Das Mammakarzinom ist die häufigste Krebserkrankung der Frau in der westlichen Welt und jede 10. Frau erkrankt im Laufe ihres Lebens daran. Dabei hat sich die Prognose in den letzten Dekaden deutlich verbessert und liegt heute bei einem 5-Jahres-Überleben von rund 85%. In der Schweiz erkranken pro Jahr rund 6000 Patientinnen am Mammakarzinom, dabei kommt es bei 900 Frauen zu einem Rückfall. Zu den prognostisch schlechten Subgruppen gehört immer noch das Tripel-negative Mammakarzinom (TNBC) mit einem Anteil von rund 15% an der Gesamtpopulation. Hier hat sich die Immuntherapie mit Immun-Checkpoint-Inhibitoren (ICI) in den letzten Jahren als eine Standardtherapie etabliert.
Breast cancer is the most common cancer in women in the western world and one in 10 women will develop the disease in the course of her life. The prognosis has improved significantly in the last decades and is now at a 5-year survival rate of about 85%. In Switzerland, approximately 6000 patients are diagnosed with breast carcinoma each year, with 900 women having a relapse. Among the prognostically poor subgroups is still the triple-negative breast carcinoma (TNBC) with a share of about 15% of the total population. Here, immunotherapy with immune checkpoint inhibitors (ICI) has established itself as a standard therapy in recent years.
Key Words: breast cancer, immunotherapy, checkpoint inhibitors (ICI)
Einführung
ICI sind monoklonale Antikörper gegen stimulierende oder inhibierende Checkpoints auf verschiedenen Immunzellen wie z.B. T-Zellen. Die beiden wichtigsten «Pathways», die in der Klinik bereits seit einigen Jahren «getargeted» werden sind CTLA4 und PDL1/PD-1. Beide Checkpoints wirken inhibitorisch auf die T-Zelle und können durch Antikörper wie Ipilimumab, Nivolumab, Pembrolizumab, Durvalumab, Atezolizumab und Avelumab gehemmt werden. Dadurch kommt es zu einer Aktivierung der T-Zelle und zum Angriff auf Tumorzellen.
Die Anzahl an Mutationen im Tumorgenom korrelieren direkt mit dem Ansprechen auf eine moderne Immuntherapie. Paradebeispiel für sogenannte immunogene Tumore sind beispielsweise das Melanom, des Bronchuskarzinom oder das Nierenzellkarzinom. Bis vor wenigen Jahren ging man davon aus, dass die für eine Immuntherapie wichtige Immunogenität beim Mammakarzinom nicht ausreichend gegeben sei. Der Subtyp des TNBC weist im Vergleich zu den anderen Subtypen aber die höchste Rate an Tumor-infiltrierenden Lymphozyten (TILs), die höchste PD-L1 Expression sowie den höchsten Tumor mutational burden (TMB) auf (1). Seit einigen Jahren kommen deshalb bei dem Subtyp des TNBC ICI immer mehr zum Einsatz. Zuerst im metastasierten Setting, nun auch in der Neoadjuvanz. Mittlerweile sind zahlreiche Untersuchungen im Gange, welche Tumoreigenschaften zu einem deutlichen Ansprechen des Mammakarzinoms unter ICI führt und wie Resistenzmechanismen entstehen. Im Tonic trial wurde erstmalig untersucht welche Kombination von Therapie, einen «kalten» Tumor wie das Mammakarzinom in einen immunogenen «heissen» Tumor überführen kann (2). In diese Phase 2 Studie wurden 67 Patientinnen mit TNBC eingeschlossen. Alle Patientinnen erhielten eine Induktion mit Nivolumab, und anschliessend entweder: Radiotherapie (3x8Gy), Cyclophosphamid, Cisplatin oder Doxorubicin. Alle Patientinnen erhielten anschliessend eine Maintenance Therapie mit Nivolumab. Die Gesamtansprechrate lag bei 20% für alle Patientinnen. Ein erhöhtes Ansprechen fand sich bei Doxorubicin in Kombination mit Nivolumab mit 35%. Im translationalen Teil der Studie konnten die Autoren zeigen, dass Gene der Inflammation und für das JAK-STAT und TNF-α Signaling herauf reguliert wurden unter/nach Doxorubicin-Therapie.
