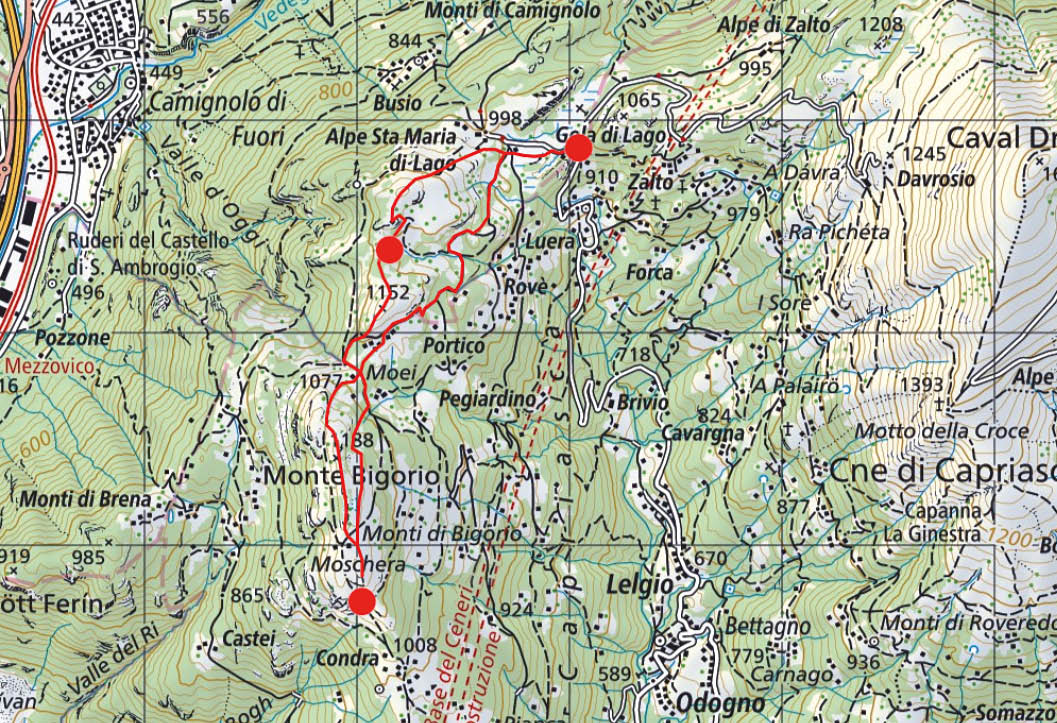
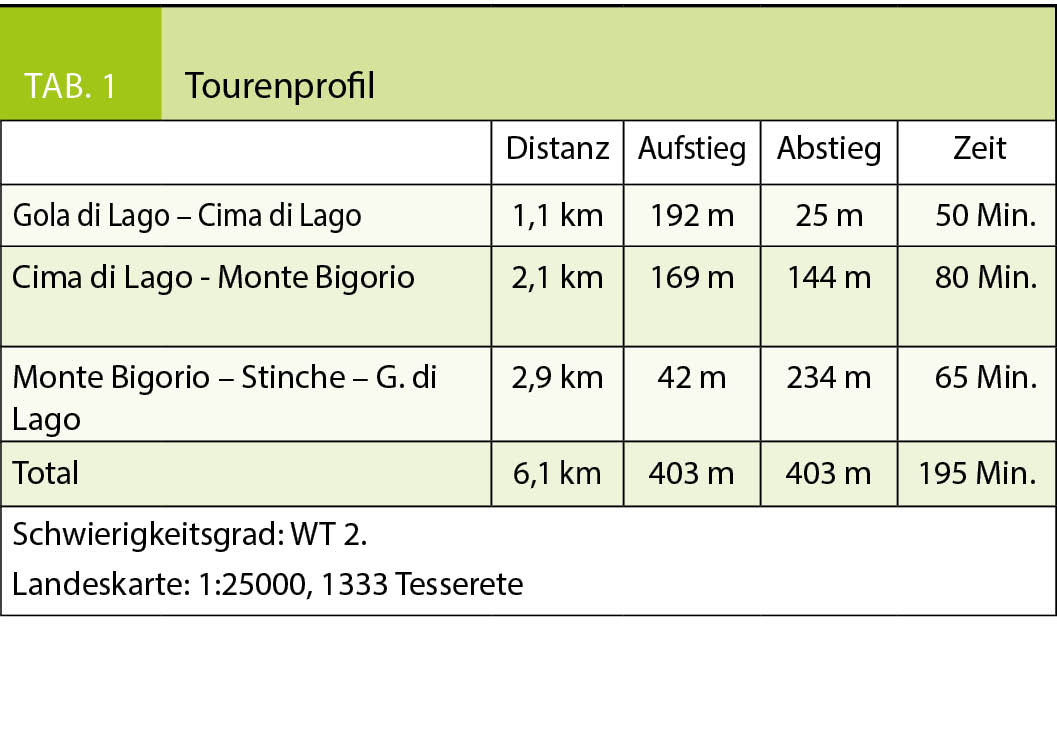
Diese Schneeschuhtour auf den Monte Bigorio entführt uns in eine glazial geprägte, vom Nordföhn oft umtoste Landschaft mit jungen Birkenwäldern, die die weiten ehemaligen Weide- und Anbauflächen der Capriasca allmählich zurückerobern. Ein Seitenarm des Tessingletschers hat nicht nur die Felsformationen am Monte Ceneri, sondern auch in der Val Capriasca glatt geschliffen und an der Gola di Lago einen glazialen Restsee hinterlassen, der mittlerweile zu einem kleinen Hochmoor verlandet ist. Der Westkamm der Val Capriasca stellte bis zum Ende des Kalten Krieges auch eine militärstrategisch wichtige Geländeformation am Passübergang des Monte Ceneri dar und wurde in den zwei Weltkriegen massiv befestigt, wie wir noch sehen werden.
