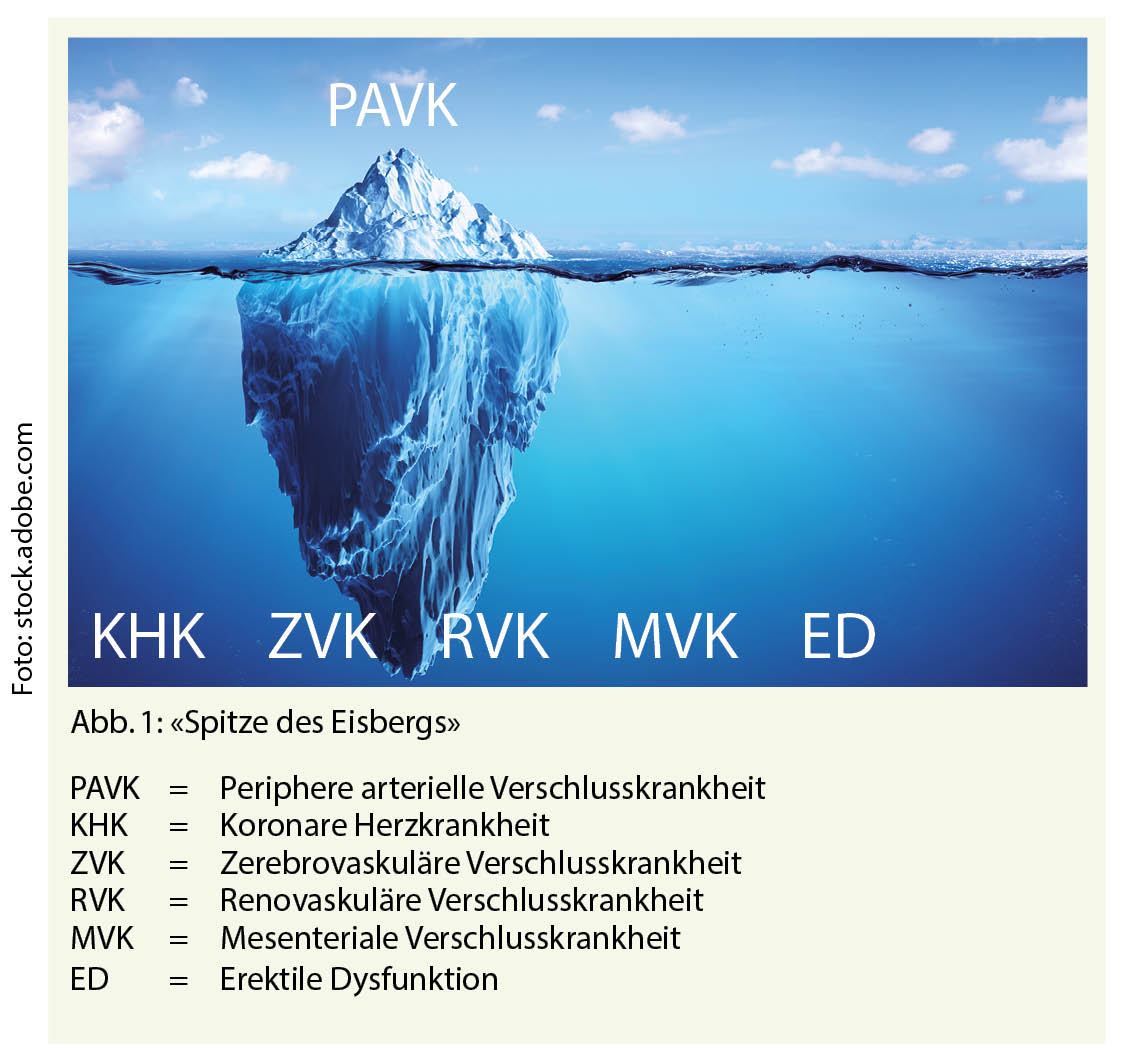
Die periphere arterielle Verschlusskrankheit beschreibt nichts anderes als einen Prozess und seine Lokalisation. Sie ist keine Krankheitsentität. In über 90% steckt eine obliterierende Arteriosklerose dahinter. Die chronische periphere arterielle Verschlusskrankheit ist damit Spitze des Eisberges einer systemischen Erkrankung. Entsprechend muss sie beurteilt und behandelt werden: «Act local, think global». Der globale Aspekt betrifft dabei das Risikofaktorenmanagement, die weiteren Manifestationsorte der obliterierenden Arteriosklerose (zerebrovaskuläre Verschlusskrankheit, koronare Herzkrankheit, renovaskuläre und mesenteriale Verschlusskrankheit, erektile Dysfunktion) sowie die allfällige gleichzeitige dilatative Spielform der Arteriosklerose, d.h. die Aneurysmata. Letztere wird leider nur zu oft vergessen, mit potentiell fatalen Folgen. Beurteilung und Behandlung der peripheren arteriellen Verschlusskrankheit bedeutet ganzheitliche Medizin in ihrem besten Sinne.
