In diesem Heft berichtet ein Pharmakologe über seine Erfahrungen zum Thema Cannabis, welches in aller Munde ist. Deshalb sollen zwei Punkte – aus politischer und medizinischer Sicht – gestreift werden.
Der Autor schreibt: «von Nutzen sein können»: wichtig ist in der Medizin auch, was allenfalls schaden könnte (primum nil nocere). Studien zeigen, dass die Legalisierung des Cannabisverkaufs zu mehr Strassentoten führt: in den USA ergab sich eine Zunahme der Strassentodesfälle in Colorado um 15% (1), und in Kanada hat sich die Prävalenz von mittelschwer verletzten Fahrern mit THC nach der Legalisierung von Cannabis mehr als verdoppelt (2). Haben die Politiker diese Studien gelesen? Hinzu kommt, dass Cannabis – wie der Autor im Artikel schreibt – verschiedene Wirkungen hat und insbesondere im Jugendalter – was der Autor nicht erwähnt – die Hirnentwicklung stört und deshalb bei Jugendlichen kontraindiziert ist (3). Eine neueste Studie legt sogar nahe, dass langjähriger Cannabiskonsum die kognitiven Leistungen von Erwachsenen schmälert (und das Hippocampusvolumen vermindert), unabhängig von Nikotin und Alkohol (4).
Zur Wirkung bei Schmerz zitiert der Autor eine Metaanalyse aus 2015. Eine neulich in der Zeitschrift Pain veröffentlichte Metaanalyse kommt hingegen zum Schluss, dass von 36 Studien (7217 Patienten; Cannabinoide (8 Studien), Cannabis (6 Studien) und CBM (22 Studien) lediglich zwei den Endpunkt (30%-ige Reduktion der Schmerzintensität) erreichten und 81% der Subgruppenanalysen negativ waren (5). Diese Daten entsprechen der klinischen Erfahrung.
Was bleibt: Cannabis ist wirksam bei Spastik und speziellen Formen der Epilepsie, jedoch wenig wirksam bei chronischen Schmerzen (Einzelfälle sind ausgenommen). Hingegen entfaltet Cannabis verschiedene nicht nur erwünschte Wirkungen im zentralen Nervensystem. Medizinisch sollten wir auf dem Boden der Wissenschaft bleiben. Die heutige politische Debatte erinnert mich an den vor 20 Jahren von den Patientenorganisationen geforderten liberaleren Umgang mit Opioiden, der heute in teuren Prozessen gipfelt. Machen wir dieselben Fehler heute mit Cannabis?
P.S. In diesem Zusammenhang sei auf den Pilotversuch des BAG im Kanton Basel hingewiesen, wo geplant ist, Cannabis nicht reguliert abzugeben. Soll Cannabis inskünftig wie Aspirin verfügbar sein?
KD Dr. med. lic. phil. Marcel Weber
marwebdr@gmail.com
1. Santaella-Tenorio J et al. Association of Recreational Cannabis Laws in Colorado and Washington State With Changes in Traffic Fatalities, 2005-2017. JAMA Intern Med 2020;180(8):1061-1068.
2. Brubacher JR et al. Cannabis Legalization and Detection of Tetrahydrocannabinol in Injured Drivers. New Engl J Med 2022 386:148-156.
3. Albaugh MD et al. Association of Cannabis Use During Adolescence With Neurodevelopment. JAMA Psychiatry 2021;78(9):1-11.
4. Meier MH et al. Long-Term Cannabis Use and Cognitive Reserves and
Hippocampal Volume in Midlife. Am J Psychiatr 2022; doi.org/10.1176/appi.ajp.2021.21060664
5. Fisher E. et al. Cannabinoids, cannabis, and cannabis-based medicine for pain management: a systematic review of randomised controlled trials. Pain 2021;162(Suppl.1):S45-S66.
Die Stosswellentherapie ist heutzutage ein fester Bestandteil der Schmerzmedizin. Extrakorporal erzeugte Stosswellen werden in unterschiedlichen Anwendungsgebieten angewendet. Dabei spielt die Unterscheidung in fokussierte und radiale Stosswelle eine wesentliche Rolle. Der vorliegende Artikel soll diese Methode der Schmerzbehandlung in der Medizin genauer erläutern.
Shockwave Therapy is today a fix component in pain medicine. Shock Waves extracorporally generated are applied in many areas. The differentiation of focussed and radial Shock waves is essential in this Therapy. In this article, the application of Shock wave therapy in pain medicine will be explained.
Key Words: pain medicine, shockwave therapy
Der Einsatz von Stosswellen ist vielfältig. Dabei sind die sogenannte Nierensteinzertrümmerung in der Urologie, sowie Wundheilungsstörungen in der Dermatologie und der Fersensporn in der Orthopädie noch die bekanntesten Anwendungsgebiete. Wie Stosswellen generiert werden und wie sie wirken, ist jedoch nicht verbreitet bekannt.
Stosswellen sind physikalisch gesehen Schallwellen. In der Atmosphäre treten sie bei explosionsartigen Vorgängen auf wie zum Beispiel bei Blitzschlag. Dies erklärt auch das typische Geräusch, welches bei der fokussierten Stosswelle auftritt. Der Fokus, der erreicht wird, ist sehr gebündelt und kann je nach applizierter Energie und Frequenz unterschiedliche Eindringtiefen erreichen. Dies ist wesentlich zur Behandlung der Schmerzquelle.
Demgegenüber sind die radialen Stosswellen, präziser ausgedrückt radiale Druckwellen, sehr viel breitflächiger. Die Eindringtiefe ist viel geringer, die Energie an der Oberfläche jedoch grösser als bei der fokussierten Stosswelle. Somit entfaltet sie die Wirkung in breitflächigem Gewebe, wohingegen die fokussierte auf zellulärer Ebene sehr gezielt ihre Wirkung entfaltet.
Stosswellen in der konservativen Schmerztherapie
In der konservativen Schmerztherapie wird mit niederenergetischen Stosswellen gearbeitet. Dies führt zur optimalen Behandlung der schmerzhaften Struktur, ohne dabei nicht betroffene Stellen zu beeinträchtigen. Die lange Zeit geglaubte Hypothese der Gewebezerstörung als Wirkmechanismus der Stosswellentherapie hat sich als nicht korrekt erwiesen. Vielmehr wird durch die Stimulation des Gewebes unter anderem die Stimulation der Mikrozirkulation angeregt. Ebenso werden Stammzellen stimuliert, sowie die Ausschüttung von NO verursacht, welche den Stoffwechsel anregen und entzündungshemmend wirken. Beim Applizieren der Stosswellentherapie an der Muskulatur kommt noch der Wirkmechanismus über das Lösen von fixierten Aktin-Myosin Filamenten hinzu. Mit demselben Prinzip wird das Lösen von myofaszialen Triggerpunkten bewirkt.
Indikationen
Eine der häufigsten Indikationen ist die Tendinitis calcarea, besser bekannt als «Kalkschulter». Ebenso die Radiale und Ulnare Epikondylopathie (Tennis-/Golferellenbogen). Der Fersensporn (Fasciitis plantaris) gehört ebenfalls zum grossen Repertoire. Weniger bekannt, jedoch in der Hand eines geübten Schmerzarztes keine aussergewöhnliche Behandlung, ist u.a. die Behandlung von Tibiakantensyndrom, Pseudarthrosen, Myofasziale Schmerzsyndrome cervikal und lumbal.
Kontraindikationen und Nebenwirkungen
Absolute Kontraindikationen sind Tumore und Thrombosen im Behandlungsgebiet. Durch die Stosswellen können Zellen oder Thromben mobilisiert werden und im restlichen Körper verteilt werden. Relative Kontraindikationen sind Blutverdünnung oder Blutgerinnungsstörung. Dies hängt mit Läsionen von kleinen Blutgefässen zusammen, welche eine Einblutung ins Gewebe bewirken können.
Die häufigste Nebenwirkung ist eine Rötung durch vermehrte Reizung im Behandlungsgebiet, welche zu Petechien im Behandlungsgebiet und sogar Erstverschlimmerung der Schmerzen führen kann. Diese Nebenwirkungen sind jedoch harmlos und verschwinden von selbst wieder.
Behandlung
Das Behandlungskonzept der Stosswellentherapie hat sich in den letzten Jahren stark gewandelt. Während zu Beginn noch mit der «Dawo’s»-, also da wo’s weh tut, Methode behandelt wurde, wird heute viel systematischer vorgegangen. Es wird nicht mehr nur das schmerzhafte Areal angegangen, sondern die dazugehörigen Triggerpunkte werden mitbehandelt. Z.B. wird beim Fersensporn nicht mehr nur das schmerzhafte Areal mit Stosswellen angegangen, sondern ebenfalls die Triggerpunkte der zugehörigen Muskulatur (Fuss- und Wadenmuskeln), wobei von der Peripherie zum Zentrum hin behandelt wird. Zusätzlich sind Kenntnisse über Satellitentrigger im Sinne eines «referred pain» zur effektiven Behandlung Voraussetzung. Bei diesen Satellitentriggern handelt es sich um Triggerpunkte, welche mit der betroffenen Struktur scheinbar nichts zu tun haben. Die Mitbehandlung dieser Punkte kann jedoch über den Erfolg der Therapie entscheidend sein.
Bei der Lokalisation der mit dem Triggerpunkt verbundenen myofaszialen Ketten kommt heute der Ultraschall zum Einsatz. Dabei ist es wesentlich, die zum Triggerpunkt gehörende Muskulatur zu erkennen, sowie deren Ansatz und deren Ursprung. Nur so kann die zur schmerzhaften Stelle gehörende periphere Faszie-Muskel-Einheit effektiv behandelt werden.
Die Wahl der eigentlichen Behandlungsmethode hängt stark vom Einsatzgebiet ab. Die Voraussetzung einer solchen differenzierten Stosswellentherapie setzt ein modernes Gerät voraus (Abb. 1). Bei diesen Geräten sind sowohl die Energie als auch die Frequenz frei einstellbar. Dies ist wesentlich, um die Eindringtiefe und die am Wirkungsort erreichte Ausbreitung des Druckes zu definieren. Ebenso sind verschiedene Handstücke mit unterschiedlichen Aufsätzen nötig, um ein effektives Resultat zu erreichen.
Anwendungsbereiche
Es werden in myofaszialen Bereichen relativ grossflächige Applikatoren eingesetzt und hauptsächlich mit radialer Stosswellentherapie gearbeitet. Dies, um ein möglichst breites Gebiet mit Stosswellen zu behandeln. Dadurch wird die Mobilität der «verklebten» Faszien erhöht. Dies kann, wie bereits erwähnt, zu einem Muskelkater ähnlichen Gefühl führen, über das der Patient vorgängig zu informieren ist. Diese Missempfindung ist spontan regredient.
Im Sehnenbereich beim muskulären Ansatz kommen speziell konstruierte Aufsätze zum Einsatz. Diese Applikatoren arbeiten mit weniger Schlagkraft, erreichen aber dieselbe Energie. Sie eignen sich daher besonders gut für die Therapie von weicheren Strukturen wie Sehnen oder aber auch von spezifischen Triggerpunkten.
Isolierte, insbesondere tiefer liegende Schmerzpunkte, werden mit der fokussierten Stosswelle behandelt. Als zu behandelnde Strukturen sind z.B. M. iliopsoas oder auch Facettengelenke zu erwähnen. Aber auch oberflächlichere Anwendungen, wie der laterale oder mediale Epikondylus, profitieren von der fokussierten Stosswelle.
Stärken und Schwächen
Die ganz offensichtlichen Vorteile dieser Behandlung sind, dass sie komplett nicht-invasiv ist und dazu noch medikamentenfrei. Nebenwirkungen, wie allergische Reaktionen oder Infektionen an der Behandlungsstelle können daher nicht auftreten. Eine Belastung durch Röntgenstrahlung ist ebenfalls nicht vorhanden. Die Kombination mit anderen physikalischen Methoden wie Physiotherapie, Massage oder kinesiologisches Taping, oder TCM (Akupunktur) eignen sich gut zur Einbindung in ein Gesamtkonzept der Behandlung von Myofaszialen Schmerzzuständen.
Eine Schwäche ist die Anzahl der Behandlungen. Obwohl invasive Methoden starke Nebenwirkungen aufweisen können, ist im Normalfall lediglich eine Behandlung notwendig (z.B. Infiltrationen mit Kortikosteroiden). Die Stosswellentherapie besteht aus vier bis sechs Sitzungen à jeweils 30 Minuten im Abstand von jeweils einer Woche. Die Behandlung kann, wenn der Therapeut zu wenig vorsichtig vorgeht, schmerzhaft sein.
Der Autor hat keinen Interessenskonflikt im Zusammenhang mit diesem Artikel deklariert.
◆ Die Stosswellentherapie ist eine vielseitige Methode der Behandlung von muskuloskelettalen Schmerzen. Dabei können alle betroffenen Körperregionen behandelt werden und es gibt praktisch keine
Kontraindikationen.
◆ Die Methode ist komplett nicht-invasiv und es kommen keine
Medikamente zum Einsatz.