Wir starten unsere Rundtour gleich beim Parkplatz auf der Passhöhe von Gola di Lago und überschreiten in westlicher Richtung den ersten Rundhöcker mit der Höhenquote 994 Meter. Wir durchqueren das kleine Tälchen jenseits des Hügels, vorbei an einem weiteren Parkplatz zum Nordhang der Cima di Lago hinüber. Über diese Flanke, die häufig dank des angreifenden Nordföhns eine eher wenig tiefe Schneedecke aufweist, gewinnen wir den Gipfel (Abb. 1).
Abb. 1: Im Aufstieg zur Cima di Lago mit Blick auf den Monte Bar und Caval Drossa
Auf der südlich des Gipfels gelegenen Felsformation steht ein Denkmal zu Ehren der Soldaten, die seit 1938 in der Grenzbrigade 9 zum Schutz der südlichen Grenze im Tessin gedient haben. Die Einheiten der Brigade und die Namen ihrer Kommandanten sind aufgeführt. Das Denkmal ist als nach Süden offene Schutzhütte mit schrägem Dach gestaltet, deren hoch aufragende Stütze zugleich als Kamin für eine offene Feuerstelle dient. Erst beim Umgehen des Felskopfes realisieren wir, dass das Denkmal auf dem zentralen Infanterieverteidigungswerk im Bereich von Gola di Lago errichtet worden ist. Es besteht aus zwei Kampfstellungen für Maschinengewehre und einem unterirdischen Unterstand mit Notausgang. Zur Sicherung dieses örtlich wichtigen Infanteriebunkers wurde im gegenüberliegenden Hügel ein Stützwerk mit zwei Maschinengewehrstellungen angelegt. Auch diese Anlage wurde mit einem allerdings kleineren Unterstand versehen. Ein Teil des Infanteriehindernisses aus Stacheldraht ist belassen worden, das das vermutlich auch zur Verminung vorgesehene Vorfeld zusätzlich sicherte.
Auf dem Weiterweg überschreiten wir den Matro di Stinche und umgehen im folgenden Sattel die Umzäunung der gleichnamigen Maiensiedlung gegen Westen (Abb. 2).
Abb. 2: Im Birkenwald zwischen der Cima di Lago und dem Matro di Stinche, im Hintergrund der Monte Tamaro
Wir passieren den Sattel gegen Osten, wenden uns bei den ersten Häusern gleich wieder gegen Süden und steigen über den Nordhang des Monte Bigorio zu dessen lang gezogenem Grat hinauf. Auch hier profitieren wir wieder von der Vorarbeit des Nordwindes und erreichen schon kurz nach der Senke wieder eine dünnere, gut spurbare Schneedecke. Es lohnt sich, den höchsten Punkt des Monte Bigorio zu überschreiten und bis zum Geländepunkt 1167.2 Meter südlich der Hütten von Moschera zu queren, da sich dort der Blick auf das gesamte Umland von Lugano und bei klarer Sicht weit über den Ceresio hinaus bis zum Appenin und den Seealpen öffnet. In der Nähe umgibt uns ein verschneiter Kranz von Bergen, von Ost nach West Caval Drossa, Monte Bar, Cima di Fojorina, Denti della Vecchia, Monte Boglia, Sighignola, Monte Generoso, Monte S. Giorgio und die Krete vom Monte Lema bis zum Tamaro. Gegen Norden erheben sich jenseits des Ceneri die Berge um die Valle Verzasca (Abb. 3).
Abb. 3: Ausblick vom Geländepunkt 1167.2 m auf die Verzascheser Berge
Nach ausgiebiger Rast wenden wir uns gegen Norden und folgen der ersten Geländerippe östlich der Krete des Monte Bigorio in eine Bachrinne, auf deren Nordseite wir den Verbindungsweg von den Monti di Cima nach Stinche erreichen. Diesmal umgehen wir die weitläufige Einfriedung von Stinche gegen Osten und benutzen weiter die Trasse des breiten Weges zurück nach Gola di Lago, wobei sich nördlich eines kleinen Hügels mit Bank ein Schlenker des Pfades abkürzen lässt (Abb. 4). Herrlich ist auch hier, wie bereits auf der gesamten Route, das Spiel des warmen Lichts der tief stehenden Sonne und der Schatten der silbern schimmernden Birkenwälder, an dem wir uns nicht satt zu sehen vermögen. Noch lange klingen in uns die leuchtenden Bilder dieses herrlichen Tages nach.