Einsatz beim lokal fortgeschrittenen oder metastasierten TNBC
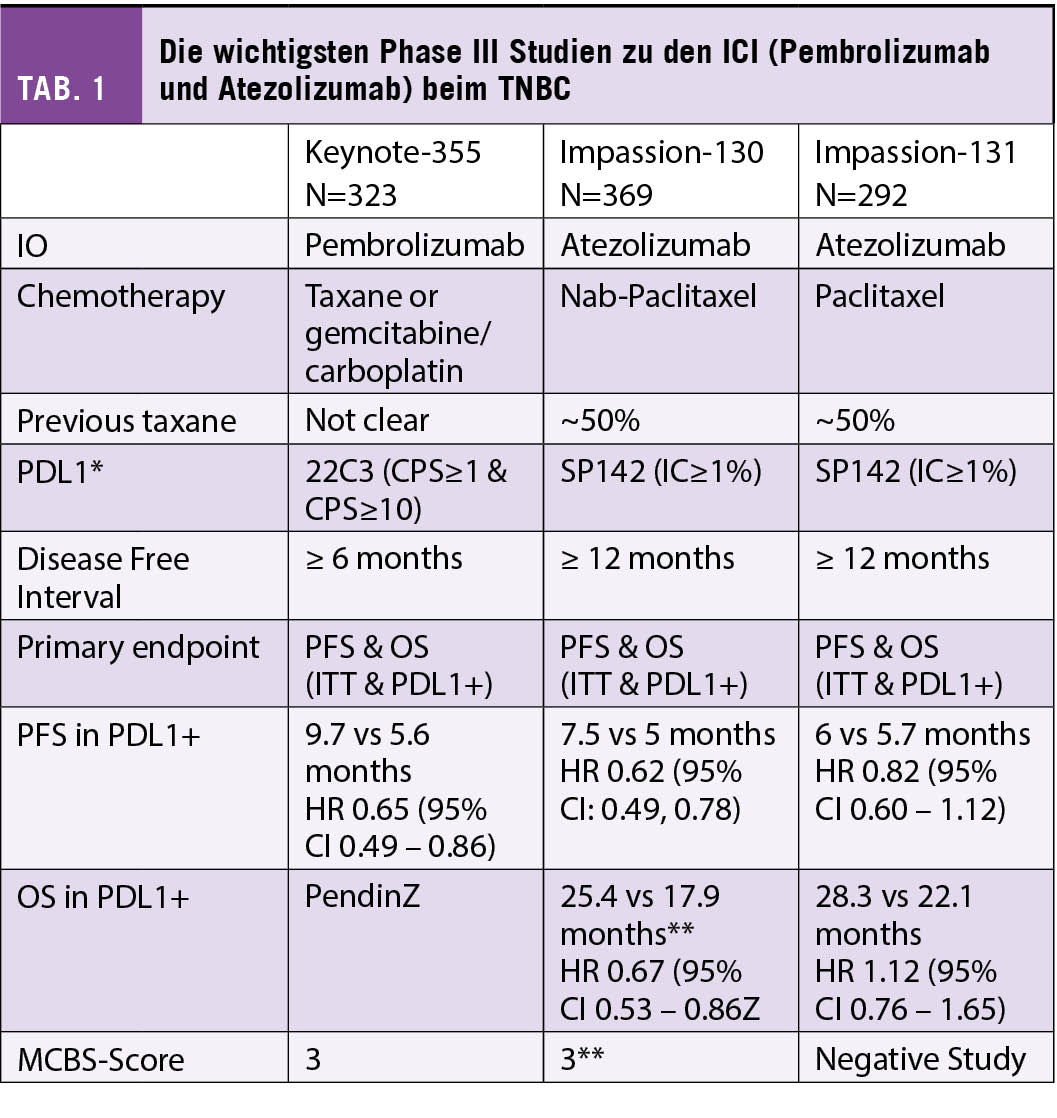
Beim metastasierten TNBC zeigten initiale Studien mit einer alleinigen Immuntherapie tiefe Ansprechraten, wobei einzelne Patientinnen einen länger anhaltenden Nutzen zeigten (3-5). Erstmals zeigte 2018 die IMpassion 130 Studie die Wirkung der Immuntherapie Atezolizumab in Kombination mit NabPaclitaxel (6). In der randomisierten Phase-III-Studie wurden 902 Patientinnen untersucht mit nicht behandeltem lokal fortgeschrittenem oder metastasiertem TNBC. Die Kombination mit Atezolizumab verbesserte das mediane PFS im Vergleich zur alleinigen Chemotherapie signifikant mit 7,2 Monaten versus 5,5 Monate in der ITT-Population (Hazard Ratio [HR] = 0,62; p < 0,001) und 7,5 Monaten versus 5,0 Monate in der PD-L1-positiven Gruppe (HR = 0,62; p < 0,001), während die PDL1-negative Gruppe nicht von der zusätzlichen Checkpoint-Blockade profitierte (5,6 Monate vs. 5,6 Monate, HR = 0,94; p = 0,52). In der PD-L1-positiven Gruppe zeigte sich zudem in der ersten OS-Interimsanalyse eine klinisch relevante Verlängerung um 9,5 Monate (25,0 vs. 15,5 Monate; HR = 0,62; noch nicht formal getestet), während auch hier die PD-L1-negative Gruppe nicht profitierte (18,9 vs. 18,4 Monate; HR = 1,02). In den verschiedenen Biomarkeranalysen war das Vorhandensein von ≥ 1% PD-L1-positiver Immunzellen im Tumor der beste prädiktive Marker für die Wirksamkeit von Atezolizumab. Eine PD-L1-Expression auf den Tumorzellen hatte keinen zusätzlichen prädiktiven Wert. Die stromale TIL-Infiltration (sTIL) und der BRCA1-Status hatten keine unabhängige prädiktive Aussagekraft.