◆ Aufgrund von ständig neuen wissenschaftlichen Erkenntnissen im Zusammenhang mit muskuloskelettalen Schmerzen und der ständigen Weiterentwicklung der Geräte ist die Stosswellentherapie eine
essentielle Methode in der modernen Schmerzmedizin.
Goertz O, Hauser J, Hirsch T, von der Lohe L, Kolbenschlag J, Stricker I et al.
Short term effects of extracorporal shock wave therapy on microcirculation.
Journal of Surgical Research 2015 ; 194 (1) : 304-311
Mani-Babu S, Morissey D, Waugh C, Screen H, Barton C.
The effectiveness of extracorporeal Shock Wave Therapy in lower limb tendinopathy: a systematic review. Am J Sports Med 2015 ; 43(3) : 752-761
Rio E, Kidgell D, Moseley GL, Gaida J, Docking S, Purdam C et al.
Tendon neuroplastic training: changing the way we think about tendon rehabilitation : a narrative review. Br J Sports Med 2016 ; 50(4) : 209-15
THC-haltige Cannabispräparate werden insbesondere bei Schmerz- und/oder Spastikpatienten eingesetzt, während CBD hauptsächlich bei den Epilepsieformen Dravet- und Lennox-Gastaut Anwendung findet. In der Schweiz gibt es zwei auf Hanfbasis zugelassene Medikamente, Sativex und Epidyolex. Nicht registrierte Cannabispräparate können als Magistralrezepturen verschrieben werden.
Cannabis preparations containing THC are primarily used to treat chronic pain and spasticity, while CBD is mainly prescribed to patients suffering from refractory epilepsy, such as Dravet and Lennox-Gastaut syndrome. In Switzerland, two cannabis-based drugs are approved by the authorities, namely Sativex and Epidyolex. Besides these, cannabis preparations can be prescribed as extemporaneous formulations.
Seit einigen Jahren mausern sich Cannabispräparate oder cannabinoidhaltige Medikamente zu möglichen Alternativen bei einer Vielzahl von Indikationen. Die medizinischen Wirkungen lassen sich vor allem auf die beiden Hauptcannabinoide THC (Tetrahydrocannabinol bzw. Dronabinol*) und CBD (Cannabidiol) zurückführen. Nur diese beiden Cannabinoide werden aktuell für therapeutische Zwecke eingesetzt, entweder als Reinstoffe oder als Bestandteil von Vielstoffgemischen (z.B. THC- oder CBD-haltige Cannabisextrakten). Der Wirkstoff THC ist besser erforscht als CBD. In zahlreichen Tier- und Humanstudien konnte nachgewiesen werden, dass THC über schmerzlindernde, antispastische, appetitfördernde, den Brechreiz unterdrückende und den Augeninnendruck senkende Eigenschaften verfügt. Dazu kommen andere zentralwirksame Eigenschaften, welche bei unterschiedlichen Erkrankungen wie Tourette-Syndrom, Tics, Restless legs, Parkinson, etc. von Nutzen sein können. Die wissenschaftliche Evidenz zur Wirksamkeit von THC ist bei obgenannten Indikationen sehr unterschiedlich. Bei vielen Erkrankungen fehlen gross angelegte Studien weitgehend; es liegen höchstens Resultate aus kleinen Studien mit geringen Patientenzahlen oder Fallberichte vor. Gute Evidenz liegt gemäss dem Standardwerk «The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research» (1) bei folgenden Einsatzgebieten vor: chronische Schmerzen bei Erwachsenen, chemotherapie-assoziierte Übelkeit und Erbrechen sowie Spastik bei Multipler Sklerose. In anderen Metaanalysen (2, 3, 4) findet sich aufgrund der Heterogenität der eingesetzten Cannabispräparate für bestimmte Indikationen nicht immer die gleiche Evidenz.
CBD wiederum wird zwar zum Teil bei gleicher Indikation wie THC eingesetzt, so vor allem bei (entzündlichen) Schmerzen und gewissen Bewegungsstörungen. Wissenschaftliche Daten zeigen aber, dass CBD vor allem antiepileptisch, antipsychotisch, entzündungshemmend und neuroprotektiv wirkt (5, 6). Dementsprechend wird CBD hauptsächlich verwendet bei therapieresistenten Epilepsieformen bei Kindern (v.a. mit Dravet- bzw. Lennox-Gastaut-Syndrom), daneben auch bei Angststörungen, Panikattacken, autistischen Spektrumsstörungen, ADHS, etc.
Cannabinoide bei chronische Schmerzen – Datenlage
In verschiedensten Untersuchungen konnte gezeigt werden, dass das Endocannabinoid-System (ECS) bzw. die beiden Cannabinoid-Rezeptoren (CB1 und CB2) bei der Schmerzverarbeitung eine Rolle spielen (7). Allerdings sind die Vorgänge sehr komplex und es scheint klar, dass die Schmerzlinderung nicht alleine durch die Aktivierung von CB1-Rezeptoren zustande kommt (8). Eine systematische Übersicht zur Klinik aus dem Jahr 2019 zeigte folgende Hauptbefunde: Die Wirksamkeit von Cannabinoiden bei chronischen Schmerzen wurde in den letzten Jahren häufig untersucht. Ein generelles Problem der Studien mit Cannabis stellt zum einen die Heterogenität der verwendeten Präparate, wie auch unterschiedliche Applikationsformen (oral vs. inhalativ) dar. In der Metanalyse zur Wirksamkeit von Cannabinoiden und chronischen Schmerzen von Whiting et al. (2015) wurden insgesamt 28 randomisierte-kontrollierte Studien berücksichtigt in der Zeit von 1975 bis 2015. Dabei waren insgesamt 2’454 Patienten eingeschlossen. Untersucht wurden verschiedene Cannabismedikamente (Medizinalhanf mit unterschiedlichem THC-Gehalt, THC- und oder THC/CBD-haltige Cannabisextrakte – meist Nabiximol-, Dronabinol wie auch das THC-Derivat Nabilon). Die Autoren kommen zum Schluss, dass Cannabinoide teilweise bis zu einer Schmerzreduktion von 30% wirksam sein können, für eine Reduktion der Symptomatik von mindestens 50 % liegt zur Zeit keine Evidenz vor.
Alle Untersuchungen zeigen aber weitere, sekundäre Wirkungsbeweise (z.B. Reduktion der durchschnittlichen Schmerzreduktion, starke oder sehr starke allgemeine Verbesserung) zugunsten der Cannabinoide (9).
Am besten untersucht wurden Cannabinoide bei neuropathischen Schmerzen. Die Effekte der verschiedenen Cannabispräparate waren ausgeprägter als bei chronischem Schmerz. Dabei ist die Wirkung von Nabiximol (Sativex) am besten untersucht. Auch bei anderen Schmerzformen wie Tumorschmerzen, Schmerzen des rheumatischen Formenkreises, Fibromyalgien u.a. werden Cannabinoide in der Praxis eingesetzt.
Zusammenfassend: Cannabinoide können chronische Schmerzen lindern, wobei vor allem Patienten mit neuropathischen Schmerzen und schmerzhafter Spastik profitieren (Whiting et al. 2015). Bei nozizeptiven Schmerzen scheinen Cannabinoide weniger wirksam zu sein (10).
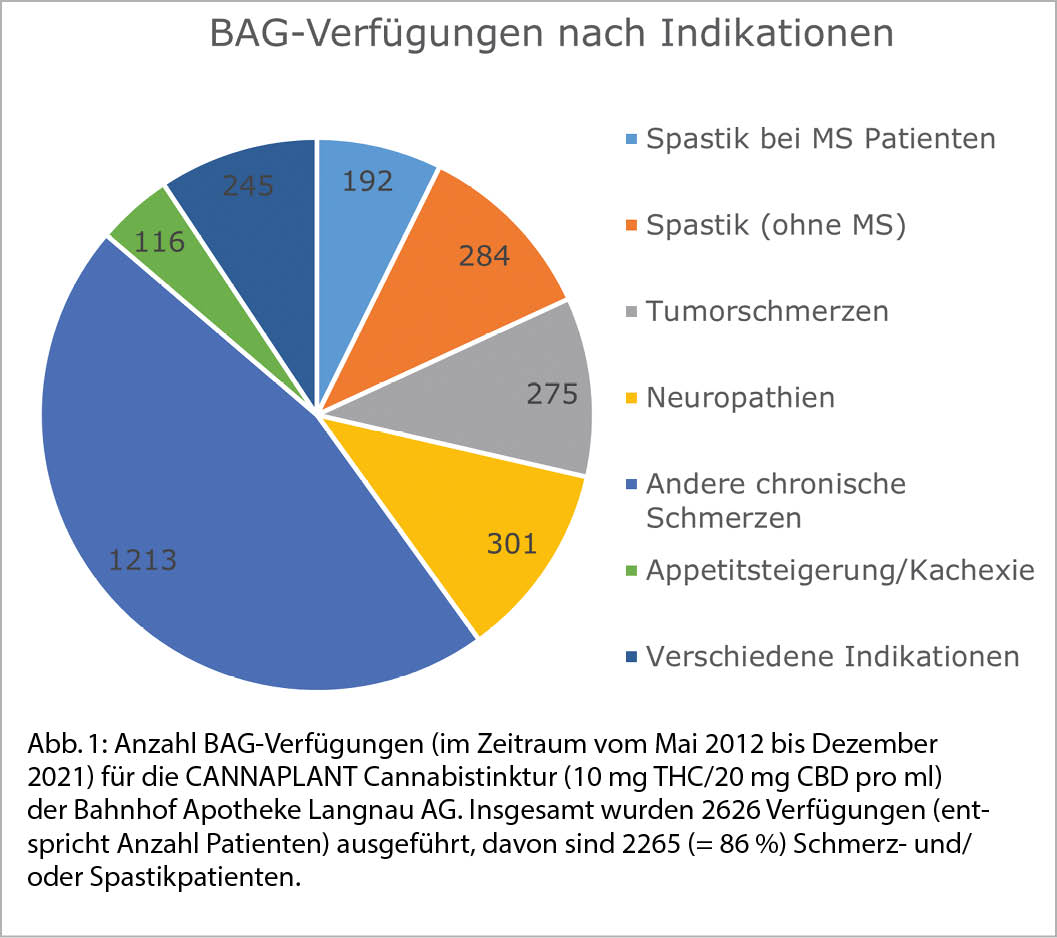
Ergänzung aus der Praxis der Bahnhof Apotheke in Langnau: In den letzten 14 Jahren erhielten mehrere Tausend Patienten ein magistral verschriebenes Cannabispräparat. Zirka ¾ davon waren THC-haltige und damit BAG-bewilligungspflichtige Präparate (Dronabinol-Lösung, Cannabistinktur, Cannabisöl). Über 80 % der Verschreibungen betrafen chronische Schmerz-/und Spastikpatienten. Nicht selten können Cannabinoide bei bestimmten Patienten im Sinne einer «add on»-Therapie (z.B. zu Opiaten) eine wirksame Ergänzung sein; manchmal wirken Cannabinoide aber auch als Monotherapie besser als etablierte Medikamente.
Dosierungen: Die Dosierung der Cannabinoide bei Schmerzpatienten ist sehr individuell. Für THC liegen die typischen Tagesdosen (aufgeteilt in 2 bis 3 Einzeldosen) zwischen 10 und 30 mg (oder selten höher) pro Tag. Für CBD (alleine oder in Kombination mit THC) können diese ein Mehrfaches betragen.
Nebenwirkungen/Abhängigkeitspotential: Sowohl THC wie auch CBD gelten als relativ nebenwirkungsarm. Typische Nebenwirkungen von THC können sein: Müdigkeit, Sedierung, Mundtrockenheit, gerötete Augen, Schwindel, Herzrasen, Übelkeit, kognitive Einschränkungen. Bei hohen Dosen von CBD werden beschrieben: Müdigkeit, Sedierung, Appetitmangel, GIT-Beschwerden, reversible Erhöhung von Lebertransaminasen (insbesondere in Kombination mit anderen die Leberfunktion beeinträchtigenden Medikamenten). In therapeutischen Dosen ist die Suchtgefahr und Abhängigkeit vernachlässigbar (sowohl für THC als auch für CBD). Ebenfalls spielen in der Praxis Toleranzentwicklung und Entzugssymptome keine grosse Rolle.
Wechselwirkungen: THC und CBD sind Substrate von CYP-Enzymen in der Leber. Bei der Kombination mit CYP-Inhibitoren und CYP-Induktoren kann allenfalls eine Dosisanpassung notwendig sein. Relevant für die Praxis ist, dass CBD potenziell CYP-Enzyme hemmen kann. Insbesondere bei höheren CBD-Dosen ist daher Vorsicht geboten bei der Kombination mit CYP-Substraten enger therapeutischer Breite, wie etwa gewissen Antiepileptika (Clobazam, Rufinamid, Topiramat) (11) und oralen Antikoagulantien vom Typ Vitamin K-Antagonisten (Phenprocoumon, Acenocoumarol) (12).