Abb. 4: Routenverlauf
Aufgepasst
In dieser Rubrik werden Berg- und Schneeschuhwanderungen vorgestellt, die in der Regel wenig bekannt sind, zu aussergewöhnlichen Orten führen und die Genugtuung einer besonderen persönlichen Leistung bieten, sei es, dass man sich am Abend nach der Arbeit noch zu einer kleinen körperlichen Anstrengung überwindet, bzw. sich in ein oder zwei Tagen abseits breit getretener Wege unvergessliche Naturerlebnisse erschliesst. Zur besseren Beurteilbarkeit des Schwierigkeitsgrades der Tourenvorschläge wird jeweils eine Einschätzung anhand der SAC-Skala für Berg- (B, EB, BG) und für Schneeschuhwanderungen (WT 1–6) gegeben. Die schwierigste Wegstelle, unabhängig von ihrer Länge, bestimmt jeweils die Gesamtbewertung der Route. Letztendlich bleibt aber jeder selbst für die Beurteilung seiner Fähigkeiten und Eignung für die vorgestellte Wanderung verantwortlich. Die Gehzeiten sind Richtwerte und gelten für normal trainierte Wanderer. Sie müssen nicht zwingend mit den Angaben auf Wegweisern übereinstimmen.
Mit einer Expositionsprophylaxe kann eine HIV-Infektion verhindert werden. In diesem Artikel werden die aktuellen Indikationen und Verfahren der Prophylaxe nach und vor sexueller HIV-Exposition präsentiert und Vor- und Nachteile abgewogen.
Das Humane Immunodefizienz-Virus (HIV) führt zu einer Schwächung des Immunsystems und kann als Spätfolge zu AIDS (Acquired Immunodeficiency Syndrome) führen. Gemäss Schätzungen leben heutzutage in der Schweiz ca. 16 000 Personen mit einer HIV-Infektion. Dank der heutzutage verfügbaren Therapieoptionen kann in den meisten Fällen (je frühzeitiger angewendet, desto besser) eine Immunschwäche zurückgebildet oder sogar ganz verhindert werden. Auch sind Menschen unter einer erfolgreichen antiretroviralen Therapie (Viren nicht nachweisbar seit 6 Monaten) nicht ansteckend (1). Trotz dieser Erfolge kommt es in der Schweiz jährlich immer noch zu ca. 400-500 neu entdeckten HIV-Infektionen (2).
Post-Expositionsprophylaxe (PEP)
Möglichst rascher Beginn der PEP
Eine rechtzeitig durchgeführte Postexpositionsprophylaxe (PEP) kann nach einer sexuellen HIV-Exposition und auch nach einer akzidentellen Verletzung z.B. von Gesundheitspersonal eine Infektion verhindern. Wichtig ist, dass die PEP so rasch wie möglich (ideal innert ein bis zwei Stunden) begonnen wird. Die Erfolgschancen einer PEP sinken bereits nach 6-8 Stunden und nach 48 Stunden lohnt sich eine Gabe nicht mehr. Falls bei der Erstkonsultation des Patienten aufgrund fehlender Informationen das Risiko nicht gut eingeschätzt werden kann, sollte im Zweifel eine PEP mit einer Startdosis eingeleitet werden. Dies gibt Zeit, in Ruhe weitere Informationen einzuholen bzw. Abklärungen zu treffen. Die Patienten können dafür auch an eine spezialisierte PEP-Notfallstelle weitergeleitet werden. Die Adressen von Beratungs- und Teststellen finden Sie unter: https://www.aids.ch/de/was-wir-tun/beratung/beratungs-und-teststellen/.
Wann eine PEP durchführen?
Eine klare Indikation für eine PEP ist ungeschützter Anal- oder Vaginalverkehr mit eine(r) PartnerIn mit bekannter HIV-Infektion ohne antiretrovirale Therapie (sprich nachweisbarer Viruslast im Blut). Bei Oralverkehr mit Ejakulation des HIV-positiven Partners im Mund sollte ebenfalls eine PEP erwogen werden. In vielen Fällen ist der HIV-Status des Partners/Partnerin nicht bekannt. Falls möglich, sollte diese(r) Partner(innen) kontaktiert und ein HIV-Test durchgeführt werden. Eine allenfalls schon begonnene PEP kann gestoppt werden, falls der Partner(in) HIV-negativ getestet wird. Wenn eine Testung des Partners nicht möglich ist, wird die Durchführung einer PEP in den folgenden Fällen empfohlen: nach einer Vergewaltigung, sowie nach ungeschütztem Anal-/Vaginalverkehr mit einem Partner(in), der einer Gruppe mit einer hohen HIV-Prävalenz angehört. In der Schweiz sind dies insbesondere Personen aus HIV-Hochprävalenzländern, Sexarbeiterinnen aus Osteuropa, Männer, die mit Männern Sex haben und Personen, die intravenös Drogen konsumieren. Im Einzelfall können auch weitere Faktoren auf ein erhöhtes Risiko hindeuten und sollten im persönlichen Gespräch besprochen werden. Eine PEP muss nicht durchgeführt werden, wenn der Partner(in) HIV-positiv ist und unter erfolgreicher antiretroviraler Therapie ist, d.h. keine Viren im Blut nachweisbar sind (3).