Da die Verfügbarkeit von NabPaclitaxel nicht überall gegeben war, wurde dieselbe Immuntherapie ein Jahr später in Kombination mit Paclitaxel in der IMpassion 131 Studie untersucht (7). Diese Studie hat zu aller Erstaunen keine Verbesserung des PFS durch die Hinzunahme von Atezolizumab in der ITT-Population (5.7 Monte versus 5.6 Monate; HR = 0.86; p = 0.86) ebenso wenig wie in der PD-L1 positiven Kohorte (6.0 Monate versus 5.6 Monate; HR = 0.86; p = 0.20) gezeigt.
Die Wirksamkeit des ICIs Pembrolizumab beim TNBC konnte in der KEYNOTE 355 Studie bestätigt werden (8). Diese randomisierte Phase III Studie untersuchte bei Patientinnen mit einem inoperablen oder metastasierten TNBC die Kombination Pembrolizumab und Chemotherapie (Gemcitabine mit Carboplatin oder Taxan mono). Die finalen OS-Daten nach 44.1 Monaten wurden am letztjährigen ESMO-Kongress präsentiert. Dabei zeigte sich bei der Subgruppe mit einem CPS ≥ 10 eine signifikante Verlängerung des OS von 16.1 Monate auf 23.0 Monate (HR = 0.73, p = 0.0093), ein PFS-Benefit von 5.6 Monate auf 9.7 Monate (HR 0.66) und eine um 12% höhere Ansprechrate.
Einsatz in der neo-adjuvanten Behandlung des TNBC
Zwischenzeitlich kommen die ICI auch zunehmend zum Einsatz in der neoadjuvanten Therapie des TNBC und stehen auch in der Schweiz kurz vor der Zulassung, wobei die Resultate der Studien z.T. unterschiedlich sind.