Verfügbare Präparate in der Schweiz: Zurzeit haben zwei cannabisbasierte Medikamente eine Swissmedic Zulassung. Zum einen der sublingual zu applizierende, BetmG-pflichtige Spray Sativex (Nabiximol), zur Anwendung bei Spastik bei MS. Zum anderen der verschreibungspflichtige Sirup Epidyolex (CBD) zur Behandlung der seltenen Epilepsieformen Dravet- und Lennox- Gastaut-Syndrom. Alle anderen zur Zeit verschreibbaren Hanfpräparate sind sogenannte Magistralrezepturen (z.B. Dronabinol-Lösung 2.5%, CANNAPLANT Cannabistinktur bzw. –Öl, Sativa-Öl, u.a.). Anders als bei den arzneimittelrechtlich zugelassenen Medikamenten gilt bei diesen individuell für den Patienten hergestellten Präparaten Therapiefreiheit, d.h. der Arzt ist nicht an eine bestimmte Indikation gebunden.
Gesetzliche Grundlagen: Die Verschreibung des von der Swissmedic zugelassenen Cannabismedikamentes Sativex geschieht analog den anderen Betäubungsmitteln. Dabei kann das Präparat einzig bei der zugelassenen Indikation Spastik bei MS-Patienten verschrieben werden, andere Indikationen sind «off label» jedoch möglich. Alle THC-haltigen (> 1 %) Magistralpräparate bedingen zurzeit noch eine BAG-Ausnahmebewilligung, d.h. der/die verschreibende Arzt/Ärztin muss zwingend ein Gesuch (zum Beispiel mittels vorhandenem Formular) einreichen, dieses wird nach Überprüfung in der Regel innerhalb weniger Tage gutgeheissen. Diese Ausnahmeregelung wurde im geltenden Betäubungsmittelgesetz geändert, der Vollzug wird voraussichtlich in der 2. Hälfte 2022 erfolgen. Was bedeutet das: künftig können THC-haltige Hanfmedikamente mit einem Gehalt > 1 % ohne BAG-Bewilligung verschrieben werden, allerdings ist eine sogenannte Begleiterhebung Pflicht. Wie diese im Detail aussehen wird, ist noch nicht bekannt.
Reine CBD-Präparate sind rezeptpflichtig, unterstehen aber nicht dem BetmG. Für alle bisher dem Chemikalienrecht unterstellte, freiverkäuflichen «CBD-Extrakte» mit einem max. THC-Gehalt von < 1 % gelten ab Herbst 2022 neue Bestimmungen. Diese Präparate (v.a. CBD-Tinkturen, CBD-Öle) müssen zwingend mit einem Vergällungsmittel versetzt werden, so dass diese nicht mehr eingenommen werden können (13). Unvergällte, nicht dem BetmG unterstellte Cannabisextrakte mit CBD sollten verschreibungsfähig sein, sind bzw. bleiben sofern diese GMP-Qualitätsanforderungen entsprechen. Cannabispräparate haben, trotz noch unbefriedigender klinischer Evidenz, bereits heute einen beachtlichen Stellenwert in der Behandlung von chronischen Schmerzen. Das Verschreiben der THC- bzw. CBD-haltigen Präparate bleibt auch in der näheren Zukunft wohl die Ausnahme, kann aber in gewissen Fällen eine wirksame Alternative und/oder Ergänzung für betroffene Patienten darstellen. Es ist zu wünschen, dass das grosse therapeutische Potenzial der Cannabinoide noch vermehrt klinisch untersucht wird, damit Cannabis evidenzbasiert die therapeutische Palette in der Schmerztherapie bereichern kann.
*Dronabinol ist der international anerkannte Freiname für THC, in der Regel ist dabei synthetisches oder halbsynthetisches THC gemeint.
Copyright by Aerzteverlag medinfo AG
Dr. pharm. Manfred Fankhauser
Bahnhof Apotheke Langnau AG
Dorfstrasse 2
3550 Langnau
034 402 12 55
manfred.fankhauser@cannabis-med.ch
Der Autor ist Inhaber und Geschäftsführer der Bahnhof Apotheke Langnau AG, welche als Pionierapotheke für medizinisches Cannabis spezialisiert ist. Die Bahnhof Apotheke Langnau AG verfügt über die notwendigen Bewilligungen, um Magistralrezepturen auf Cannabisbasis herstellen zu dürfen
1. National Academies of Sciences, Engineering and Medicine (2017): The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research. Washington, DC: The National Academies Press. https://doi.org/10.1722/24625
2. Whiting PF, Wolff RF, Deshpande S et al.Cannabinoids for Medical Use: A Systematic Review and Meta-analysis. JAMA. 2015; 313(24): 2456-73.
3. Allan GM, Finley CR, Ton J et al. Systematic review of systematic reviews for medical cannabinoids: Pain, nausea and vomiting, spasticity and harms. Can Fam Physician. 2018; 64 (2): e78-e94.
4. Stockings E, Campbell G, Hall WD et al. Cannabis and cannabinoids for the treatment of people with chronic noncancer pain conditions: a systematic review and meta-analysis of controlled and observational studies. Pain 2018; 159 (10): 1932-1954.
5. Iffland K, Grotenhermen F. An Update on Safety and Side Effects of Cannabidiol: A Review of Clincal Data und Relevant Animal Studies. Cannabis Cannabinoid Res. 2(1), 2017; 139-154.
6. Bih CI, Chen T, Nunn AVW et al. Molecular Targets of Cannabidiol in Neurological Disorders. Neurotherapeutics. 2015; 12(4): 699-730.
7. Lötsch J, Weyer-Menkhoff I et al. Current evidence of cannabinoid-based analgesia obtained in preclinical and human experimental settings. Eur J Pain. 2018; 22(3): 471-84. Doi: 10.1002/ejp.1148.
8. Agarwal N, Pacher P et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors: Nat. Neurosci. 2007; 10(7): 870-9. Doi: 10.1038/nn1916.
9. Hoch E, Friemal CM, Schneider M. (2019). Cannabis. Potenzial und Risiko. Eine wissenschaftliche Bestandesaufnahme, 295.
10. Fitzcharles MA, Baerwald C et al. Efficacy, tolerability and safety of cannabinoids in chronic pain associated with rheumatic diseases (fibromyalgia syndrome, back pain, osteoarthritis, rheumatoid arthritis); A systematic review of randomized controlled trials. Schmerz. 2016; 30(1): 47-61.doi:10.1007/s00482-015-0084-3.
11. Gaston TE, Bebin EM, Cutter GR et al. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia 2017; 58 (9): 1586-1592.
12. Grayson L, Vines B, Nichol K et al. An interaction between warfarin and cannabidiol, a case report. Epilepsy Behav Case Rep. 2017; 10-11.
13. BBl: Bundesblatt 2022 668 Allgemeinverfügung der Anmeldestelle Chemikalien zum Inverkehrbringen von CBD-haltigem Duftöl, gestützt auf Artikel 10 Absätze 1 und 2 in Verbindung mit Artikel 1, Absätze 1-3 PrSG, 24.3.2022.
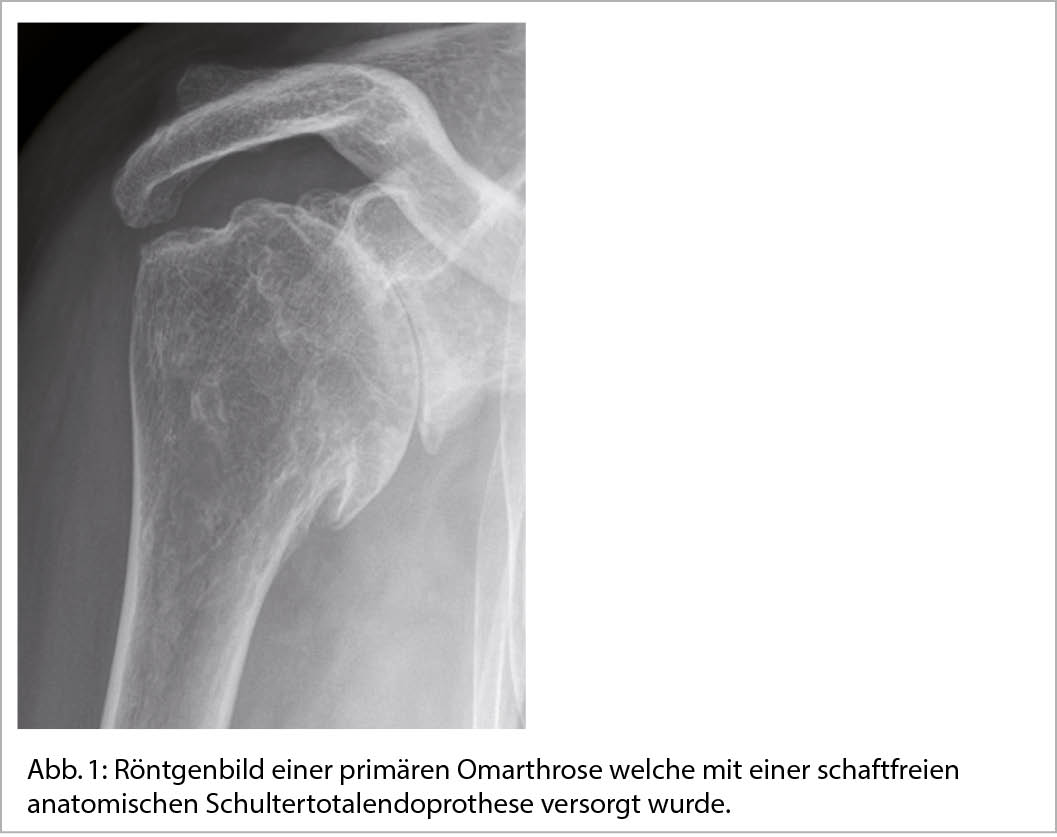
Die Schultergelenksarthrose kann in eine primäre Arthrose und eine sekundäre Arthrose eingeteilt werden. Während bei der primären Arthrose mit Ausnahme des eventuell höheren Alters und des damit einhergehenden Verschleisses keine eindeutige Ursache genannt werden kann, ist die sekundäre Arthrose auf einen klaren Grund zurückzuführen der eine frühzeitige Degeneration des Schultergelenkes auslöst. Stellt sich in der Anamnese und in der klinischen Untersuchung der Verdacht einer Schultergelenksarthrose, benötigt man zur weiteren Diagnostik eine Bildgebung in Form von Röntgenaufnahmen. Initial steht eine schmerzlindernde Kortisoninfiltration und anti-entzündliche medikamentöse Therapie im Vordergrund, welche durch physiotherapeutische Massnahmen begleitet werden sollte. Bei therapierefraktären Beschwerden und grossem Leidensdruck besteht die Indikation zur operativen Versorgung mittels einer Schulterendoprothese. Hierdurch kann sowohl im kurz- sowie langfristigen Verlauf eine deutliche Schmerzreduktion und Funktionsverbesserung erzielt werden und somit die Lebensqualität der Patienten verbessert werden. Zeigt sich eine fortgeschrittene Dezentrierung des Oberarmkopfes oder ein instabiles Drehzentrum ist die Versorgung mittels einer inversen Schulterendoprothese indiziert. Moderne Planungsverfahren erlauben die individuelle Anpassung der Schulterendoprothese an die individuelle Anatomie des Schulterblattes und Oberarmkopfes sowie die Körperhaltung der Patientinnen und Patienten.
Osteoarthritis of the shoulder joints can be divided into primary osteoarthritis and secondary osteoarthritis. While in the case of primary osteoarthritis, with the possible exception of older age and the associated wear and tear, no clear cause can be named, secondary osteoarthritis is due to a clear reason that triggers premature degeneration of the shoulder joint. If the history and clinical examination raise the suspicion of shoulder joint osteoarthritis, imaging in the form of X-rays is required for further diagnosis. Initially, the focus is on pain-relieving cortisone infiltration and anti-inflammatory drug therapy, which should be accompanied by physiotherapeutic measures. In cases where the symptoms are refractory to therapy and the patient is suffering greatly, there is an indication for surgical treatment with a shoulder arthroplasty. This can lead to a significant reduction in pain and an improvement in function in both the short and long term, thus improving the patient’s quality of life. If there is advanced decentering of the humeral head or an unstable center of rotation, treatment with a reverse shoulder arthroplasty is indicated. Modern planning procedures allow the shoulder arthroplasty to be individually adapted to the anatomy of the scapula and humeral head as well as the patient’s posture.
Schulterbeschwerden sind der dritthäufigste Vorstellungsgrund unter den muskuloskelettalen Beschwerden in der primären Gesundheitsversorgung. In 21% der durch den Hausarzt aufgrund von persistierenden Schulterschmerzen überwiesenen Patienten wird in der radiologischen Bildgebung eine Omarthrose diagnostiziert (1). In Zweidrittel der Fälle liegt den degenerativen Veränderungen des Knorpels und des angrenzenden Knochens sowie des periartikulären Weichteilgewebes kein spezifischer Auslöser zugrunde. Die Prävalenz dieser sogenannten primären Arthrose steigt mit zunehmendem Alter. Während 15% bereits in der 6. Lebensdekade betroffen sind, steigt der Anteil bei Patienten über 70 Jahren auf über 25% (2). Beeinflussbare Risikofaktoren für die primäre Arthrose stellen Übergewicht, Rauchen sowie systemische Erkrankungen wie arterielle Hypertonie dar (Abb. 1) (3).