Ablauf einer PEP
Die eidgenössische Kommission für sexuelle Gesundheit (EKSG) empfiehlt grundsätzlich eine Kombinationstherapie aus Tenofovir/Emtricitabin (Truvada®)1x/d plus Raltegravir (Isentress®) 400 mg 2x/d. Als Alternative zum Raltegravir kann auch Dolutegravir (Tivicay®) 50 mg 1 x /d verwendet werden oder aber mit Ritonavir (Norvir®) 100 mg 1 x /d verstärktes Darunavir (Prezista®) 800 mg. Bei bekannter Resistenzlage des Virus sollten die Medikamente nach Rücksprache mit einer Fachärztin für Infektiologie entsprechend abgestimmt werden.
Vor dem/beim Start der PEP ist es sinnvoll, einen HIV-Test beim Patienten durchzuführen, um eine allfällig bereits bestehende HIV-Infektion auszuschliessen. Weiter wird eine Bestimmung von Blutbild, Leber- und Nierenwerten empfohlen, um einen Ausgangswert zu haben. Grundsätzlich empfiehlt es sich auch, den Hepatitis-B-Status zu überprüfen (Impfschutz, bzw. Ausschluss einer aktiven Hepatitis B mittels HBs-Antigen-Test). Der Grund ist, dass einerseits die Hepatitis B ebenfalls sexuell übertragen wird und daher auch ein Risiko für eine Ansteckung bestehen kann. Andererseits kann es bei aktiver Hepatitis B nach dem Absetzen des Tenofovir (Truvada®) zu einem Hepatitis-Flare kommen. Bei nicht geimpften Personen ist der Beginn einer aktiven Immunisierung angezeigt.
Die Dauer der Prophylaxe für einen maximalen Nutzen ist nicht bekannt, pragmatisch wird empfohlen, diese über 4 Wochen fortzusetzen. Die weiteren klinischen Nachkontrollen verlaufen je nach Problemen und Beschwerden des Patienten (generell nach 2 und 4 Wochen). Laborkontrollen sind nur bei vorhandener klinischer Indikation notwendig. Frühestens 6 Wochen nach Abschluss der PEP sollte ein erneuter HIV-Test (4. Generation -> kombinierter Test Antikörper und p24-Antigen) durchgeführt werden, um eine HIV-Infektion ausschliessen zu können (4).
Eine HIV-PEP ist auch eine sinnvolle Gelegenheit, über weitere sexuell übertragbare Erkrankungen wie Syphilis, Gonorrhoe und Chlamydien zu sprechen bzw. im Verlauf auch zu testen. Eine PEP kann auch ein guter Anlass sein, um mit dem Patienten über eine PrEP zu sprechen.
Prä-Expositionsprophylaxe (PrEP)
Für wen eignet sich eine PrEP?
Die HIV-Prä-Expositionsprophylaxe eignet sich für Personen, die ein erhöhtes Risiko haben, sich mit HIV anzustecken. Dazu gehören z.B. Angehörige einer Gruppe mit hoher HIV- Prävalenz (gemäss WHO > 3%), Analsex mit inkonsistentem Kondomgebrauch, kürzliche STD-Infektion (speziell Syphilis, LGV), Chem-Sex (Sex unter Drogeneinfluss) und St.n. (mehrfacher) PEP. Letztendlich finden sich medizinische, wie auch individuelle, persönliche Kriterien für eine PrEP, die in einer individuellen Beratung besprochen werden sollten und eine gemeinsame Entscheidung getroffen werden soll, ob eine PrEP gestartet werden soll. Keine Indikation für eine PrEP sind Personen in einer monogamen Beziehung mit einer HIV-positiven Person, welche unter ART mind. 6 Monate supprimiert war und Personen in einer monogamen Beziehung mit einer Person, die HIV-negativ getestet wurde (5).
Einnahmemöglichkeiten
Für die PrEP wird die Kombination aus zwei Wirkstoffen (Tenofovir und Emtricitabin (TDF/FTC) angewendet. Die Schutzwirksamkeit ist vergleichbar mit Kondomen (bei korrekter Anwendung) und zeigt eine Risikoreduktion von 44-90% je nach Studie und Anwendungsmodus (Studien gibt es v.a. zur täglichen Einnahme) (6).
