Weltweit treten bei einem Sechstel aller Paare Unfruchtbarkeitsfaktoren auf. Bei der Hälfte ist der Mann betroffen, die genaue Ursache bleibt dabei bei 40% unbekannt. Die Samenqualität kann von zahlreichen Faktoren beeinflusst werden, wie ein ungünstiger Energiestoffwechsel, ein unvorteilhaftes Milieu und/ oder oxidativer Stress, Rauchen, Adipositas u.a.m. Ein Schutzeffekt ist für die Antioxidantien Vitamin C, Selen, Zink, Coenzym Q10 und L-Carnitin am besten untersucht.
Der therapeutische Effekt von antioxidativer Nahrungsergänzung wie Proxeed®Plus auf die Spermienqualität bei männlicher Infertilität wurde in zahlreichen randomisierten klinischen Studien erwiesen. Jedoch sind Daten aus dem mitteleuropäischen Umfeld untervertreten. In einer Schweizer Studie wurde der therapeutische Effekt einer gezielten antioxidativen Nahrungsergänzung (Proxeed® Plus) auf die Spermienqualität von Männern mit Oligoasthenoteratospermie und unerfülltem Kinderwunsch (Temime RB et al. Clin Res Trials, 2020; (7)1-6) untersucht im Hinblick auch die Auswirkungen einer Vitamin- und Antioxidantien-Supplementierung auf Spermaparameter und Schwangerschaftsraten.
Experimentelles
Studiendesign
Die prospektive Beobachtungsstudie über 15 Monate verglich das Spermiogramm vor und nach 3-monatiger Supplementation mit Proxeed®Plus, mit Probenahme nach 3–5-tägiger Abstinenz. Statistisch signifikant wurden p-Werte < 0.05 eines Wilcoxon-Rang-summen-Tests