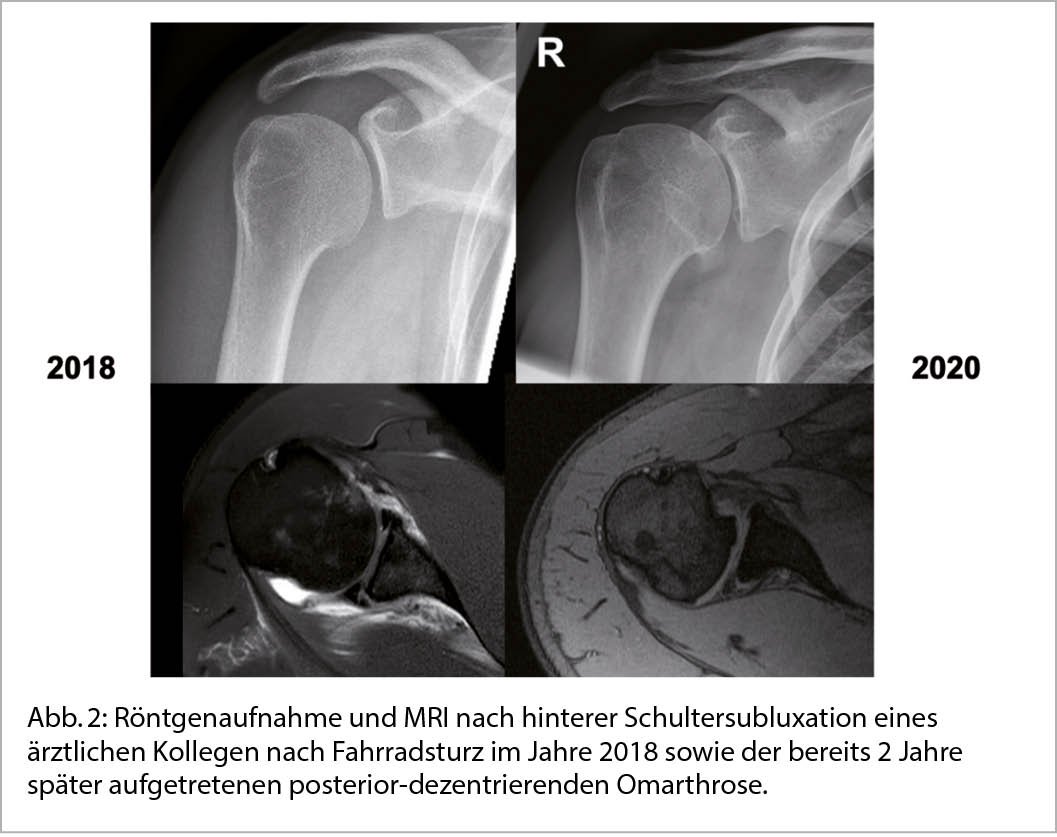
Die sekundäre GHA entstehen auf Basis einer klar definierten Ursache wie z.B. einer Gelenksinstabilität, Fehlstellung nach Trauma, oder einer medikamenteninduzierten Nekrose (Abb. 2).
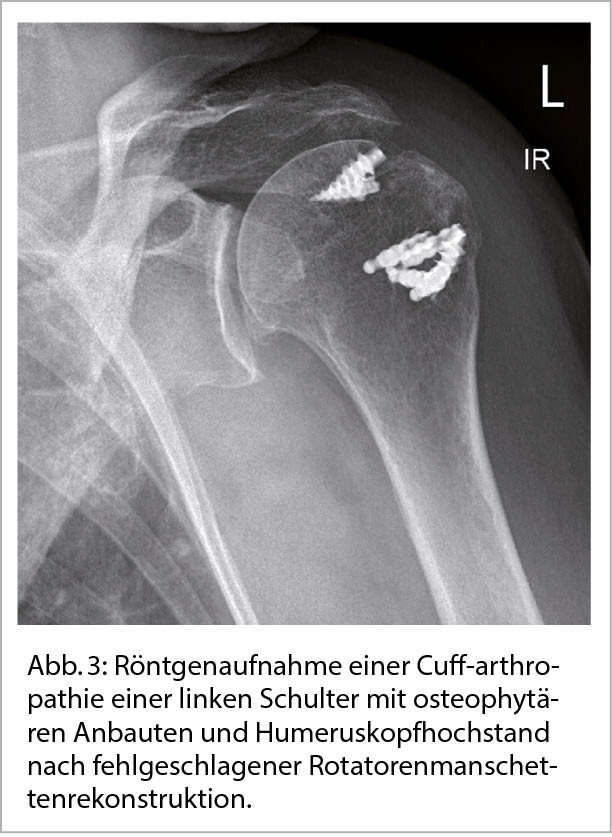
Eine sehr häufige Form der sekundären Schultergelenksarthrose ist die sogenannte Cuff-Arthropathie bei Patienten mit ausgeprägten Rotatorenmanschettenrupturen und konsekutivem Humeruskopfhochstand (Abb. 3) (4).
Tritt eine primäre Arthrose frühzeitig auf, so sollte nach einer versteckten Ursache geforscht werden. Kürzlich konnten wir z.B. in einer Studie zeigen, dass häufig vergleichsweise junge Männer betroffen sind, die Sportarten mit einer hohen Belastung der Schultergelenke ausüben wie z.B. Kraftsport und Kampfsport. Vermeintlich kann eine repetitive Überlastung und ein unausgeglichenes Training zu einer Dysbalance der schulterstabilisierenden Kräftepaare führen und in weiterer Folge eine posterior-exzentrische Arthrose begünstigen (3).
Diagnostik
Die Erstvorstellung erfolgt zumeist aufgrund eines zunehmenden Schulterschmerzes, der je nach Art der vorliegenden Arthrose als Belastungs-, Ruhe-, oder Nachtschmerz auftreten kann.
Während bei einer primären Omarthrose häufig die zunehmende Steife des Gelenkes einen Funktionsverlust bedingt, so kann bei einer Cuff-Arthropathie der Verlust der Rotatorenmanschette zu einer Pseudoparalyse der Schulter führen mit mangelnder aktiver (jedoch passiv erhaltener) Elevationsfähigkeit des Armes. Bei fortgeschrittenen Omarthrosen kann bei der klinischen Untersuchung häufig eine Krepitation in der Schulter bei passiver Bewegung wahrgenommen werden. Die primäre Diagnosesicherung erfolgt über eine konventionelle radiologische Bildgebung des Schultergelenks in 2 Ebenen (antero-posterior und axiale Aufnahme). Klassische radiologische Zeichen der Omarthrose umfassen osteophytäre Anbauten am Pfannenrand oder Oberarmkopf, Gelenkspaltverschmälerung, sowie subchondrale Zystenformationen und Sklerosierung. Die Beurteilung der Schweregrade nach radiologischen Gesichtspunkten korreliert häufig nicht mit der klinischen Symptomatik der Patienten weshalb eine sorgfältige klinische Untersuchung unumgänglich ist (5).
Nicht-operative Therapie
Nach Diagnosestellung ist eine gründliche Aufklärung über das vorliegende Krankheitsbild und das zu erwartende Voranschreiten der degenerativen Gelenksveränderungen notwendig. Die Entscheidung über die Therapie ist von den Schmerzen und der Funktions- bzw. Alltagseinschränkung abhängig unter Berücksichtigung des Aktivitätsniveaus und eventueller Begleiterkrankungen. Initial ist ein konservativer Therapieversuch empfohlen. Die symptomatische Schmerztherapie steht dabei im Vordergrund. Abhängig von den patientenspezifischen Risikofaktoren sind nicht-steroidale antiinflammatorische Medikamente über einen begrenzten Zeitraum Mittel der Wahl. Opioide sollten aufgrund des limitierten Effekts bei Gelenksschmerzen und ungünstigem Nebenwirkungsprofil, sowie erhöhtem Suchtpotential, nur in Ausnahmefällen verschrieben werden (5). Bei Schmerzexazerbation kann eine intraartikuläre Kortisoninfiltration unter Beachtung der Kontraindikationen eine deutliche, wenngleich häufig leider zeitlich begrenzte, Verbesserung der Schmerzsymptomatik und Schulterfunktion herbeiführen (6). Alternativ können intraartikuläre Infiltrationen mit Hyaluronsäure durchgeführt werden (7). Die klinische Wertigkeit von Hyaluronsäureinfiltrationen gegenüber Kortisoninfiltrationen muss noch in weiteren Vergleichsstudien näher untersucht werden (8). Weitere Forschungsschwerpunkte der intraartikulären Infiltrationstherapie sind derzeit Platelet-Rich Plasma- (PRP) und Zell-Therapien. Aktuell kann bei nicht ausreichender Datenlage jedoch noch keine generelle Therapieempfehlung gegeben werden (9). Wichtig ist eine Aufklärung über das Risiko einer iatrogenen Gelenksinfektion bei Gelenkinfiltrationen, wobei es sich um eine sehr seltene aber schwerwiegende Komplikation handelt. Um das Risiko eines periprothetischen Gelenksinfektes zu vermindern ist zudem von einer Gelenkinfiltration drei Monate vor einer geplanten Endoprothesenimplantation abzuraten (10).
Die Physiotherapie ist die zweite feste Säule der konservativen Therapie. Das Ziel ist die bestmögliche Erhaltung der Schulterbeweglichkeit und Kraft sowie die Schmerzreduktion. Im Fokus steht die Kräftigung der Humeruskopf-zentrierenden Rotatorenmanschette, sowie ausgleichende Entspannungsübungen zur Entlastung der periskapulären Muskelgruppen (11). Aktive Bewegung im Alltag und Sport mit wenig Belastung für die Schultern sind empfehlenswert, während Sportarten mit hoher schulterspezifischer Belastung gemieden werden sollten (3).
Operative Therapie:
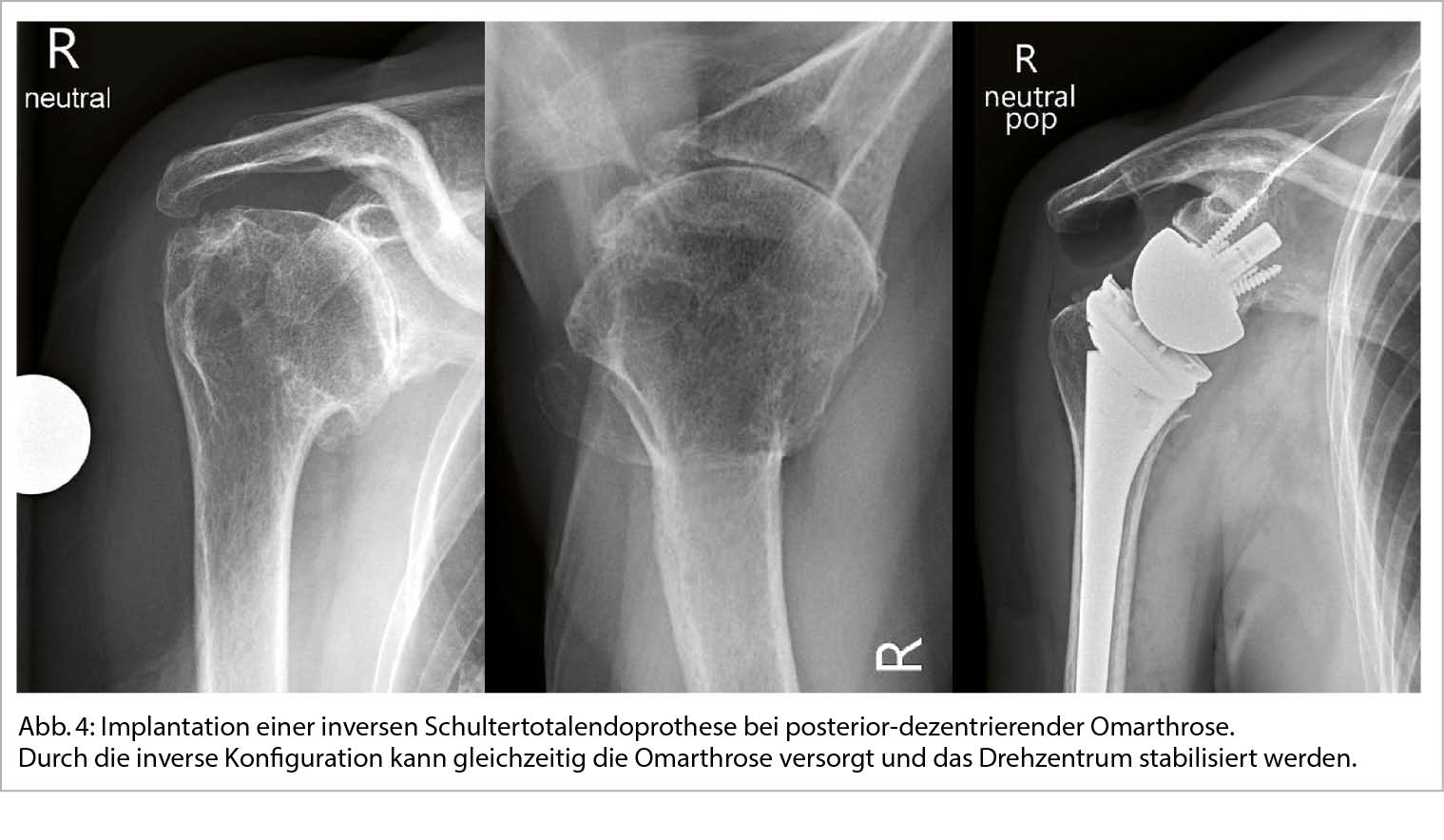
Ist die konservative Therapie ausgeschöpft kann durch die Implantation einer Schulterendoprothese eine zuverlässige Verbesserung der Beschwerden erreicht werden. Die Indikationen zur operativen Versorgung besteht bei therapieresistenten Schmerzen oder unzureichend kompensierbaren Einschränkungen des Bewegungsausmasses sowie zunehmendem Leidensdruck der Patienten. Eine endoprothetische Versorgung kann als anatomische (Total)endoprothese (aTSA) unter Erhalt der ursprünglichen Anatomie oder als inverse Totalendoprothese (rTSA) zur Stabilisierung des Drehzentrums erfolgen (Abb. 4) (12).