Grundsätzlich gibt es zwei Möglichkeiten die PrEP einzunehmen: eine tägliche Einnahme oder event-basiert. Bei der täglichen Einnahme wird 1 Tablette/Tag eingenommen. Der Vorteil ist eine Routine bei der Medikamenteneinnahme; ein konstanter Schutz, verzeiht eher eine vergessene/verspätete Tabletteneinnahme und die meisten Studien wurden mit der täglichen Einnahme durchgeführt. Nachteile sind eine höhere Medikamentenexposition und darum evtl. mehr UAW. Die event-basierte Einnahme bietet sich bei eher wenigen Partnern/zeitlich beschränktem erhöhtem HIV-Risiko an (z.B. Urlaub). Vorteile sind eine niedrigere Medikamentenexposition und damit möglicherweise weniger Nebenwirkungen. Nachteile sind, dass das Schema gut verstanden und korrekt angewendet werden muss, da es weniger Fehler verzeiht und die Schutzwirkung nur sehr eingeschränkt durch Studiendaten belegt ist. Eine event-basierte Einnahme ist nicht möglich bei Frauen (Medikamentenspiegelaufbau langsamer in vaginaler Schleimhaut) und chronische Hepatitis-B-Infektion (mögliches Risiko eines Flare-Ups bei Absetzen).
Abklärungen vor Beginn der PrEP
Vor dem Start einer PrEP muss die Person HIV-negativ getestet worden sein und es sollten folgende Tests durchgeführt werden:
HIV-Serologie, Hepatitis-Serologien (Ausschluss Hep B-Infektion: anti-HBc + anti-HBs (wenn nicht geimpft), HAV-Antikörper (bei unklarem Impfstatus), HCV-Antikörper)
Niere: Kreatinin/eGFR, im Urin Gesamtprotein und Kreatinin
Auch sollte der Impfstatus überprüft werden für Hepatitis A + B, sowie HPV und falls nicht (vollständig) geimpft, sollten die Impfungen noch ergänzt werden.
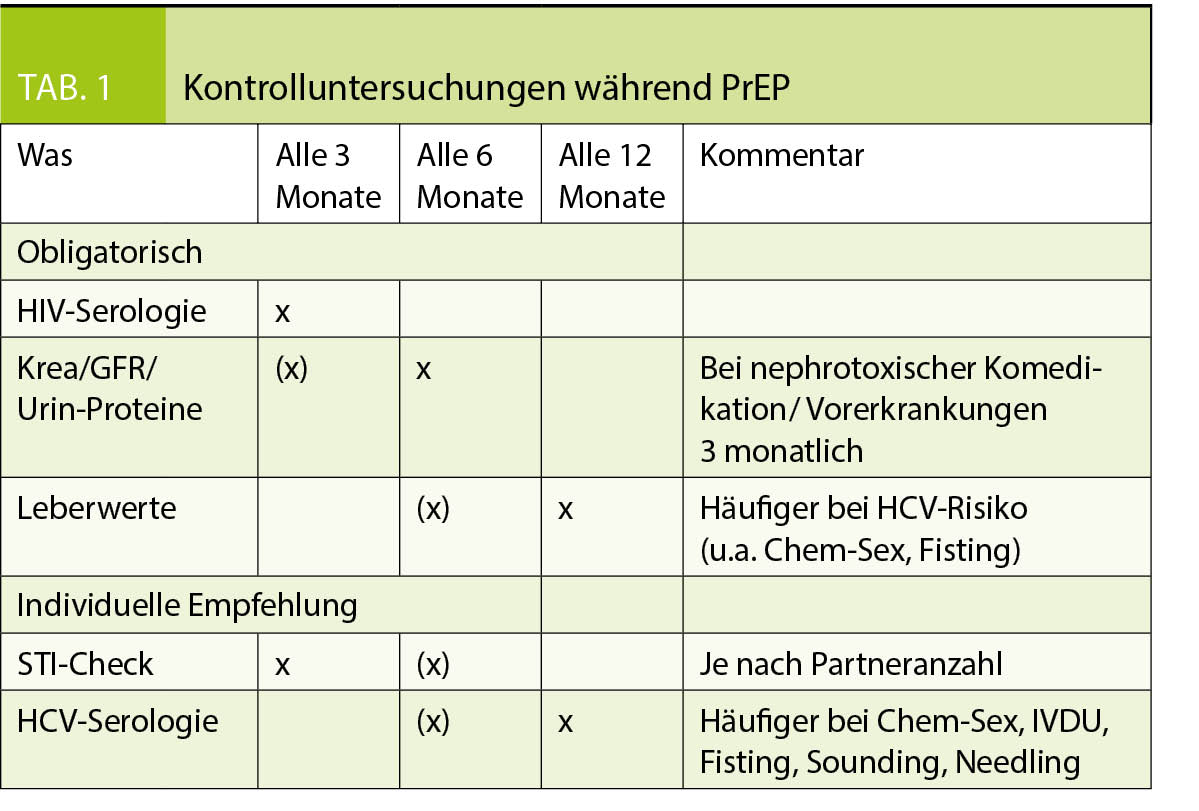
Eine PrEP sollte nicht durchgeführt werden bei einer HIV-Infektion, signifikanten Proteinurie und/oder einer Nierenfunktion mit GFR < 50 ml/min. Bei chronischen Nierenerkrankungen mit einer GFR > 50 ml/min, Osteoporose oder sonstigen Vorerkrankungen, die das Risiko für Nebenwirkungen der PrEP erhöhen, muss dies sorgfältig mit den Personen besprochen werden und das Nutzen-Risiko-Profil analysiert werden und falls eine PrEP durchgeführt wird allenfalls auch engmaschigere Kontrollen. Bei einer chronischen Hepatitis B kann eine PrEP durchgeführt werden, allerdings nur mit täglicher Einnahme und nicht event-basiert. In diesem Fall lohnt sich die Zuweisung zu einem Infektiologen. Während der PrEP-Einnahme empfehlen wir die folgenden Kontrollen gemäss der Tabelle 1.