In der KEYNOTE 522 Studie wurden Patientinnen mit einem TNBC im Stadium II und III (T1c, N1-2 oder T2-4, N0-2) randomisiert und neo-adjuvant mit 4 Zyklen Carboplatin AUC 5 in Kombination mit wöchentlich Taxol gefolgt von 4 Zyklen Doxorubicin/Epirubicin in Kombination mit Cyclophosphamid behandelt +/- der Immuntherapie Pembrolizumab (9). Postoperativ wurde Pembrolizumab/Placebo für weitere 9 Zyklen fortgeführt. Dabei zeigte sich nach 39.1 Monaten Follow-up ein signifikant besseres Event-free-survival (EFS) von 84.5% gegenüber 76.8% mit der zusätzlichen Immun-Checkpoint-Blockade sowie eine Reduktion des Risikos für Tod um 37% (HR = 0.63). Die OS-Daten sind aktuell noch nicht reif. Bezüglich der pathologischen Komplettremission (pCR) zeigte sich ein Nutzen von 13.6% (51.2% versus 64.8%) unabhängig vom PD-L1 Status. Dass die pCR sich in das Gesamtüberleben übertragen lässt, wissen wir bereits aus früheren Studien. (10). In der KEYNOTE 522 Studie ist nun auch sehr schön zu sehen, dass die pathologische Komplettremission sowohl in der Patientengruppe mit ICI als auch in der Placebo-Gruppe, mit einem deutlich besseren EFS nach 3 Jahren assoziiert ist (Abb. 1).
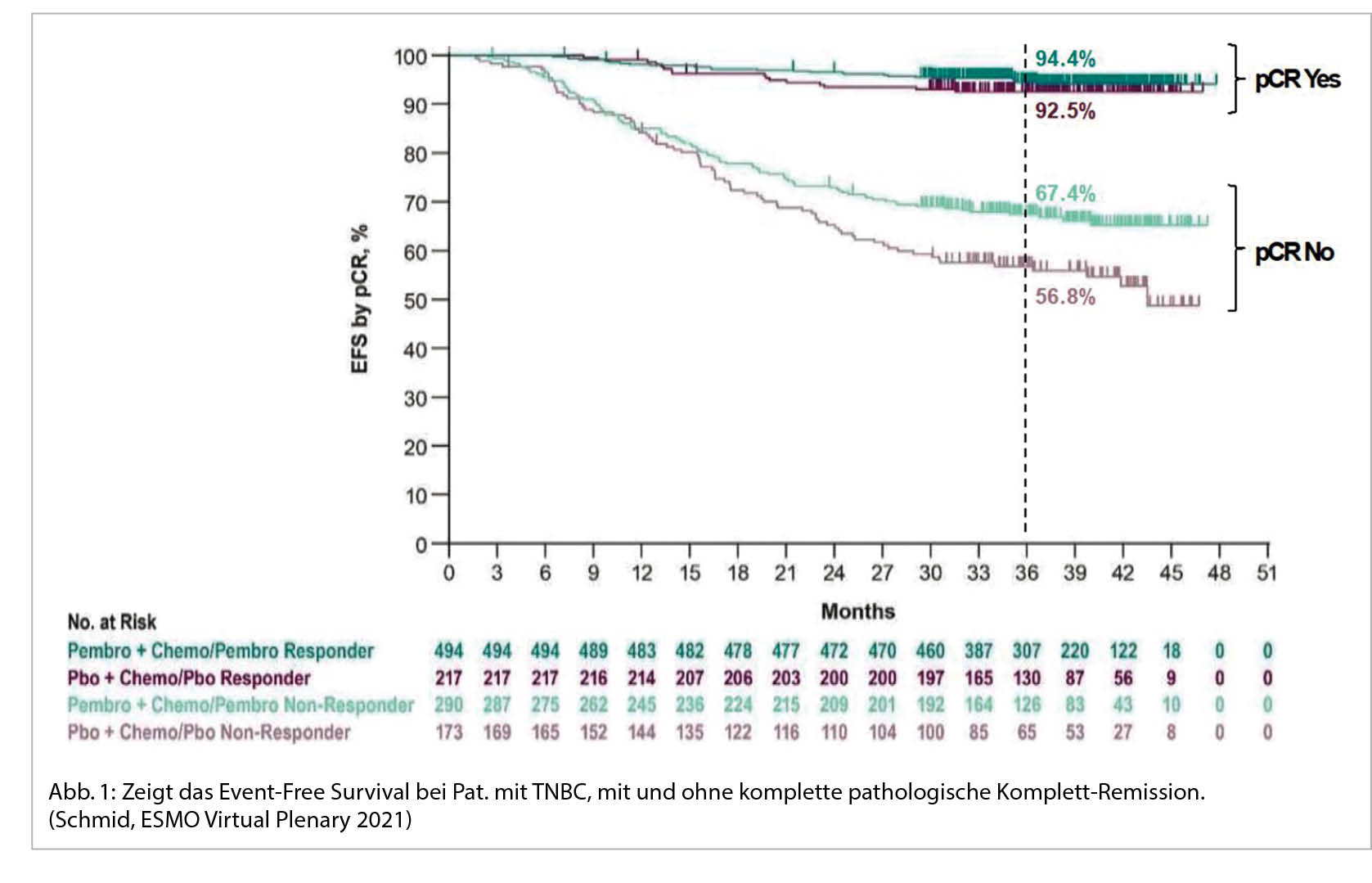
Die Analyse des Gesamtüberlebens ist noch nicht final, allerdings ist Pembrolizumab in einigen Ländern bereits in dieser Indikation zugelassen, z.B. USA (FDA).