Wichtige Strukturen, die zur Zentrierung des Humeruskopfes beitragen sind knöchern die Schultergelenkspfanne sowie weichteilig die Muskeln und Sehnen der Rotatorenmanschette. Exzentrische Omarthrosen aufgrund einer schwerwiegenden Pfannendeformität und statischer Dezentrierung des Oberamkopfes oder aufgrund einer Rotatorenmanschetteninsuffizienz mit instabilem Gelenksdrehzentrum sprechen für die Implantation einer inversen Schulterendoprothese. Der Anteil der implantierten inversen Endoprothesen ist in den letzten Jahren aufgrund der vielfältigen Indikationserweiterungen und nicht zuletzt aufgrund der sehr guten und verlässlich erzielbaren Ergebnisse kontinuierlich gestiegen. Während die inverse Endoprothese durch das stabilere Drehzentrum überzeugt, ist bei der anatomischen Endoprothese das durchschnittlich zu erwartende postoperative Bewegungsausmass grösser. Bei Verlust der Stabilität des Drehzentrums im Verlauf nach Implantation einer anatomischen Schultertotalendoprothese aufgrund einer sekundären Rotatorenmanschetteninsuffizienz oder eines zunehmenden exzentrischen Pfannenabriebes ist der Wechsel auf eine inverse Prothese möglich (13).
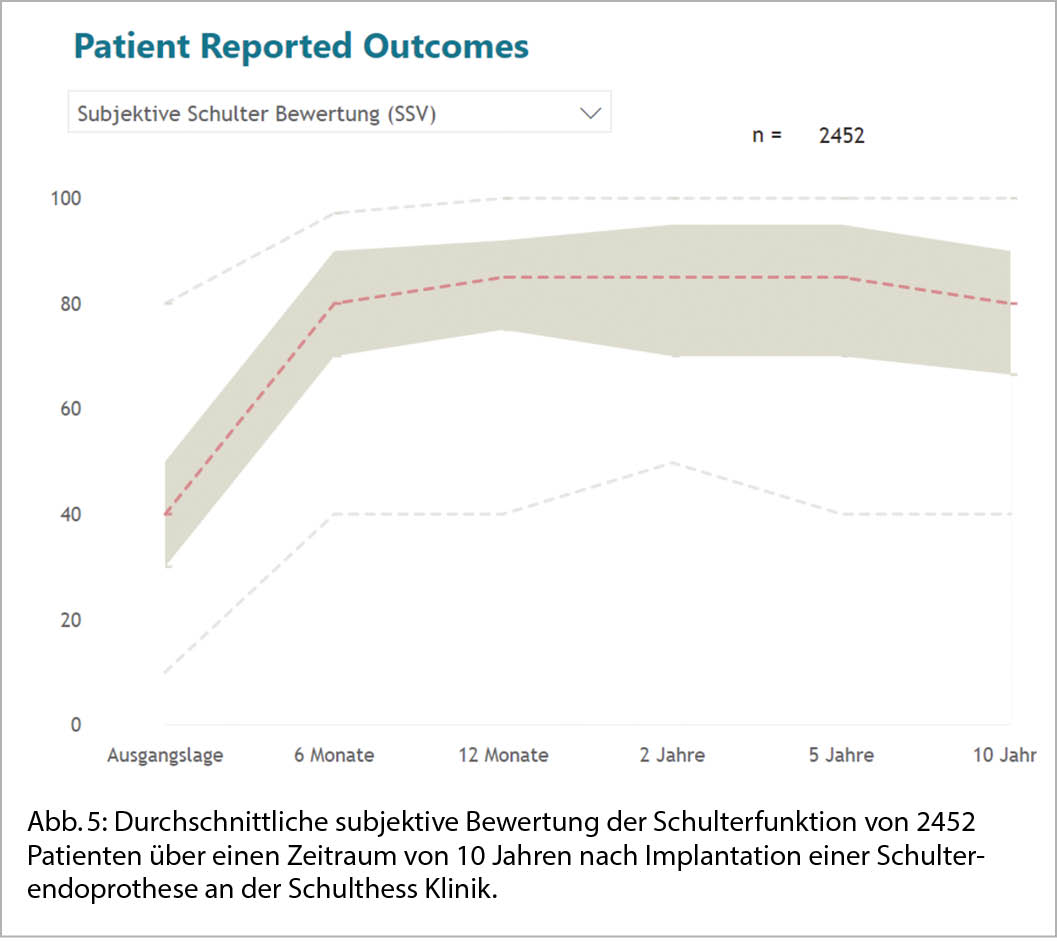
Generell, kann unabhängig von der Art der gewählten Schulterendoprothese bei Patientinnen und Patienten, die unter einer der verschiedenen Formen der Omarthrose leiden, eine zuverlässige Verbesserung der Schmerzen und des Bewegungsumfanges erreicht werden und über einen langen Zeitraum die Lebensqualität deutlich verbessert werden (Abb. 5).
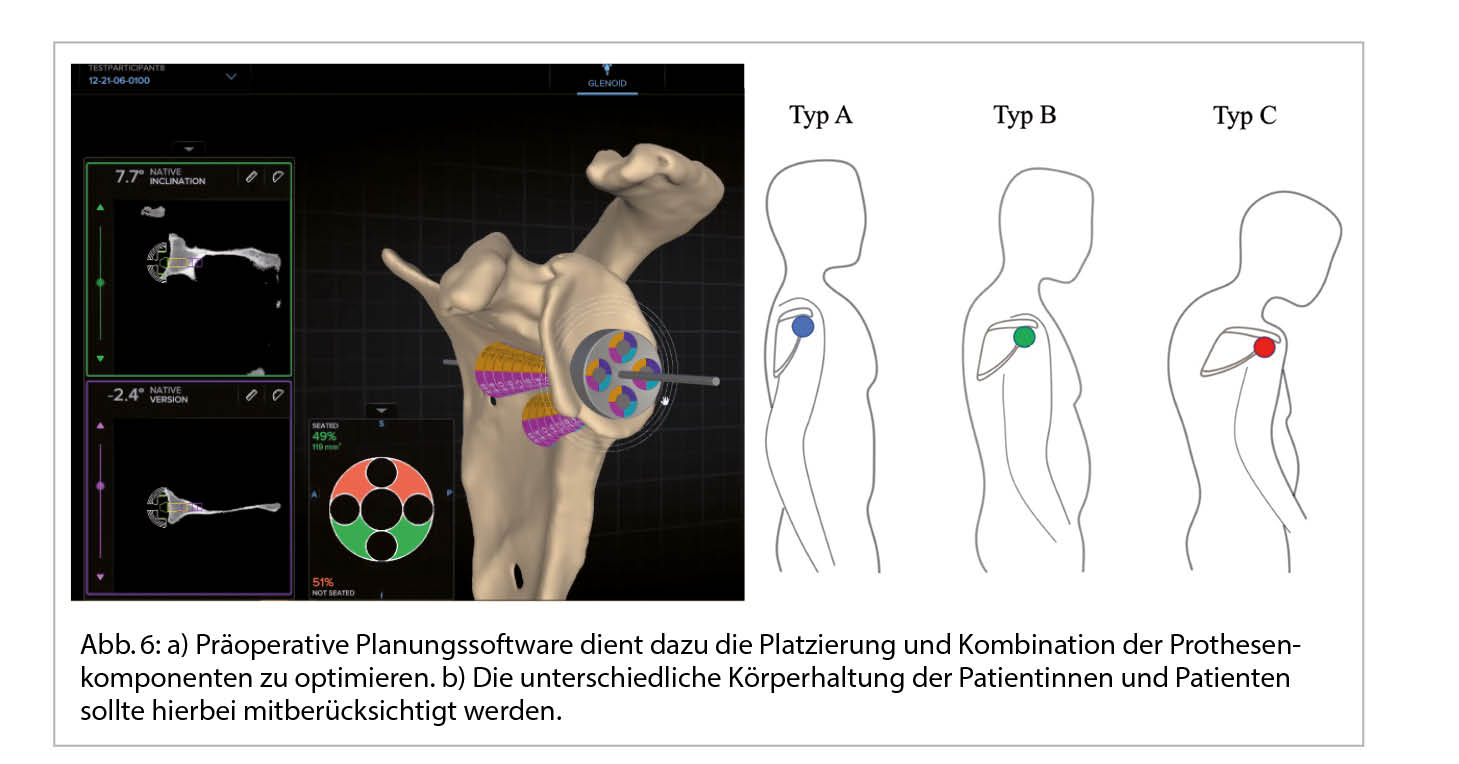
Präoperative Planungssoftware wird genutzt um die Platzierung und Kombination der Prothesenkomponenten zu simulieren und so intra- und postoperative Komplikationen aufgrund von Fehlimplantation und mechanischer Konflikte zu vermeiden.[14] In eigenen Arbeiten konnten wir zudem auf die Wichtigkeit der Miteinbeziehung der Patienten-individuellen Körperhaltung auf die präoperative Planung von Schulterendoprothesen zur Verbesserung des Bewegungsausmasses hinweisen (15). Eine angepasste Auswahl von Implantatkomponenten scheint in der Lage zu sein, die negativen Auswirkungen von häufig vorkommender kyphotischer Fehlhaltung auf das erzielbare Bewegungsausmass abzuschwächen (Abb. 6) (16).
Postoperativ erfolgt je nach Knochenqualität, Beschaffenheit der Sehnen und Art der Endoprothese typischerweise eine Ruhigstellung für 1-6 Wochen sowie eine schmerz- und funktionsadaptierte physiotherapeutische Beübung. Nach der Schulterendoprothesenimplantation werden die Patienten in regelmässigen Zeitabständen im behandelnden Zentrum nachuntersucht um mögliche Verschleisserscheinungen, Lockerung und andere auftretende Probleme frühzeitig zu erkennen. Bei Verdacht auf einen periprothetischen Infekt, Fraktur oder generellen Funktionsausfall sowie Schmerzzunahme sollte eine Überweisung an den Operateur erfolgen.
Copyright by Aerzteverlag medinfo AG
Prof. Dr. med. univ. Philipp Moroder
Schulter- und Ellenbogenchirurgie,
Schulthess Klinik, Zürich
Lengghalde 2
8008 Zürich
moroder.info@kws.ch
Prof. Moroder ist in der Entwicklung neuer Schulterendoprothesensysteme tätig (Arthrex Inc. und Medacta Corporate).
◆ Schulterschmerzen und Funktionsverlust im Alter sind häufig auf eine primäre oder sekundäre Omarthrose zurückzuführen.
◆ Initial ist ein nicht-operativer Therapieversuch empfohlen mit Physiotherapie, zeitlich limitierter Einnahme von entzündungshemmender Medikation, und Gelenksinfiltration.
◆ Ist die konservative Therapie ausgeschöpft kann durch die Implantation einer Schulterendoprothese eine zuverlässige Verbesserung der Beschwerden erreicht werden.
◆ Eine Abstimmung der Endoprothesenkomponenten auf die individuell vorliegende Art der Omarthrose, Schulteranatomie und Körperhaltung trägt zur Optimierung der klinischen Ergebnisse bei.