Medikamente
Die PrEP-Medikamente waren bis jetzt nicht zugelassen in der Schweiz und wurden off-label gegeben (Möglichkeiten: Beziehen des Truvada via SwissPrEPared-Programm1, Generikum aus z.B. Deutschland oder via Internetbestellung aus dem Ausland. Gerade ganz aktuell wurde nun Truvada® für die PrEP in der Schweiz zugelassen, wie sich dies nun preislich gestaltet, ist aktuell noch unklar.
Dr. med. Julia Notter
Klinik für Infektiologie/Spitalhygiene
Kantonsspital St. Gallen
Die Autorin hat in Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
Die HIV PEP und PrEP bieten gute Möglichkeiten, HIV-Neuinfektionen wirksam zu verhindern und so neben dem frühen Therapiestart bei Neuinfektionen die HIV-Neuansteckungen weiter zu senken.
Eine PEP muss so rasch wie möglich begonnen werden. Ihre Erfolgschancen sinken bereits nach 6-8 Stunden. Zum Einsatz kommen
Tenofovir/Emtricitabin und entweder Raltegravir oder Dolutegravir oder mit Ritonavir verstärktes Darunavir.
Eine PrEP eignet sich für Personen, die ein erhöhtes Risiko haben, sich mit HIV anzustecken, nicht aber für Personen in einer monogamen Beziehung mit einer HIV-positiven Person, welche unter ART mind. 6 Monate supprimiert war
Neu ist Truvada® für die PrEP in der Schweiz zugelassen.
1. Roger AJ, Cambiano V, Bruun T et al. Risk of HIV transmission through condomless sex in serodifferent gay couples with the HIV-positive partner taking suppressive antiretroviral therapy (PARTNER): final results of a multicentre, prospective, observational study. Lancet 2019; 393 (10189): 2428-38.
2. Bundesamt für Gesundheit. HIV, Syphilis, Gonorrhoe und Chlamydiose in der Schweiz im Jahr 2018: eine epidemiologische Übersicht. BAG Bulletin 2019; 41: 10-20.
3. Eidgenössische Kommission für sexuelle Gesundheit (EKSG). Notfall HIV-Exposition – PEP kann die richtige Antwort sein. BAG Bulletin 2014; 48: 834-5.
4. Eidgenössische Kommission für sexuelle Gesundheit (EKSG). Reassessment of the Diagnostics Window Period for HIV Diagnostics. August 2018. Erhältlich unter: https://www.bag.admin.ch/dam/bag/de/dokumente/mt/p-und-p/richtlinien-empfehlungen/eksg-rec-window-period-hiv-diagnosis.pdf.download.pdf/eksg-rec-window-period-hiv-diagnosis.pdf
5. Eidgenössische Kommission für sexuelle Gesundheit (EKSG). Empfehlungen der Eidgenössischen Kommission für sexuelle Gesundheit (EKSG) zur HIV Prä-Expositionsprophylaxe (PrEP) in der Schweiz. BAG Bulletin 2016; 4:77-79.
6. Molina JM, Capitant C, Spire B et al.: On-Demand Preexposure Prophylaxis in Men at High Risk for HIV-1 Infection. New England Journal of Medicine 2015; 373 (23): 2237–2246
Die Opioidkrise in den USA gipfelte 2015 in einem starken Anstieg von Opioidabhängigen und damit verbundenen Todesfällen. Ein aggressives Marketing mit einer Opioid-verharmlosenden Strategie einiger Pharmafirmen führte dazu, dass schnell anflutende Opioide einer breiteren Patientenklientel verschrieben wurden. In diesem ersten Teil des Artikels werden der Stellenwert einer Opioidtherapie sowie das rationale und praktische Vorgehen bei einer analgetischen Therapie mit Opioiden in Bezug auf Schweizerische Verhältnisse kritisch diskutiert. In einem zweiten Teil werden hierzulande zugelassene Opioide vorgestellt, Überlegungen zur Opioid-Sicherheit in der Schweiz präsentiert sowie die Eingangsfrage nach einer drohenden Opioidkrise in der Schweiz eingehend erörtert.