definiert. Ziel der Studie war, die Untersuchung des therapeutischen Effekts von gezielter antioxidativer Nahrungsergänzung (Proxeed® Plus) auf die Spermienqualität von Männern mit Oligoasthenoteratozoospermie und unerfülltem Kinderwunsch.
Studienmedikation
Die Studienmedikation (Proxeed®Plus) bestand aus: L-Carnitin (1000 mg), Fructose (1 g), Zitronensäure (50 mg), Coenzym Q10 (20 mg), Vitamin C (90 mg), Zink (10 mg), Folsäure (200 μg), Vitamin B12 (1.5 μg), Selen (50 μg).
Patientencharakteristika
Der BMI war im Durchschnitt 25.9 mit 17.3% adipösen Patienten. 71.1% der Paare hatten eine primäre und 32.6% eine sekundäre Unfruchtbarkeit. 42.3% der Partnerinnen hatten selbst eine verminderte Fruchtbarkeit. Anamnetisch diagnostiziert war Diabetes in 7.6% und Asthma in 5.7% der Fälle. 23% der Studienteilnehmer hatten Varikozele, 9.6% operierte Leistenhernien und 7.6% eine Orchitis-Anamnese.
Studien-Endpunkte
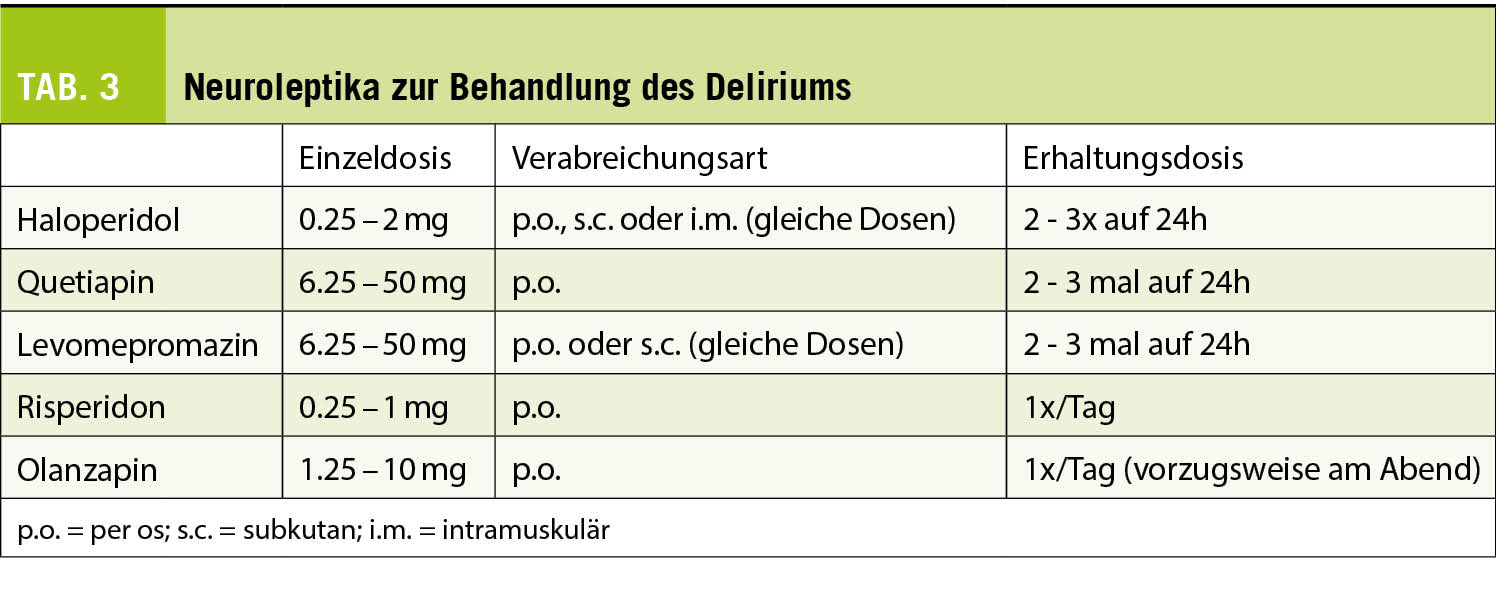
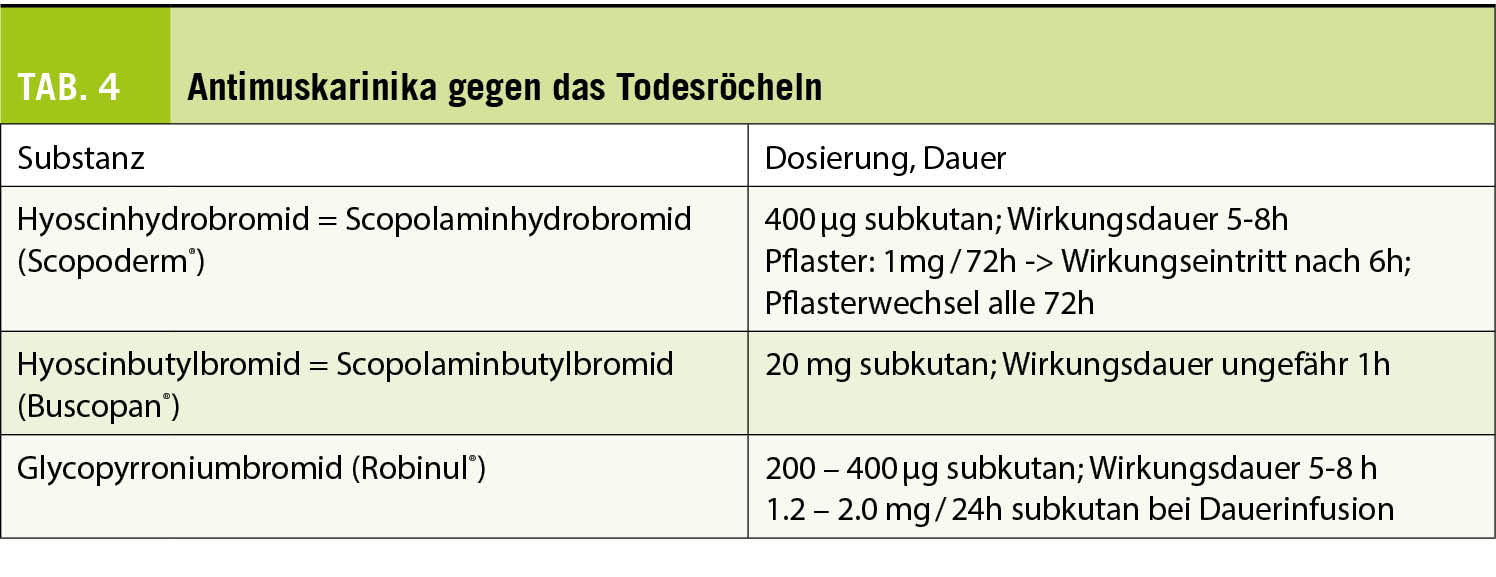
Primärer Endpunkt: Veränderung der Spermienqualität nach Proxeed®Plus-Supplementation gemäss WHO-Kriterien 2010.
Sekundärer Endpunkt: Schwangerschaftsraten nach Proxeed®Plus Supplementation.
Resultate und Diskussion
Nach gründlichen urologischen und genetischen Untersuchungen (11 bakterielle Infektionen mit Antibiotika-Verordnung, zwei neue Varikozele-Diagnosen, ein Fall von CFTR-Polymorphismus) blieben 34.6% der Unfruchtbarkeiten idiopathisch.
