandura, 1977; Lorig & Holman, 2003). Eine Massnahme zur Bekämpfung der Fatigue-Problematik ist ein zweimal täglicher Spaziergang mit einer Dauer von 15 Minuten. Die Zuversicht bezüglich der Umsetzung der Massnahme würde wie folgt erfragt «Wie zuversichtlich sind Sie auf einer Skala von 0 (gar nicht zuversichtlich) bis 10 (absolut zuversichtlich), dass Sie während Ihres Aufenthaltes zweimal täglich während 15 Minuten spazieren werden?» Liegt der Wert unter sieben, soll nach der Literaturempfehlung eine geeignetere Massnahme gesucht werden (Lorig & Holman, 2003).
Resultate der Machbarkeitsstudie
Im Oktober 2021 haben wir den Gesprächsleitfaden bei insgesamt 15 Betroffenen in je zwei Pflegeberatungen über den Zeitraum von vier Wochen getestet. Dabei wurden strukturelle (Art und Anzahl Problemfelder, Ziele, Zuversicht) und Prozessziele (Gesprächsdauer, Vor-Nachbereitungszeit, Zufriedenheit) gemessen.
Krebsbetroffene gaben im Distress Thermometer als Mittelwert eine Belastung von 4.8 (Spannbreite (SP) 1 – 9) an. Die meistgenannten Problemfelder waren Schmerz (n = 10), gefolgt von Erschöpfung, Schlaf, Essen, Ängste und Traurigkeit (je n = 7). Darauf basierend wurden die meisten Ziele im Bereich der Schmerz- und Fatigue-Bekämpfung und der Ernährung (je n = 5) gesetzt. Im Durchschnitt wurden im Erstgespräch pro betroffene Person 1.9 Ziele und 4.3 Massnahmen gesetzt. In einem Zweitgespräch wurde evaluiert, dass 84 Prozent aller gesetzten Ziele erreicht wurden.
Auf der Prozessebene dauerte ein Erstgespräch im Mittel 46 Minuten (Min.) (SP 20 – 60 Min.). Die Nachbereitungszeit des Erstgespräches ergab einen Mittelwert von 14 Min. (SP 5 – 30 Min.). Die Zufriedenheit der Pflegefachpersonen mit der Beratung lag im Durchschnitt bei 81 %, während die der Betroffenen bei 97 % lag.
Erreichte Ziele als Garant für eine gesteigerte Lebensqualität?
Dieses Projekt zeigt auf, wie ein forschungsbasierter Gesprächsleitfaden in Pflegeberatungen zur Zielerreichung führen kann. Die meistgenannten Problemfelder vom Distress Thermometer sind in den Zielsetzungen ersichtlich. Da die Mehrzahl aller Ziele realisiert wurde, sind demnach mit den Betroffenen erreichbare Ziele mit geeigneten Massnahmen gesetzt worden. Die hohen Zufriedenheitswerte von Pflegefachpersonen und Krebsbetroffenen zeigen, dass der Gesprächsleitfaden im Praxisfeld gut akzeptiert wurde.
Inwieweit das Erreichen von anspruchsvollen Zielen in der Pflegeberatung die Lebensqualität von Krebsbetroffenen in der onkologischen Rehabilitation beeinflusst, kann nicht abschliessend beantwortet werden. So wird die Lebensqualität durch viele Kontextfaktoren beeinflusst, die sich nur schwer in einer Forschungssituation kontrollieren lassen. Nichtsdestotrotz half uns die systematische Entwicklung des Gesprächsleitfadens, kontextuelle Faktoren mit der Forschungsevidenz und der Theorie ideal zu verbinden. Der Gesprächsleitfaden dient somit einer Förderung des Selbstmanagements der Krebsbetroffenen, was sich ebenfalls positiv auf die Lebensqualität auswirken kann.
Verena Witzig-Brändli, MScN / wissenschaftliche Mitarbeiterin, OST – Ostschweizer Fachhochschule
Nadja Wyrsch, cand. MScN / Pflegeexpertin, Kliniken Valens, Standort Gais
Nadia Gadmer, dipl Pflegefachfrau HF / Onkologische Beraterin, Zürcher RehaZentren, Klinik Davos
Ursula Blättler, BScN, cand. MAS Onkologiepflege / Expertin Onkologiepflege, Klinik Adelheid
Anne de Graaf, BScN / Pflegeexpertin, Zürcher RehaZentren, Klinik Davos
Andrea Kobleder, Prof. Dr. phil / Co-Leiterin Kompetenzzentrum Onkologie, OST – Ostschweizer Fachhochschule
Myrta Kohler, Prof. Dr. phil / Leiterin Pflegeentwicklung, Leiterin Kompetenzzentrum Rehabilitation und Gesundheitsförderung, Kliniken Valens, OST – Ostschweizer Fachhochschule (Korrespondenzautorin: myrta.kohler@ost.ch)
Erstpublikation des Artikels: Zeitschrift Onkologiepflege 3/2022
Glasgow, R. E., Davis, C. L., Funnell, M. M., & Beck, A. (2003). Implementing practical
interventions to support chronic illness self-management. Joint Commission Journal on
Quality and Safety, 29(11), 563–574. https://doi.org/10.1016/s1549-3741(03)29067-5
Howell,D., Mayer,D.K., Fielding,R., Eicher,M. , Verdonck-de Leeuw, I. M., Johansen,
C., the Global Partners for Self-Management in Cancer (2020). Management of Cancer and Health After the Clinic Visit: A Call to Action for Self-Management in Cancer Care. Journal of the National Cancer Institute, 113(5), 523–531.
https://doi.org/10.1093/jnci/djaa083
Levack, W. M. M., Weatherall, M., Hay-Smith, E. J. C., Dean, S. G., McPherson, K., & Siegert, R. J. (2015). Goal setting and strategies to enhance goal pursuit for adults with acquired disability participating in rehabilitation. The Cochrane Database of
Systematic Reviews, 2015(7), CD009727.
https://doi.org/10.1002/14651858.CD009727.pub2
Mayrhofer, L., Mrak, L., Kobleder, A., & Kohler, M. (2021). Die Rolle von Pflegefach-
personen in der onkologischen Rehabilitation. Pflege, 1–9.
https://doi.org/10.1024/1012-5302/a000810r