Die Opioidkrise in den USA gipfelte 2015 in einem starken Anstieg von Opioidabhängigen und damit verbundenen Todesfällen durch Überdosierung (2016: 42.000) (1, 2). Die meisten Opfer waren abhängig geworden von zunächst legal verschriebenen kurzwirksamen Opioiden. Chronischer Schmerz ist häufig: 2018 waren hiervon in den USA 25 Millionen Menschen betroffen (1, 2). Ein aggressives Marketing mit einer Opioid-verharmlosenden Strategie einiger Pharmafirmen führte dazu, dass schnell anflutende Opioide einem breiteren Patientenklientel verschrieben und die Indikationen für diese Analgetikaklasse, welche vormals schweren oder präfinalen Krankheitsverläufen vorbehalten waren, auf leichtere, nozizeptive Schmerzen ausgeweitet wurden. Des Weiteren waren die «Pill Mills», also Kliniken oder Apotheken, welche niederschwellig Opioide abgeben, wegbahnend für die Opioidkrise (3, 4).
Zahlreiche so abhängig gewordene Patienten wichen in der Folge auf billigere und z.T. auch illegal erworbene Opioide wie Fentanyl oder Heroin aus. Heute konsumieren geschätzt eine Million US-Amerikaner Heroin. Bei 80 Prozent von ihnen soll die Sucht mit legal oder illegal erworbenen Schmerzmitteln begonnen haben (1). Weltweit sollen 2016 gemäss WHO 275 Mio. Menschen opioid-abhängig sein, der grösste Teil davon abhängig von illegalen Drogen (2). Täglich sterben in den USA 130 Menschen an einer Überdosis eines verschriebenen Opioids. Als Reaktion wurde am 26. Oktober 2017 in den USA der medizinische Notstand ausgerufen.
Es stellt sich nun die Frage, ob eine Globalisierung der Opioid-Krise droht. Besteht eine ähnliche Gefahr auch für die Schweiz? In den USA hatten die Pharmariesen leichteres Spiel als hierzulande: Regularien sind laxer, das Versicherungswesen anders strukturiert. Zudem versuchen Mediziner dort häufiger, unrealistische Therapieziele wie z.B. das Versprechen auf völlige Schmerzfreiheit bei chronischen Schmerzerkrankungen zu erfüllen. Und nicht zuletzt wird in den USA betont auf pharmakologische Therapieoptionen gesetzt; für ein multimodales und interdisziplinäres Therapieregime fehlt häufig das Geld.
Schauen wir uns vor diesem Hintergrund also die aktuelle Handhabung der Opiodverschreibung in der Schweiz an. Eine USA-analoge Opioidkrise scheint sich hier bisher nicht abzuzeichnen: Die Anzahl der an Opioid- Überdosis verstorbenen Menschen ist in der Schweiz von 2000 bis 2016 signifikant gesunken (5). Doch auch wie im restlichen Europa sind die Verbrauchzahlen von Opioiden seit Publikation der WHO- Schmerzleiter 1986 gestiegen. Zwischen 1985 und 2015 ist der schweizerische Opioidverbrauch von 18 zu 421 mg/Person/ Jahr angestiegen. Dies macht die Schweiz zum weltweit siebtgrössten Opioidkonsumenten (6).
Opioide: Nur ein Puzzleteil in einer multimodalen Schmerztherapie
Der Stellenwert einer Opioidtherapie im Rahmen eines analgetischen Therapieregimes ist unbestritten; idealerweise kommen hier gezielt Substanzen zum Einsatz, welche in ihrer Pharmakokinetik und Galenik auf das zugrundeliegende Schmerzsyndrom eingehen. So können orale, buccale oder transdermale Applikationsformen, retardierte oder rasch freisetzende Substanzen gewählt und auch kombiniert werden. Im Therapieverlauf wird die Indikation dann wiederholt reevaluiert, die Pharmakotherapie den aktuellen Bedürfnissen angepasst und möglicherweise auch rotiert werden, um Gewöhnung und Dosiseskalation zu vermeiden.
Ein breites Spektrum an Nicht-Opioidanalgetika und Koanalgetika steht uns zur Verfügung und sollte primär oder additiv zum Zuge kommen. Evidenzbasiert sind hier vor allem der Einsatz von Antidepressiva und Antiepileptika bei chronisch neuropathischem Schmerz − diese Substanzen erzielen bei vielen Schmerzerkrankungen hervorragende Ergebnisse und die Datenlage darf als sehr gut bezeichnet werden. Auch Nicht-Opioidanalgetika wie Paracetamol, Metamizol und NSAR werden breit eingesetzt, wobei auch für diese Analgetikaklassen weitestgehend Langzeitstudien zur Sicherheit bei chronischer Anwendung fehlen. Trotz breitem Einsatz bestehen zahlreiche Risiken auch unter diesen Substanzklassen (7).
Weitere Säulen eines analgetischen Therapiemanagements werden zum Zuge kommen. Multimodale Konzepte berücksichtigen Optionen wie physikalische Massnahmen, Physiotherapie, die interventionelle Schmerztherapie sowie ein breites Spektrum an verhaltenstherapeutischen Massnahmen und Coping- Instruktionen. Ein breites Abstützen auf mehrere Therapiesäulen sollte helfen, den Analgetikabedarf zu minimieren. Ziel einer multimodalen Schmerztherapie bleibt dabei – ausserhalb palliativer Indikation − die Wiederherstellung und Erhaltung der Funktionalität im Alltag. Dabei gilt als realistisches und erfolgreiches Therapieziel, wenn bei der Hälfte der chronischen Schmerzpatienten eine Schmerzreduktion um 50% erreicht wird.