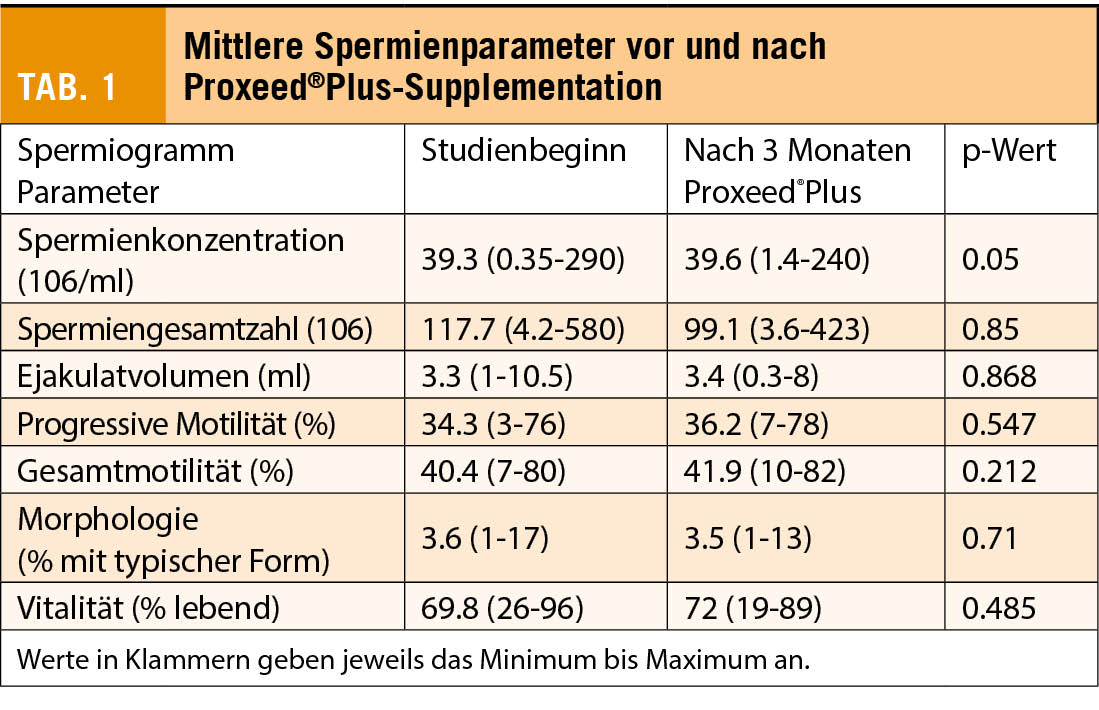
Die Einnahme von Proxeed®Plus über 3 Monate führte zu einer Normalisierung der Spermienkonzentration bei 23% der Patienten und der progressiven Motilität bei 22.1% der Patienten.
Über die gesamte Studienpopulation gesehen, ergab die statistische Analyse eine signifikant erhöhte Spermienkonzentration (p = 0.05 (Tab. 1). Eine Subgruppenanalyse zeigte, dass bei idiopathischer Unfruchtbarkeit der therapeutische Effekt von Proxeed®Plus am grössten war (n = 18), verglichen mit Patienten mit Varikozelen (n = 12) oder Infektionen (n = 11). Bei der Spermienkonzentration trat bei 61.1% eine Verbesserung auf.
Die progressive Motilität verbesserte sich bei 50% der Patienten. Die begrenzte Stichprobenzahl erlaubt keine statistische Analyse.
Während den 15 Studienmonaten traten 16 Schwangerschaften auf (30.7% aller Paare), davon 5 spontan und 11 mittels assistierter Reproduktionstechniken. Die Verträglichkeit der Proxeed®Plus-Supplementation war gut: Es trat nur ein Fall von gastrointestinaler Verstimmung (1.9%) auf.
Fazit
Proxeed®Plus ist empfohlen für Männer in der Familienplanung, die ihre Fruchtbarkeit optimieren möchten, für Männer, die sich auf eine Behandlung wie IVF, ICSI vorbereiten und eine bestmögliche Spermienqualität anstreben sowie Männer, die durch eine Spermienproduktionsstörung ein eingeschränktes Spermiogramm haben und ihre natürliche Fertilität optimieren möchten. Proxeed®Plus ist nicht zur Diagnose, Behandlung, Heilung oder Vorbeugung von Krankheiten bestimmt.
Quelle: Temime RB, Fahrni AC, Attilah L, Feki A. Variation of sperm parameters after metabolic and antioxidant supplementation in infertility patients with Oligoasthenoteratospermia. Clin Res Trials, 2020; (7)1-6.
riesen@medinfo-verlag.ch