Seit 2-3 Monaten weist der 74 jährige Max Muster eine einseitige Wadenclaudicatio nach einer Gehdistanz (ebenaus, in normalem Gehtempo) von 300-400 m auf. Beim Stillstehen verschwinden die Beschwerden nach einer kurzen Zeit von 1-2 Minuten. Beim Bergaufgehen oder Lastentragen, also bei vermehrter Belastung, sind die Beschwerden verstärkt, beim Bergabgehen geringer oder gar nicht vorhanden. Die schmerzfreien Intervalle beim Weitergehen sind bei identischer Belastung relativ konstant und es besteht keine wesentliche Abhängigkeit von einer Tagesform.
Es gibt wohl kaum eine typischere Anamnese in der Medizin als die Claudicatio oder «Schaufensterkrankheit». Allerdings ist die Symptomatik nicht immer lehrbuchmässig, sodass sie manchmal durch gezieltes Befragen herausgeschält werden muss.
Differentialdiagnose
Claudicatio ist aber nicht gleichbedeutend mit peripherer arterieller Verschlusskrankheit. Herr Muster weist also nicht prima vista ein arterielles Problem auf. Die Claudicatio spinalis kann sehr ähnliche Symptome hervorrufen. Zur Unterscheidung helfen nicht selten nur feine Unterschiede in der Anamnese. Der Patient mit einer Spinalkanalstenose hat dabei die Beschwerden oft beim Bergauf- wie Bergabgehen. Er neigt dazu, nicht einfach nur stehenzubleiben, sondern kauert nieder, bückt sich oder setzt sich zumindest auf eine Bank, um den Spinalkanal weit zu machen. Auch verschwinden die Beschwerden in der Regel beim Stillstehen nicht so schnell, die schmerzfreien Intervalle sind gerne variabel und abhängig von der Tagesform. Auch die (eher seltene) Claudicatio venosa verhält sich ähnlich. Bedingt durch eine venöse Abflussbehinderung, z.B. im Bereich der Beckenvenen, kommt es zu einem teils berstenden Spannungsschmerz, der bei Belastungsende dazu tendiert, länger anzuhalten als die Claudicatio arteriosa. Oft haben diese Patientinnen und Patienten das Bedürfnis abzuliegen und die Beine hoch zu lagern.
Max Muster gibt keine Symptome an, die Verdacht auf eine nichtvaskuläre Claudicatio erregen, so dass anamnestisch von einer peripheren arteriellen Verschlusskrankheit ausgegangen werden darf.
Einteilung
In unseren Breitengraden hat sich eher die Einteilung nach Fontaine, im angelsächsischen Sprachraum diejenige nach Rutherford etabliert. Praktisch relevant ist aber in erster Linie die Unterscheidung in Claudicatio oder kritische Ischämie (Ruheschmerzen und Nekrose). Bei ersterer geht es lokal um Lebensqualität, bei letzterer um Extremitätenerhalt.
Ursachen
Die chronische periphere arterielle Verschlusskrankheit ist in weit über 90% bedingt durch eine Arteriosklerose (1). Seltenere Auslöser sind Krankheitsbilder wie Vaskulitiden (z.B. Thrombangiitis obliterans), zystische Adventitiadegeneration, traumatische Gefässverletzungen oder fibromuskuläre Dysplasie. Auf diese und auf die möglichen Ursachen einer akuten Extremitätenischämie wird in diesem Artikel nicht weiter eingegangen.