1. Tran G, Fascia D, Askew J et al. The prevalence of glenohumeral joint osteoarthritis in a primary care shoulder pain population referred for radiographs. Rheumatology 2021; 61: 1290-1292. doi:10.1093/rheumatology/keab867
2. Oh JH, Chung SW, Oh CH et al. The prevalence of shoulder osteoarthritis in the elderly Korean population: association with risk factors and function. Journal of Shoulder and Elbow Surgery 2011; 20: 756-763. doi:https://doi.org/10.1016/j.jse.2011.01.021
3. Plachel F, Akgün D, Imiolczyk JP et al. Patient-specific risk profile associated with early-onset primary osteoarthritis of the shoulder: is it really primary? Arch Orthop Trauma Surg 2021. doi:10.1007/s00402-021-04125-2. doi:10.1007/s00402-021-04125-2
4. Ibounig T, Simons T, Launonen A et al. Glenohumeral osteoarthritis: an overview of etiology and diagnostics. Scandinavian Journal of Surgery 2020; 110: 441-451. doi:10.1177/1457496920935018
5. Boselli KJ, Ahmad CS, Levine WN. Treatment of Glenohumeral Arthrosis. The American Journal of Sports Medicine 2010; 38: 2558-2572. doi:10.1177/0363546510369250
6. Metzger CM, Farooq H, Merrell GA et al. Efficacy of a single, image-guided corticosteroid injection for glenohumeral arthritis. Journal of Shoulder and Elbow Surgery 2021; 30: 1128-1134. doi:10.1016/j.jse.2020.08.008
7. Merolla G, Sperling JW, Paladini P et al. Efficacy of Hylan G-F 20 versus 6-methylprednisolone acetate in painful shoulder osteoarthritis: a retrospective controlled trial. MUSCULOSKELETAL SURGERY 2011; 95: 215-224. doi:10.1007/s12306-011-0138-3
8. Colen S, Geervliet P, Haverkamp D et al. Intra-articular infiltration therapy for patients with glenohumeral osteoarthritis: A systematic review of the literature. Int J Shoulder Surg 2014; 8: 114-121. doi:10.4103/0973-6042.145252
9. Rossi LA, Piuzzi NS, Shapiro SA. Glenohumeral Osteoarthritis: The Role for Orthobiologic Therapies: Platelet-Rich Plasma and Cell Therapies. JBJS Reviews 2020; 8: e0075. doi:10.2106/jbjs.Rvw.19.00075
10. Richardson SS, Schairer WW, Sculco TP et al. Comparison of Infection Risk with Corticosteroid or Hyaluronic Acid Injection Prior to Total Knee Arthroplasty. J Bone Joint Surg Am 2019; 101: 112-118. doi:10.2106/jbjs.18.00454
11. Bennell KL, Buchbinder R, Hinman RS. Physical therapies in the management of osteoarthritis: current state of the evidence. Current Opinion in Rheumatology 2015; 27
12. Berliner JL, Regalado-Magdos A, Ma CB et al. Biomechanics of reverse total shoulder arthroplasty. Journal of Shoulder and Elbow Surgery 2015; 24: 150-160. doi:https://doi.org/10.1016/j.jse.2014.08.003
13. Jo YH, Kim DH, Lee BG. When should reverse total shoulder arthroplasty be considered in glenohumeral joint arthritis? Clin Shoulder Elb 2021; 24: 272-278. doi:10.5397/cise.2021.00633
14. Verborgt O, Vanhees M, Heylen S et al. Computer Navigation and Patient-specific Instrumentation in Shoulder Arthroplasty. Sports Medicine and Arthroscopy Review 2014; 22: e42-e49. doi:10.1097/jsa.0000000000000045
15. Moroder P, Akgun D, Plachel F et al. The influence of posture and scapulothoracic orientation on the choice of humeral component retrotorsion in reverse total shoulder arthroplasty. J Shoulder Elbow Surg 2020; 29: 1992-2001. doi:10.1016/j.jse.2020.01.089
16. Moroder P, Urvoy M, Raiss P et al. Patient Posture Affects Simulated ROM in Reverse Total Shoulder Arthroplasty: A Modeling Study Using Preoperative Planning Software. Clin Orthop Relat Res 2022; 480: 619-631. doi:10.1097/corr.0000000000002003
Für Patienten ist aufgrund der in der Krankenversicherung vorgesehenen Beteiligung des Patienten an den entstandenen Kosten (Franchise, Selbstbehalt) massgebend, ob die Behandlung seiner Gesundheitsschädigung durch die Krankenversicherung oder die Unfallversicherung übernommen wird. In diesem Artikel wird ein Überblick darüber gegeben, welche Gesundheitsschädigungen durch die Unfallversicherung gedeckt sind.
Due to the patient’s participation in the costs incurred as provided for in the health insurance (franchise, deductible), it is decisive whether the treatment of the health impairment is covered by the health insurance or the accident insurance. This article gives an overview of which health impairments are covered by accident insurance.
Key Words: accident definition, bodily injury similar to that sustained in an accident, occupational disease, causality
Das Bundesgesetz über die Unfallversicherung (UVG) sieht vor, dass Versicherungsleistungen gemäss diesem Gesetz bei Berufs-, Nichtberufsunfällen, Berufskrankheiten oder unfallähnlichen Körperschädigungen gewährt werden (Art. 6 UVG). Die soziale Krankenversicherung gewährt hingegen Leistungen bei Krankheit, soweit dafür keine Unfallversicherung aufkommt auch bei Unfall sowie bei Mutterschaft (Art. 1a des Bundesgesetzes über die Krankenversicherung; KVG).
Unfall
Wann von einem Unfall im Sinne des Unfallversicherungsrechts auszugehen ist, wird im Bundesgesetz über den Allgemeinen Teil des Sozialversicherungsrechts (ATSG) definiert. Als Unfall gilt die plötzliche, nicht beabsichtigte, schädigende Einwirkung eines ungewöhnlichen äusseren Faktors auf den menschlichen Körper, die eine Beeinträchtigung der körperlichen, geistigen oder psychischen Gesundheit oder den Tod zur Folge hat (Art. 4 ATSG). Demgegenüber ist Krankheit jede Beeinträchtigung der körperlichen, geistigen oder psychischen Gesundheit, die nicht Folge eines Unfalls ist und die eine medizinische Untersuchung oder Behandlung erfordert oder eine Arbeitsunfähigkeit zur Folge hat (Art. 3 Abs. 1 ATSG). Ob der Unfallbegriff erfüllt ist, ist eine Rechtsfrage und deshalb von der Rechtsanwendung zu beantworten, es handelt sich dabei um keine medizinische Frage (1). Um einen Unfall gemäss Gesetz anerkennen zu können, müssen alle aufgeführten Kriterien erfüllt sein. Insbesondere im Zusammenhang mit der Frage, ob ein ungewöhnlicher äusserer Faktor gegeben ist, besteht eine reiche Rechtsprechung des Bundesgerichts (2). In der ärztlichen Arbeit ist die Beurteilung, ob ein Unfall im Sinne des Gesetzes vorliegt, häufig schwierig, sodass der Begriff des Unfalls vermieden werden sollte.
Aus der Definition des Unfalles ist ersichtlich, dass auch eine gewisse Verbindung zwischen dem Unfallereignis sowie der Gesundheitsschädigung gegeben sein muss. Das Bundesgericht und die juristische Lehre definieren dazu, dass eine Leistungspflicht eines Unfallversicherers gegeben ist, wenn zwischen dem Unfallereignis und dem Gesundheitsschaden ein natürlicher, wie auch ein adäquater Kausalzusammenhang vorhanden ist (3).
Mit der natürlichen Kausalität wird der tatsächlich erklärbare Zusammenhang zwischen Ursache und Beschwerdebild geprüft. Dabei werden naturwissenschaftliche und technische Aspekte diskutiert. Es handelt sich um eine medizinische beziehungsweise naturwissenschaftliche Fragestellung. Das Bundesgericht hält hierzu fest, dass Ursachen im Sinne des natürlichen Kausalzusammenhangs alle Umstände sind, ohne deren Vorhandensein der eingetretene Erfolg nicht als eingetreten oder nicht als in der gleichen Weise bzw. nicht zur gleichen Zeit eingetreten gedacht werden kann. Entsprechend dieser Umschreibung ist für die Bejahung des natürlichen Kausalzusammenhangs nicht erforderlich, dass ein Unfall die alleinige oder unmittelbare Ursache gesundheitlicher Störungen ist; es genügt, dass das schädigende Ereignis zusammen mit anderen Bedingungen die körperliche oder geistige Integrität der versicherten Person beeinträchtigt hat, der Unfall mit anderen Worten nicht weggedacht werden kann, ohne dass auch die eingetretene gesundheitliche Störung entfiele (3).
Dieser natürliche Kausalzusammenhang muss mit dem Beweisgrad der überwiegenden Wahrscheinlichkeit nachgewiesen sein. Die blosse Möglichkeit eines Zusammenhanges genügt nicht, damit der Unfallversicherer leistungspflichtig wird (3). Nach dem Beweismass der überwiegenden Wahrscheinlichkeit gilt ein Beweis als erbracht, wenn für die Richtigkeit der Sachbehauptung nach objektiven Gesichtspunkten derart gewichtige Gründe sprechen, dass andere denkbare Möglichkeiten vernünftigerweise nicht massgeblich in Betracht fallen (4). Das Gericht hat mit anderen Worten jener Sachverhaltsdarstellung zu folgen, die es von allen möglichen Geschehensabläufen als die wahrscheinlichste würdigt (5).
Als Abgrenzung dazu ist die adäquate Kausalität zu sehen. Diese wird geprüft, wenn als Folge eines Unfalls kein organisch nachweisbarer Befund vorliegt und trotzdem weiterhin Beschwerden beklagt werden (6). Als Beispiel können persistierende Nackenbeschwerden ohne Nachweis einer strukturell objektivierbaren Veränderung durch eine Röntgen- oder MRT-Untersuchung nach einem leichten Heckauffahrunfall erwähnt werden. Gegenüber der natürlichen Kausalität handelt es sich nicht um eine logische Kausalitätstheorie, sondern um eine wertende Zurechnungstheorie. Entsprechend ist die Frage nach dem adäquaten Kausalzusammenhang im Gegensatz zur Frage nach dem natürlichen Kausalzusammenhang eine juristische Fragestellung (1). Ein Ereignis gilt gemäss Rechtsprechung dann als adäquate Ursache eines Erfolges, wenn die betreffende Ursache nach dem gewöhnlichen Lauf der Dinge und der allgemeinen Erfahrung geeignet ist, den eingetretenen Erfolg zu bewirken, sodass der Eintritt des Erfolgs als durch die fragliche Tatsache allgemein begünstigt erscheint (3). Das Bundesgericht hat zwei verschiedene Kriterienkataloge entwickelt, die die Rechtsanwendung je nach vorliegender Situation bei der Frage nach dem adäquaten Kausalzusammenhang zu prüfen hat (7).
Auch wenn ein Unfallversicherer seine Leistungspflicht bejaht hat, bedeutet dies nicht in jedem Fall eine lebenslange Leistungsübernahme. Ein Unfallversicherer hat beispielsweise seine Leistungen einzustellen, wenn zwischen den Beschwerden einer versicherten Person und dem Unfallereignis kein natürlicher Kausalzusammenhang mehr gegeben bzw. dieser mit überwiegender Wahrscheinlichkeit weggefallen ist. Dabei sind insbesondere die Konstellationen zu erwähnen, bei welchen das Ereignis ein Körperteil schädigt, welches bereits von einem, häufig der versicherten Person unbekannten, Vorzustand (Abnützung oder Krankheit) betroffen ist. Damit die Leistungspflicht zeitlich unbeschränkt besteht, ist bei gegebenem Vorzustand eine dauernde oder richtunggebende Verschlimmerung als Folge des Unfalls notwendig. Sobald ein Gesundheitszustand denjenigen erreicht, wie er unmittelbar vor dem Unfall bestanden hatte (Status quo ante) oder wie er sich nach dem schicksalsmässigen Verlauf eines krankhaften Vorzustandes auch ohne Unfall früher oder später eingestellt hätte (Status quo sine), hat der Unfallversicherer seine Leistungen einzustellen (8). Als Beispiel für eine dauernde oder richtunggebende Verschlimmerung kann ein Sturz auf ein Knie genannt werden, nach welchem eine Gonarthrose festgestellt, jedoch auch eine vordere Kreuzbandruptur diagnostiziert wird. Mit und ohne operative Versorgung ist davon auszugehen, dass durch die vordere Kreuzbandruptur der Verlauf einer Gonarthrose negativ beeinflusst wird. Demgegenüber ist von einer zeitlich begrenzten Verschlimmerung auszugehen, falls durch eine Kontusion ohne zusätzliche Band- oder Meniskusläsion eine Gonarthrose symptomatisch wird. Je nach Heftigkeit der Prellung ist die Unfallversicherung einige Wochen bis wenige Monate leistungspflichtig. Es liegt in der Natur des Verschleissleidens, dass unabhängig eines Unfalls Schmerzen auftreten können.