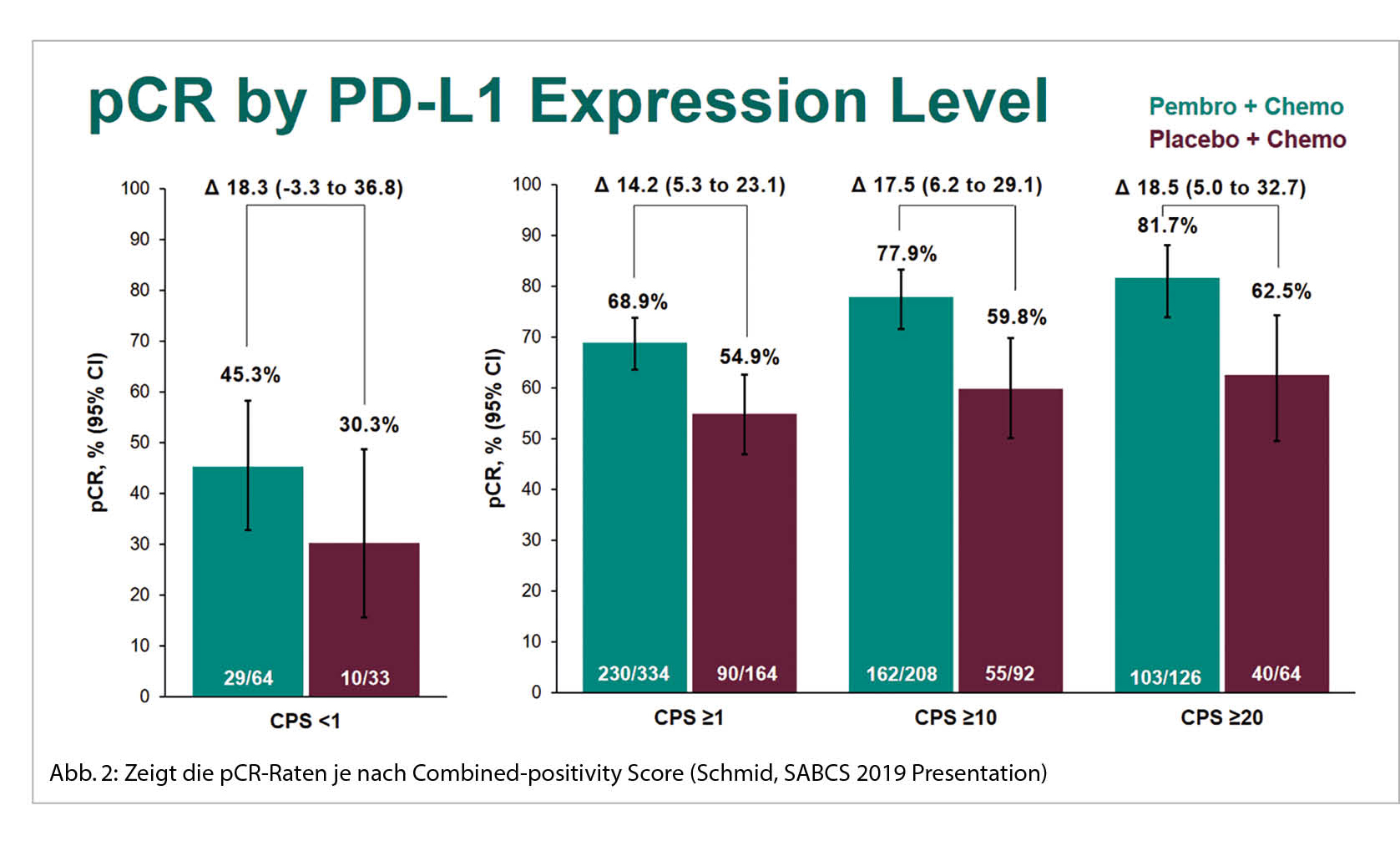
Die pCR bei der PD-L1 positiven Subgruppe war mit der zusätzlichen ICI mit 68.9% höher als mit Placebo 54.9%. Aber auch in der PD-L1 negativen Gruppe war ein Anstieg der pCR-Rate von 30.3% auf 45.3% zu sehen. Somit ist der Nutzen der ICI unabhängig vom PD-L1 Status. Es zeigt sich hier zudem, dass der PD-L1 Status prognostisch ist für den Outcome beim TNBC (Abb. 2).