Epidemiologie
Max Muster ist nicht allein. Immer noch sind die Herz-Kreislaufkrankheiten in der Schweiz und weltweit die häufigste Todesursache (2). Ab einem Alter von 70 Jahren weisen 15-20% der Bevölkerung eine periphere arterielle Verschlusskrankheit auf. Allerdings wird nur etwa ein Drittel davon symptomatisch und Männer sind ca. 4 x häufiger betroffen als Frauen (3).
Risikofaktoren
Die zwei wichtigsten und zugleich nicht behandelbaren Risikofaktoren werden häufig vergessen. Es sind dies Alter und Genetik. Die behandelbaren Katalysatoren des Prozesses der Arteriosklerose sind natürlich Nikotinabusus, arterielle Hypertonie, Hyperlipidämie, Diabetes mellitus und Hyperurikämie. Nikotinabusus ist dabei ein besonders aggressiver Beschleuniger der peripheren arteriellen Verschlusskrankheit (während dies in der Gewichtung für die zerebrovaskuläre Verschlusskrankheit die Hypertonie und für die koronare Herzkrankheit die Hyperlipidämie sind).
Diagnostik
Das wichtigste diagnostische Instrument wurde bereits erwähnt. Es ist dies die Anamnese! Diese ermöglicht wie oben beschrieben eine Weichenstellung hinsichtlich Differentialdiagnose zu einer nichtvaskulären Claudicatio. Sie lässt aber auch eine erste Lokalisationsdiagnostik zu. Die Beschwerden tauchen nämlich immer eine Etage tiefer auf als die Obstruktionslokalisation liegt. So weisen Patienten mit Obstruktion im Bereich der Beckenstrombahn eine Oberschenkelclaudicatio, solche mit Obstruktion im Bereich der Oberschenkelstrombahn eine Waden- und diejenigen mit Unterschenkelarterienobstruktionen eine Fusssohlenclaudicatio auf. Eine Gesässclaudicatio tritt bei Obstruktionen von Aorta, A. iliaca communis oder A. iliaca interna auf.
Die nächsten diagnostischen Schritte umfassen beidseitige Blutdruckmessung (Obstruktion im Bereich der oberen Extremitäten?), Pulspalpation und Auskultation (Gefässströmungsgeräusche). Während die Bestimmung des Knöchel-Arm-Indexes («ankle-brachial-index»; ABI) noch in der Praxis möglich ist, werden die weiteren Untersuchungen in der Regel beim Spezialisten vorgenommen (Oszillographie, Laufbandergometrie, Lichtreflexionsrheographie, transkutane Sauerstoffdruckmessung und vor allem Farbduplexsonographie). Computertomographische Angiographie (CTA) und MR-Angiographie (MRA) sind heute eher selten notwendig, insbesondere wenn eine kathetertechnische Therapie erfolgen soll. Dabei erfolgt die zusätzliche Bildgebung nämlich in Form einer Angiographie im Rahmen des Eingriffes (digitale Subtraktionsangiographie in PTA-Bereitschaft; PTA = Perkutane transluminale Angioplastie). Somit können potentielle Nebenwirkungen von CTA und MRA minimiert werden (Strahlenexposition; Kontrastmittelnebenwirkungen: allergische Reaktion, Nephrotoxizität bei CTA; sog. nephrogene systemische Fibrose bei MRA). Sicher im Patienteninteresse ist auch die Tatsache, dass bei einer Angiographie in PTA-Bereitschaft beim erfahrenen Spezialisten deutlich weniger Kontrastmittel verwendet werden muss als beispielsweise in der Kardiologie (meist unter 50 ml).
Spitze des Eisberges
Herr Muster hat nicht nur ein lokales Problem. Herr Muster hat ein systemisches Leiden. Die periphere arterielle Verschlusskrankheit ist also nur eine Indikatorerkrankung. Entsprechend dürfen die anderen Manifestationsorte der obliterierenden Arteriosklerose nie vergessen werden: Zerebrovaskuläre Verschlusskrankheit, koronare Herzkrankheit, renovaskuläre Verschlusskrankheit, mesenteriale Verschlusskrankheit, erektile Dysfunktion.