Unfallähnliche Körperschädigung
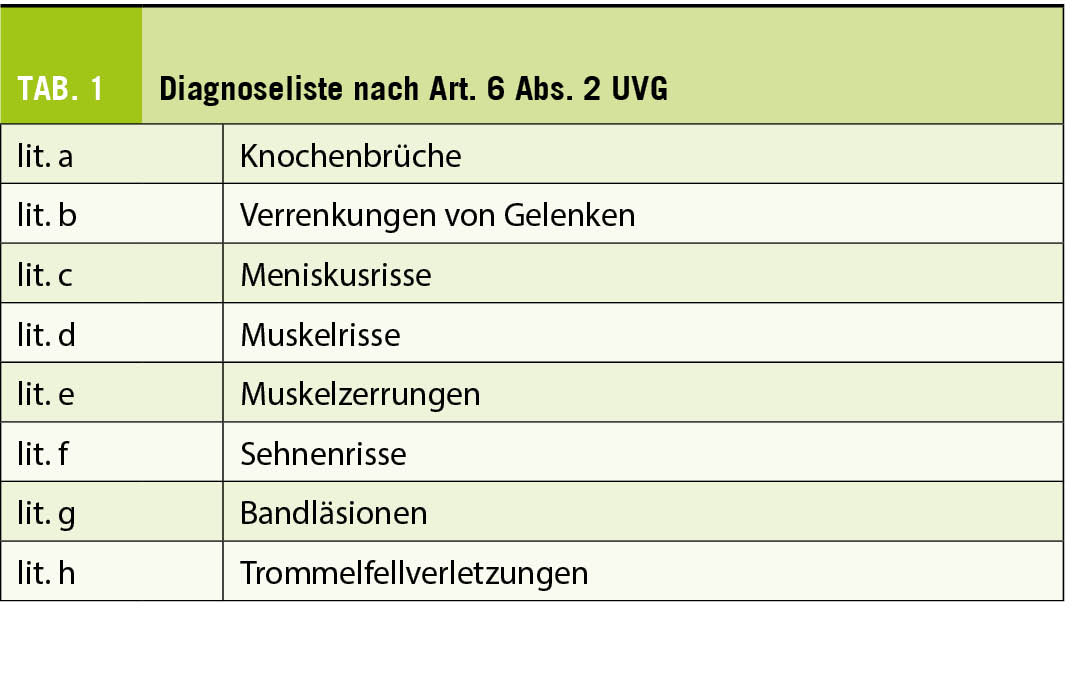
Neben der Gewährung von Versicherungsleistungen bei Berufsunfällen und Nichtberufsunfällen erbringt die Unfallversicherung ihre Leistungen auch bei einer abschliessenden Liste von acht Diagnosen, sofern sie nicht vorwiegend, das heisst zu mehr als 50 % (9), auf Abnützung oder Erkrankung zurückzuführen sind (Art. 6 Abs. 2 UVG) (Tab. 1).
Die Voraussetzung für eine Leistungspflicht ist das Vorliegen einer der genannten Diagnosen. Zentral in der versicherungsmedizinischen Bewertung ist die Auseinandersetzung mit der Pathogenese der im Einzelfall zu diskutierenden Pathologie. Sofern die Betrachtung der Pathogenese zu der Konklusion führt, es bestehe eine vorwiegend auf Abnützung oder Erkrankung zurückzuführende Pathologie, ist die Zuständigkeit des Unfallversicherers zu verneinen (10).
Berufskrankheit
Abschliessend ist der Unfallversicherer bei Berufskrankheiten leistungspflichtig. Hierbei ist zu beurteilen, ob es sich um eine Krankheit handelt, die bei der beruflichen Tätigkeit ausschliesslich oder vorwiegend durch schädigende Stoffe oder bestimmte Arbeiten verursacht worden ist (Art. 9 Abs. 1 UVG). Der Bundesrat hat eine Liste dieser Stoffe und Arbeiten sowie der arbeitsbedingten Erkrankungen erstellt (Anhang 1 zur Verordnung über die Unfallversicherung; UVV). Nach dem Bundesgericht ist eine «vorwiegende» Verursachung von Krankheiten durch schädigende Stoffe oder bestimmte Arbeiten nur dann gegeben, wenn diese mehr wiegen als alle anderen mitbeteiligten Ursachen, mithin im gesamten Ursachenspektrum mehr als 50 % ausmachen. «Ausschliessliche» Verursachung hingegen meint praktisch 100 % des ursächlichen Anteils der schädigenden Stoffe oder bestimmten Arbeiten an der Berufskrankheit (11). Aber auch andere Krankheiten, von denen nachgewiesen wird, dass sie ausschliesslich oder stark überwiegend durch berufliche Tätigkeit verursacht worden sind, gelten als Berufskrankheiten (Art. 9 Abs. 2 UVG). Dabei muss die Berufskrankheit zu mindestens 75 % durch die berufliche Tätigkeit verursacht worden sein. Vor allem diese Krankheiten, welche nicht gelistet sind, führen im Praxisalltag wiederholt zu Diskussionen. Zur Anerkennung einer Berufskrankheit gemäss Art. 9 Abs. 2 UVG ist eine vierfach höhere Inzidenzrate in der spezifischen Berufsgruppe gegenüber der Allgemeinheit gefordert, um das Kriterium der ausschliesslichen oder stark überwiegenden Verursachung durch die berufliche Tätigkeit bejahen zu können (12, 13).
Copyright bei Aerzteverlag medinfo AG
Dr. med. Josef Grab
MAS Versicherungsmedizin
EMBA HSG Insurance und Financial Services
Facharzt für Chirurgie, Mitglied FMH
Suva
Fluhmattstrasse 1
6004 Luzern
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
◆ Die Unfallversicherung übernimmt Leistungen bei Unfällen, unfallähnlichen Körperschädigungen und Berufskrankheiten.
◆ Die Rechtsanwendung ist bei der Prüfung der Leistungspflicht in vielen Fragen auf eine medizinische Beurteilung angewiesen. Insbesondere obliegt es der Ärzteschaft zu Fragen der natürlichen Kausalität Stellung zu nehmen.
◆ Der Unfallbegriff ist im allgemeinen Teil des Sozialversicherungsgesetzes definiert. Ob dieser erfüllt ist, ist eine Rechtsfrage und deshalb von der Rechtsanwendung zu beantworten. Es handelt sich dabei um keine medizinische Frage.
◆ Die Bejahung der Leistungspflicht durch einen Unfallversicherer bedeutet nicht in jedem Fall eine lebenslange Leistungsübernahme.
Rechtsprechung und Literatur:
1. Urteil des Bundesgerichts (BGer) 8C_298/2016 vom 30.11.2016 E. 5.2
2. zum ungewöhnlichen äusseren Faktor: BGE 134 V 72 E. 4.1
3. BGE 129 V 177 E. 3
4. BGE 140 III 610 E. 4.1
5. BGE 138 V 218 E. 6
6. BGE 140 V 356 E. 3.2
7. BGE 115 V 133 und BGE 134 V 109
8. Urteil des BGer 8C_589/2017 vom 21.2.2018 E. 3.1
9. BGE 146 V 51 E. 8.2.2.1
10. Koch H, Henseler S. Zur versicherungsmedizinischen Bewertung des Art. 6 Abs. 2 und der hiermit angegebenen Listendiagnosen im Bundesgesetz über die Unfallversicherung (UVG) der Schweiz. Der medizinische Sachverständige. 2020;116 (4):187-92.
11. BGE 117 V 354 E. 2a
12. BGE 116 V 136 E. 5c
13. Urteil des BGer 8C_746/2012 vom 29.10.2012 E. 5
Niereninsuffizienz ist ein wichtiger kardiovaskulärer Risikofaktor, welcher in der neuesten ESC- und AGLA-Risikobeurteilung direkt unabhängig von anderen Risikofaktoren für ein hohes (eGFR 30-60), respektive sehr hohes Risiko (eGFR <30 ml/min/1.73m2) klassifiziert. Die Früherkennung, Identifikation und Progressionshemmung eines Nierenschadens ist deshalb nicht nur wichtig für die Vermeidung einer Nierenersatztherapie, sondern auch zur Vermeidung einer entsprechenden kardiovaskulären Morbidität und Mortalität. Nach der länger zurückliegenden Einführung der ACE-I und ARB kam zur Progressionsverlangsamung in den 2010er Jahren die Korrektur der Azidose hinzu; in der aktuellen Dekade sind nun weitere vielversprechende Substanzklassen auf dem Markt verfügbar: die SGLT2I und ein nichtsteroidaler Mineralokortikoidrezeptor-Antagonist.
Renal insufficiency is an important cardiovascular risk factor, which in the most recent ESC and AGLA risk assessment is classified as high (eGFR 30-60) or very high risk (eGFR <30 ml/min/1.73m2), independent of other risk factors. Early detection, identification, and progression of renal damage is therefore important not only for avoiding renal replacement therapy but also for preventing related cardiovascular morbidity and mortality. Following the more recent introduction of ACE-I and ARB, correction of acidosis was added to progression slowing in the 2010s; in the current decade, additional promising classes of agents are now available on the market: the SGLT2I and a nonsteroidal mineralocorticoid receptor antagonist
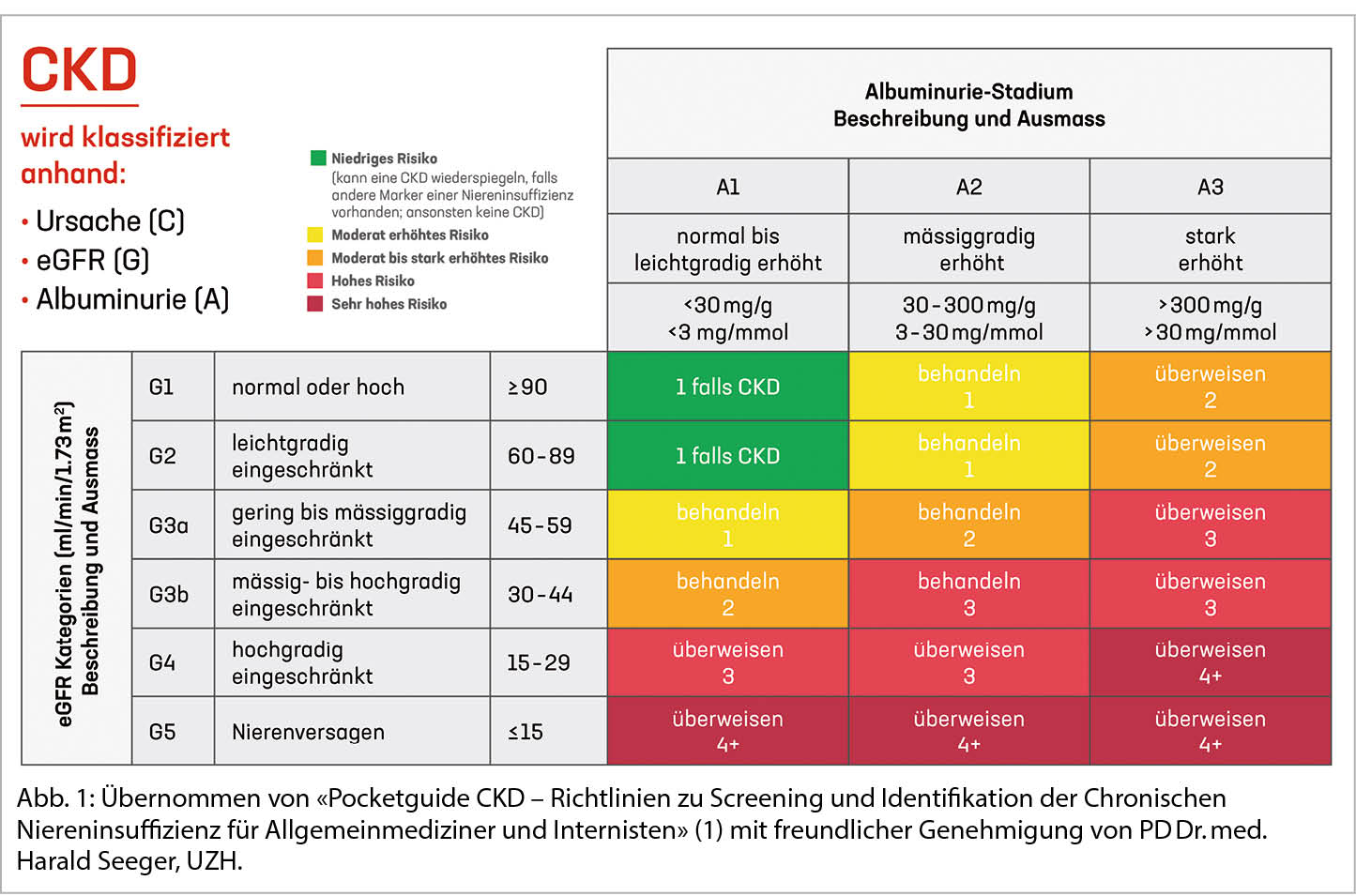
Die Schweizerische Gesellschaft für Nephrologie publizierte neulich einen Pocketguide zum Screening und Identifikation einer CKD. Besonders gefährdete Personen mit arterieller Hypertonie, Diabetes mellitus und Herz Kreislauf Erkrankungen sollten mindestens einmal jährlich auf das Vorliegen einer CKD getestet werden (1). Die CKD Diagnose ist bestätigt, wenn über 3 Monate eine eGFR <60 ml/min/1.73 m2 und/oder eine mässig erhöhte Albuminurie (ACR >30 mg/mmol) nachgewiesen werden kann. Die Abbildung 1 zeigt die CKD-Kategorien anhand von eGFR und Albuminurie, farblich codiert das Risiko für das Fortschreiten der CKD, die Kontrollfrequenz sowie ob eine Überweisung zur nephrologischen Abklärung empfohlen wird.