Auch die IMpassion 031 Studie konnte 2020 ähnliche Resultate zeigen (11). Hier wurde Atezolizumab zur neoadjuvanten Chemotherapie dazugegeben, wobei im Vergleich zur vorgängigen Studie wöchentliches NabPaclitaxel anstelle von Carboplatin und Paclitaxel verwendet wurde. Auch hier wurden Patientinnen mit TNBC im Stadium II und III eingeschlossen. Nach einem Follow-Up von 20.6 Monaten zeigte sich ebenfalls ein Anstieg der pCR-Rate um 17%, von 41% mit Placebo auf 58 % mit Atezolizumab (p = 0.0044).
Die GeparNuevo Studie bestätigt 2019 den Nutzen der neoadjuvanten ICI beim TNBC (12). In dieser Phase II Studie wurden ebenfalls Patientinnen mit einem TNBC im Stadium II und III (cT1b-cT4a-d) eingeschlossen. Hier wurde die Immuntherapie Duvalumab kombiniert mit wöchentlichem NabPaclitaxel gefolgt von Epirubicin/Doxorubicin und Cyclophosphamid. Durvalumab wurde 2 Wochen vor Start der Chemotherapie begonnen. Die pCR-Rate mit Durvalumab war 53.4% im Vergleich zu 44.2% unter Placebo (p = 0.048). Vor Therapiestart wurden die sTILs, der PD-L1-Status sowie Ki67 bestimmt. sTILs und das Grading waren unabhängige Prädiktoren für das Erreichen einer pCR. Bei PD-L1 negativen Tumoren zeigte sich ein Anstieg der pCR Rate von 30.0% auf 54.3% (p = 0.061) und bei den PD-L1 positiven Tumoren von 44.4% auf 58.0% (p = 0.445). Es gab einen Trend für eine erhöhte Response-Rate bei der PD-L1 positiven Gruppe.
Im Februar 2022 wurde zudem die NeoTRIP Studie veröffentlich (13). Eine Phase III Studie, welche 3-wöchentliches Atezolizumab in Kombination mit Carboplatin AUC 2 und NabPaclitaxel (Tag 1 und 8) für 8 Zyklen neoadjuvant gefolgt von der Operation sowie adjuvant 4 Zyklen einer Anthrazyklin-haltigen Chemotherapie untersucht hat. Erstaunlicherweise zeigte sich hier zwar eine numerisch höhere Rate an pCR (44.4% versus 48.6%) mit der zusätzlichen ICI in allen Subgruppen, wobei dies aber keine statistische Signifikanz erreichte. Die Ursache für dieses nicht signifikante Resultat ist nicht abschliessend geklärt. Eine mögliche Erklärung sind die unterschiedlichen Chemotherapieregime und Chemotherapiesequenzen. Zudem wird auch der unterschiedliche Steroidbedarf unter der Chemotherapie diskutiert. Auch ist ein eventueller Wirksamkeitsunterschied zwischen dem PD1-Inhibitoren Pembrolizumab und den PD-L1-Inhibitoren Atezolizumab und Durvalumab eine mögliche Erklärung. Was die unterschiedlichen Resultate beim metastasierten TNBC sowie in der Neoadjuvanz sicherlich aussagen, ist die Notwendigkeit weiterer Untersuchungen, um die Patientengruppe, die von einer zusätzlichen Checkpoint-Blockade profitiert, besser bestimmen zu können.