Immer wieder wird zudem vergessen, dass die Arteriosklerose neben einer obliterierenden auch eine dilatative Spielform kennt und nicht selten in Kombination auftritt. Der Ausschluss, insbesondere eines Bauchaortenaneurysmas oder eines thorakalen Aneurysmas, ist unbedingt notwendig, zumal sich dieses Damoklesschwert ja meist mittels fehlender Symptomatik zu kaschieren weiss und es sich um eine nichtbelastende, kurze und simple Ultraschalluntersuchung handelt.
Prognose
Was droht Max Muster?
Das Risiko, dass die schmerzfreie Gehstrecke im Verlauf bis hin zu Ruheschmerzen und Nekrose zunimmt, beträgt 25-30%. Das 10-Jahres-Risiko einer Major-Amputation liegt bei 2-3%. Hätte Herr Muster bereits eine kritische Extremitätenischämie würde das 1-Jahres-Risiko bereits bei ca. 25% liegen (4, 5).
Herr Musters Risiko eines schweren kardiovaskulären Ereignisses (Myokardinfarkt, apoplektischer Insult) liegt bei 13% (im Vergleich zu 5% bei einer Vergleichspopulation im selben Zeitraum)(6). Mit allen Mitteln, und das ist etwas vom Zentralsten bei diesen Patienten, muss der Patient also vor Hirn- und Herzschlag bewahrt werden. Aus diesem Grunde rate ich immer zu einer Farbduplexsonographie der extrakraniellen Hirngefässe und zu einer kardiologischen Standortbestimmung.
Therapie («act local, think global»)
Prinzipiell stehen Herrn Muster drei therapeutische Wege offen: Konservatives Vorgehen, Kathetertechnik (PTA) oder Gefässchirurgie. Nochmals: Es handelt sich um einen Eingriff zur Verbesserung der Lebensqualität, also einen fakultativen Eingriff («man kann»). Dies im Gegensatz zur obligaten Behandlung bei kritischer Ischämie («man muss»). Obwohl die Langzeitresultate bezüglich Funktion und Lebensqualität zwischen Kathetertherapie und überwachtem Gehtraining nicht signifikant verschieden scheinen, tendiert man in unserer Konsumgesellschaft zu einer Kathetertherapie als First-line Massnahme, da oftmals das Bedürfnis nach einer schnellen Linderung der Beschwerden besteht und die Kombination von PTA und Gehtraining im Langzeitverlauf doch etwas bessere Ergebnisse ergibt als Gehtraining allein.
Bei Herrn Muster wird also meist eine Angiographie in PTA-Bereitschaft erfolgen und die Obstruktion mittels Ballon allein oder Ballon und Stent behandelt. Dabei können sowohl Ballon wie Stents medikamentenbeschichtet sein, was zu etwas besseren Langzeitresultaten bei allerdings auch höheren Kosten führt. Hilfsmittel während des Eingriffes können Thrombektomie- oder Endarterektomiekatheter sein, vor allem bei komplexen Läsionen oder langstreckigen Verschlüssen. Scheitert der kathetertechnische Behandlungsversuch kann immer noch ein gefässchirurgisches Verfahren (Endarterektomie; Bypasschirurgie) diskutiert werden, wobei bei einer Claudicatio wegen des Kalibers des Eingriffes im Gegensatz zu einer kritischen Ischämie noch grössere Zurückhaltung gefordert ist als beim Kathetereingriff.
Von überragender Bedeutung aber ist die systemische Therapie des Patienten, die auf den ganzen Gefässbaum abzielt.
Die lebenslange thrombozytenaggregationshemmende Therapie bei symptomatischer Arteriosklerose wird heute als unbestritten angesehen. Acetylsalicylsäure 100 mg/die ist bei uns die weitverbreitete Behandlung, obwohl wahrscheinlich 75mg/die ausreichend wären. Eine Alternative stellt Clopidogrel 75 mg/die dar, was insbesondere bei Patienten mit peripherer arterieller Verschlusskrankheit sogar etwas wirksamer zu sein scheint (7). Eine orale Antikoagulation ist nur dann berechtigt, wenn eine andere Indikation dazu besteht (z.B. Vorhofflimmern, thromboembolisches Krankheitsbild) (8). Eine Kombination von Acetylsalicylsäure 100 mg/die und Rivaroxaban 2 x 2,5 mg/die stellt neuerdings eine Erweiterung des therapeutischen Armamentariums bei arteriosklerotischen Hochrisikopatientinnen und -patienten dar (9, 10).