Da es im Falle einer akuten Nierenschädigung, bei Fieber oder extremer körperlicher Anstrengung zum eGFR-Abfall oder zur reversiblen Proteinurie/Albuminurie kommen kann, wird durch die Frist von mindestens drei Monaten eine frühzeitige CKD-Diagnosestellung vermieden. Bei einem raschen eGFR Verlust oder massiver Proteinurie sollte die Zuweisung zur fachärztlichen Beurteilung jedoch nicht verzögert werden.
Proteinurie und Albuminurie Bestimmung
Traditionell versteht sich unter der Nierenfunktion die glomeruläre Filtration. Die Klassifikation der chronischen Nierenkrankheit basiert jedoch nicht nur auf der renalen Filtrationsfähigkeit, sondern beinhaltet auch den Marker eines glomerulären Schadens, die Albuminurie. Sie ist einer der wichtigsten Prädiktoren für eine Verschlechterung der Nierenfunktion und des Nierenfunktionsverlusts (2). Aus diesem Grund sollte für das Screening und die richtige Einstufung in die CKD-Risiko-Kategorie immer eine Albuminurie quantifiziert werden. Da die 24h Urinsammlung für Patienten aufwändig und oft wegen ungenauer Sammlung schwer zu interpretieren ist, ist empfohlen, die Albuminurie in einer Urinportion gleichzeitig mit Urin-Kreatinin zu messen. Die Albumin-Kreatinin Ratio liefert eine für den klinischen Alltag gut anwendbare Abschätzung der Albuminurie und kann aufgrund der konstanten täglichen Kreatininausscheidung auch zur Verlaufsbeurteilung hinzugezogen werden. Im Falle einer konsistent stark erhöhten Albuminurie (ACR >300mg/mmol) wird eine Überweisung an Nephrologen/in empfohlen.
Bei Abklärung einer Nierenerkrankung lohnt sich immer auch, die gesamt Proteinurie (PCR – Protein-Kreatinin Ratio) analog zur Albumin-Kreatinin Ratio zu bestimmen. Eine grosse Abweichung von diesen zwei Werten weist auf eine nicht-glomeruläre Proteinurie hin, die weiter abgeklärt werden sollte (vor allem wenn PCR >> ACR und PCR >100 mg/mmol).
Metabolische Azidose
Die Aufrechterhaltung des Säuren-Basen-Haushalts ist eine der wichtigsten Rollen der Nieren. Bei chronischer Nierenerkrankung resultiert die verminderte Fähigkeit Säuren auszuscheiden zu einer positiven H+ Bilanz. Der Grund dafür ist vor allem die verminderte renale Ammoniak Ausscheidung; die Ausscheidung von titrierbarer Säure ist erst bei stark eingeschränkter Nierenfunktion (eGFR <15 ml/min/1.73 m2) reduziert (3). Die Prävalenz einer metabolischen Azidose steigt bei eGFR Abnahme <40 ml/min/1.73 m2 (4), wenn die kompensatorische Ammoniak Produktion der verbleibenden Nephrone nicht mehr ausreicht.
Eine metabolische Azidose ist bei CKD-Patienten mit einem erhöhten Todesrisiko sowie anderen negativen Outcomes verbunden (Fortschreiten der CKD, Knochen- und Muskelabbau, Hyperkaliämie). Aus diesem Grund wird gemäss Richtlinien vorgeschlagen, bei CKD eine Behandlung mit oralem Bicarbonat zu etablieren um die Serum Bicarbonat Konzentration >22 mmol/l aufrechtzuhalten (5). Der Benefit einer Behandlung basiert jedoch nur auf Daten kleinerer Studien, es fehlt immer noch an guter Evidenz von grossen randomisierten klinischen Studien. Einige Observationsstudien weisen sogar auf eine U Beziehung zwischen Serum Bicarbonat und Mortalität hin, sodass übermässig hohe Bicarbonat-Werte vermieden werden sollten (6).
Neue renoprotektive Pharmakotherapie
SGLT2I
Der Durchbruch von Hemmern des Natrium-Glukose Transporters 2 (SGLT2I) änderte in den letzten Jahren die Richtlinien in vielen Fachdisziplinen; die Praxisänderung ist vergleichbar mit dem Einzug der Angiotensin-Konvertase Hemmer und Angiotensin-Rezeptorblocker. Bei Personen mit Diabetes mellitus Typ 2 (T2D), CKD und GFR >30 ml/min/1.73 m2 werden SGLT2I als Erstlinientherapie zusammen mit Metformin universell empfohlen (5). Der protektive Effekt von SGLT2I scheint auch bei eGFR <30 ml/min/1.73 m2 erhalten zu bleiben (obwohl im kleineren Ausmass) – im Gegenteil zu Metformin, müssen SGLT2I bei CKD Progression deshalb nicht abgesetzt werden. Der kardiovaskuläre und renoprotektive Effekt von SGLT2I ist durch die bessere Diabetes Einstellung alleine nicht erklärbar. So zeigte die DAPA-CKD Studie, dass auch Patienten ohne T2D profitieren: in dieser Studie wurden Patienten mit CKD (Durchschnitts eGFR 43 ml/min/1.73 m2, median ACR 107 mg/mmol) mit oder ohne T2D (32.5%) für entweder Dapagliflozin oder Placebo randomisiert. Nach einem medianen Follow-up von 2.4 Jahren wurde die Studie wegen klarer Wirksamkeit frühzeitig gestoppt. Dapagliflozin führte zu einem 39% tieferen Risiko (absolute Risikoreduktion 5.3%) des kombinierten primären Endpunktes (eGFR Abnahme um 50%, Nierenfunktionsverlust, Tod aus renalen oder kardiovaskulären Ursachen). Man musste nur 19 Patienten behandeln, um einen primären Endpunkt zu verhindern (Anzahl der notwendigen Behandlungen – NNT (7). Aufgrund dieser überzeugenden Daten wurde Dapagliflozin neulich auch bei Personen mit CKD (eGFR >25 ml/min/1.73 m2) mit/ohne Diabetes als Ergänzungstherapie zu einer maximal tolerierten Dosis von ACE-I oder ARB zugelassen, die Listung auf der Spezialitätenliste wird in Kürze erwartet. Ähnlich wie bei ACE-I oder ARB können SGT2I initial einen leichten eGFR Abfall verursachen; dieser ist meistens funktionell, hämodynamisch bedingt und deshalb reversibel und sollte nicht zum Absetzen von SGLT2I führen. Zu den häufigsten Nebenwirkungen von SGLT2I zählen genitale Mykosen; einige Studien haben auch ein erhöhtes Risiko von Amputation der unteren Extremitäten aufgezeichnet. Eine seltene Komplikation der SGLT2I-Therapie ist die (euglykämische) diabetische Ketoazidose, die vor allem bei Stresszuständen mit einer verminderten Kohlenhydratzufuhr vorkommen kann (CAVE Operationen), weshalb bei SGLT2I, wie auch bei Metformin «sick day rules» gelten und die Therapie vorübergehend pausiert werden sollte.
Finerenon
Der nichtsteroidale Antagonist des Mineralokortikoidrezeptors Finerenon hat sich als ein neues Medikament zur Verzögerung der CKD Progression bei Patienten mit T2D in zwei grossen doppelt-randomisierten Studien bewährt (FIDELIO-DKD und FIGARO-DKD). In FIDELIO-DKD führte Finerenon zu einem um 18% reduzierten Risiko (absolute Risikoreduktion 3.3%) den primären renalen Endpunkt zu erreichen (Nierenversagen, eGFR Abnahme von 40% und Tod aus renaler Ursache). Die Anzahl der notwendigen Behandlungen (NNT) um einen primären Endpunkt zu verhindern beträgt 30 (7). In der am kardiovaskulären Risiko orientierten FIGARO-DKD Studie zeigte die Finerenon Gruppe ein um 13% tieferes Risiko (absolute Risikoreduktion 1.8%) eines kombinierten primären Endpunktes (kardiovaskulärer Tod, nicht-fataler Myokard Infarkt und Schlaganfall oder Hospitalisation für Herzversagen). Die Anzahl der notwendigen Behandlungen (NNT) um einen primären Endpunkt zu verhindern beträgt hier 56 (8). Erwartungsgemäss wurde in der Finerenon Gruppe öfters eine Hyperkaliämie beobachtet. Eine Listung auf der Spezialitätenliste durch das BAG ist noch nicht erfolgt.
Abkürzungen:
ACE-I: Angiotensin-Converting-Enzym-Hemmer
ACR: Albumin-Kreatinin Ratio
ARB: Angiotensin 1-Rezeptorblocker
CKD: Chronic Kidney Disease, chronische Nierenkrankheit
eGFR: estimated Glomerular Filtration Rate
NNT: Number Needed to Treat, Anzahl der notwendigen Behandlungen
PCR: Protein-Kreatinin Ratio
SGLT2I: Hemmer des Natrium-Glukose Transporters 2
T2D: Diabetes mellitus Typ 2
Copyright bei Aerzteverlag medinfo AG
Dr. med. Dusan Harmacek
Assistenzarzt
Klinik für Nephrologie und Transplantationsmedizin
Kantonsspital St. Gallen
Rorschacherstrasse 95
9007 St. Gallen
dusan.harmacek@kssg.ch
Dr. med. Christian Bucher
Leitender Arzt
Klinik für Nephrologie und Transplantationsmedizin
Kantonsspital St. Gallen
Rorschacherstrasse 95
9007 St. Gallen
christian.bucher@kssg.ch
Die Klinik für Nephrologie und Transplantationsmedizin war Untersuchungszentrum der beiden Finerenone-CKD Studien und empfing Beratungshonorare von Astra Zeneca AG. C.B. vertrat die Klinik (Advisoryboard Dapagliflozin), empfing keine Gelder.
◆ Es ist wichtig, das Auftreten einer CKD zu verhindern, eine CKD
frühzeitig zu erkennen und Patienten mit CKD optimal zu betreuen.
◆ Zur Diagnose einer CKD und zum Screening bei Risikopersonen wird neben der eGFR immer eine Albuminurie mitbestimmt.
◆ Etablierte Therapien um das Fortschreiten einer CKD zu verhindern respektive zu verlangsamen: RAAS-Blocker bei ACR >3 mg/mmol
und NaBicarbonat zur Korrektur der Azidose bei Serumbicarbonat
<22 mmol/l.
◆ Neu soll bei eGFR >25 ml/min ein für CKD zugelassener SGLT2I
eingesetzt werden mit oder ohne Diabetes mellitus Typ 2.
◆ Finerenon kann bei Typ 2 Diabetikern erwogen werden.
1. Seeger H, de Seigneux S, Cippà P. Pocketguide CKD – Richtlinien zu Screening und Identifikation der Chronischen Niereninsuffizienz für Allgemeinmediziner und Internisten [Internet]. 2021; [Abgerufen am 06.03.2022].
Erhältlich unter: https://www.swissnephrology.ch/wp/wp-content/uploads/
2021/11/161121_SGN_Pocketguide_CKD_Web_A4_d.pdf
2. Packham DK, Alves TP, Dwyer JP, Atkins R, de Zeeuw D, Cooper M, et al. Relative Incidence of ESRD Versus Cardiovascular Mortality in Proteinuric Type 2 Diabetes and Nephropathy: Results From the DIAMETRIC (Diabetes Mellitus Treatment for Renal Insufficiency Consortium) Database. Am J Kidney Dis. 2012 Jan;59(1):7
5–83.
3. Raphael KL. Metabolic Acidosis in CKD: Core Curriculum 2019. Am J Kidney Dis. 2019 Aug;74(2):263–75.
4. Moranne O, Froissart M, Rossert J, Gauci C, Boffa J-J, Haymann JP, et al.
Timing of Onset of CKD-Related Metabolic Complications. J Am Soc Nephrol. 2009 Jan;20(1):164–71.
5. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management
of Chronic Kidney Disease. Kidney Int Suppl. 2013 Jan;3(1):1–150.
6. Kovesdy CP, Anderson JE, Kalantar-Zadeh K. Association of serum bicarbonate
levels with mortality in patients with non-dialysis-dependent CKD. Nephrol Dial Transplant. 2008 Dec 4;24(4):1232–7.
7. Heerspink HJL, Stefánsson B V., Correa-Rotter R, Chertow GM, Greene T,
Hou F-F, et al. Dapagliflozin in Patients with Chronic Kidney Disease. N Engl
J Med. 2020 Oct 8;383(15):1436–46.
8. Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P, et al. Effect of
Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N Engl
J Med. 2020 Dec 3;383(23):2219–29.
9. Pitt B, Filippatos G, Agarwal R, Anker SD, Bakris GL, Rossing P, et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N Engl
J Med. 2021 Dec 9;385(24):2252–63.