Zusammenfassung und Diskussion
Im Bereich des TNBC ist die Immuntherapie mittlerweile vor allem im metastasierten Setting eine Standardtherapie, welche die Prognose deutlich verbessert hat. In der neo-adjuvanten Therapielinie ist die pCR Rate durch Atezolizumab und Pembrolizumab deutlich erhöht. Die Gesamtüberlebensrate war in der GeparNuevo Studie (Phase II) signifikant verbessert. Ob nach einem Erreichen einer pCR eine weitere Therapie mit ICI erforderlich ist, bleibt momentan Gegenstand der Forschung. Inwieweit die Wahl des Chemotherapie-Backbones, z.B. Nab-Paclitaxel, Paclitaxel, Carboplatin oder Gemcitabine, eine Rolle spielt bleibt noch zu beurteilen. Bezüglich der anderen Subtypen (Luminal und HER2-angereichert) bleiben grössere Studien abzuwarten. Single-Agent Studien mit Pembrolizumab oder Atezolizumab waren vom Ansprechverhalten eher enttäuschend. Auch hier wäre das Ziel das «Tumormikroenvironment» entsprechend zu einem inflammatorischen Zustand umzuwandeln, um ein Ansprechen auf ICI zu bewirken.
Copyright bei Aerzteverlag medinfo AG
Abteilung für Onkologie und Hämatologie
Kantonsspital Baselland
Rheinstrasse 26
4410 Liestal
angela.kohler@ksbl.ch
Zentrum Onkologie und Hämatologie
Tumorzentrum Baselland
Kantonsspital Baselland
Rheinstrasse 26
4410 Liestal
Schweiz
marcus.vetter@ksbl.ch
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Beitrag deklariert.
1. Tutt A et al. Immunotherapy and targeted therapy combinations in metastastic breast cancer. Lancet Oncol 2019;20:e175-86.
2. Voorwerk L et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med 2019 Jun;25(6):920-928.
3. Emens LA et al. Long-term clinical outcomes and biomarker analyses of Atezolizumab therapy for patients with metastatic triple-negative breast cancer: a phase 1 study. JAMA Oncol 2019;5:74-82
4. Dirix LY et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1B javelin solid tumor study. Breast Cancer Res Treat 2018;167:671-86
5. Nanda R et al. Pembrolizumab in patients with advanced triple-negative breast cancer: phase Ib KEYNOTE-012 study. J Clin Oncol 2016;34:2460-7
6. Schmid P et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 2018 Nov 29; 379(22): 2108–2121
7. Miles D et al. Primary results from Impassion131, a double-blind, placebo-controlled, randomized phase III trial of firstline paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann Oncol 2021 Aug;32(8):994-1004.
8. Cortes J, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020;396(10265):1817-1828.
9. Schmid P, et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N Engl
J Med 2020 Feb 27;382(9):810-821.
10. Spring LM, et al. Pathologic Complete Response after Neoadjuvant Chemotherapy and Impact on Breast Cancer Recurrence and Survival: A Comprehensive
Meta-analysis. Clin Cancer Res 2020 Jun 15;26(12):2838-2848.
11. Mittendorf E., et al. Neoadjuvant atezolizumab in combination with sequential nab-
paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy
in patients with early-stage triple-negative breast cancer (Impassion031): a randomized, double-blind phase 3 trial. Lancet 2020 Oct 10;396(10257):1090-1100.
12. Loibl S., et al. A randomized phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: clinical results and biomarker analysis of GeparNuevo study. Ann Oncol 2019;30(8);1279-1288.
13. Gianni L, et al. Pathologic complete response (pCR) to neoadjuvant treatment with or without atezolizumab in triple-negative, early high-risk and locally advanced breast cancer: NeoTRIP Michelangelo randomized study. Ann Oncol 2022 May;33(5):534-543.