Von grosser Bedeutung ist last but not least die Behandlung der Risikofaktoren. Die Grundlagen, die von den Betroffenen, weil anstrengend, gerne übersprungen werden, sind natürlich die nichtmedikamentösen Massnahmen wie Nikotinstopp, gesunde Ernährung, Gewichtsreduktion und körperliche Aktivität. Es wäre eine grundsätzliche ethische und gesundheitspolitische Diskussion wert, ob solche Massnahmen vor Beginn von pharmakologischen Therapien bei den Patienten bis zu einem gewissen Masse nicht eingefordert werden sollten. Das vorwiegend pharmakologische Risikofaktorenmanagement betrifft die arterielle Hypertonie (Zielblutdruck < 140/90 mmHg), den Diabetes mellitus (Ziel-HbA1c < 7%) und die Hyperlipidämie (Ziel-LDL C < 1,4 mmol/l) (11).
Der Patient mit peripherer arterieller Verschlusskrankheit benötigt wie alle arteriosklerotischen Patienten eine langfristige Begleitung und Führung, frei nach dem Prinzip «gouverner, c’est prévoir». Herr Muster wird Ihnen also erhalten bleiben.
Ich danke Herrn Dr. med. N. Wahli, Mellingen, für die kritische Durchsicht des Manuskriptes.
Copyright bei Aerzteverlag medinfo AG
Innere Medizin und Angiologie FMH
HerzGefässKlinik Bethanien
Toblerstrasse 51
8044 Zürich
info@angio.ch
Der Autor hat in Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
1. Aboyans V, Ricco JB, Bartelink MEL, Björck M, Brodmann M, Cohnert T,
Collet JP; 2017 ESC Guidelines on the Diagnosis and Treatment of Peripheral
Arterial Diseases. The Task Force for the Diagnosis and Treatment of Peripheral Arterial Diseases of the European Society of Cardiology (ESC) and of the European Society for Vascular Surgery (ESVS). Eur Heart J. 2018 Mar 1; 39(9):763–816.
2. www.bag.admin.ch/bag/de/home.html
3. Criqui MH, Fronek A, Barrett-Connor E, Klauber MR, Gabriel S, Goodman D. The prevalence of peripheral arterial disease in a defined population. Circulation. 3/1985 1985; 71(3):510–5.
4. Cambou JP, Aboyans V, Constans J, Lacroix P, Dentans C, Bura A. Characteristics and outcome of patients hospitalised for lower extremity peripheral artery disease in France: the COPART Registry. European journal of vascular and endovascular surgery: the official journal of the European Society for Vascular Surgery. May 2010;39(5):577–85.
5. Adam DJ, Beard JD, Cleveland T, et al. Bypass versus angioplasty in severe ischaemia of the leg (BASIL): multicentre, randomised controlled trial. Lancet. Dec 3 2005;366(9501):1925–34.
6. Sigvant B, Lundin F, Wahlberg E. The risk of disease progression in peripheral arterial disease is higher than expected: a meta-analysis of mortality and
disease progression in peripheral arterial disease. Eur J Vasc Endovasc Surg. 2016;62:1642–51.
7. Gerhard-Herman MD, Gornik HL, Barrett C, et al. 2016 AHA/ACC Guideline on the Management of Patients With Lower Extremity Peripheral Artery Disease:
A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2017 Mar 21;135 (12):
e726–e779.
8. Rastan A, Dopheide J, Baumgartner I. Claudicatio intermittens bei peripherer
arterieller Verschlusskrankheit: Teil 1. Swiss Medical Forum 2020; 20(49-50): 724-728.
9. Branch KR, Probstfield JL, Eikelboom JW, et al. Rivaroxaban With or Without
Aspirin in Patients With Heart Failure and Chronic Coronary or Peripheral Artery Disease. Circulation. 2019;140:529–37.
10. Anand SS, Eikelboom JW, Dyal L, et al. Rivaroxaban Plus Aspirin Versus Aspirin in Relation to Vascular Risk in the COMPASS Trial. J Am Coll Cardiol 2019; 73:3271–80.
11. www.agla.ch/de