Rationale Opioidtherapie nach Indikation
Unbestritten und wahrscheinlich am wenigsten problematisch ist der Einsatz von Opioiden bei palliativen Patienten. Hier sind Schmerzreduktion und Verbesserung der Lebensqualität oberstes Therapieziel und eine allfällige Abhängigkeit tritt in diesem Kontext eher in den Hintergrund. Karzinomschmerzen zählen zu den etablierten Indikationen für Opioide. Auf diese zielte die Publikation der WHO Schmerzleiter, gemäss der in einem Stufenschema zunächst Nicht- Opioid Analgetika, dann leichte und letztlich potente Opioide verabreicht werden. Nicht-Opioid Analgetika, Antiepileptika, Antidepressiva und Steroide werden bei Bedarf und indikationsgerecht in allen Stufen kombiniert (8).
Die Langzeitanwendung von Opioiden bei nichttumorbedingten Schmerzen hingegen stellt behandelnde Ärzte und die Betroffenen vor zahlreiche Herausforderungen. Hier gilt es, transparent und indikationsgerecht in Zusammenarbeit mit dem Patienten und den beteiligten Spezialisten eine sichere und wirksame Medikation zu etablieren. Von beeinträchtigenden, chronischen nichttumor-bedingten Schmerzen waren 2013 7,4% einer repräsentativen Bevölkerungsstichprobe betroffen (9). Nichttumorbedingte Schmerzen führen bei einem grossen Patientenanteil zu Einschränkung von physischem und psychischem Wohlbefinden, der Lebensqualität, der Arbeitsfähigkeit sowie zu hohen direkten und indirekten Gesundheitskosten. Als Reaktion wurden auch in Europa schwache und starke opioidhaltige Analgetika vermehrt und über einen längeren Zeitraum verschrieben.
Die Langzeitanwendung von opioidhaltigen Analgetika bei nichttumorbedingten Schmerzen wird bei einer Diskrepanz zwischen breiter klinischer Anwendung und gleichzeitig lückenhaft vorhandener Evidenz kritisch diskutiert (9, 10, 11).
Opioidhaltige Analgetika gelten als eine medikamentöse Therapieoption bei kurzfristiger, d.h. ein bis drei Monaten währenden Behandlung von Arthroseschmerzen, diabetischer Neuropathie, postherpetischer Neuralgie sowie chronischem Rückenschmerz. Von einer Langzeittherapie (>26 Wochen) profitiert nur ein Viertel aller Patienten.
Mögliche Indikationen für eine Langzeittherapie mit opioidhaltigen Analgetika, zu denen eine ausreichende Evidenz besteht, umfassen Schmerzen bei Arthrose, diabetischer Polyneuropathie, Postzosterneuralgie und chronischen Rückenschmerzen. Für andere Schmerzsyndrome fehlt der Expertenkonsens und eine Behandlung müsste als individueller Therapieversuch bewertet werden.
Als Kontraindikationen gelten primäre Kopfschmerzen, Opioid-Abhängigkeit, Fibromyalgie-Syndrom, entzündliche Darmerkrankungen, chronische Pankreatitis sowie funktionelle und psychische Störungen mit dem Leitsymptom Schmerz. Der niedrigste Evidenzlevel existiert für die Behandlung von Schmerzen nach Gehirnläsionen, nach Wirbelfrakturen bei manifester Osteoporose, bei rheumatischen Erkrankungen ausser rheumatoider Arthritis, chronischen postoperativen Schmerzen, Schmerzen bei peripherer arterieller Verschlusskrankheit, bei Dekubitus oder Kontrakturen bei pflegebedürftigen Patienten. In diesen Fällen kann allenfalls ein individueller Behandlungsversuch mit Opioiden unternommen werden (9).
In der Betrachtung zu Langzeitstudien mit dem Thema opioidhaltige Analgetika bei nichttumorbedingten Schmerzen müssen neben dem Studiendesign und dem Beobachtungszeitraum folgende Endpunkte berücksichtig werden: Wirksamkeit (in Bezug auf Ausmass der Schmerzreduktion, verbessertem Befinden und Erhalten der Funktionalität), Verträglichkeit (Anzahl der Patienten, die die Studie wegen unerwünschter Wirkungen abbrechen mussten) und Sicherheit (Anzahl der schweren unerwünschter Wirkungen und Anzahl der Todesfälle).
Praktisches Vorgehen einer Therapie mit opioidhaltigen Analgetika bei nichttumorbedingten Schmerzen
Bei der Behandlung chronischer Schmerzpatienten gibt es vieles zu beachten. Die Wahl des eingesetzten Pharmakons richtet sich dabei nach der vorliegenden Erkrankung und der wissenschaftlichen Evidenz zum Opioideinsatz in diesem Kontext, den Begleiterkrankungen, eventuellen Kontraindikationen, der individuellen Erfahrung des Patienten mit bisher eingesetzten Analgetika und dessen Präferenzen. Meist wird das Opioid nicht als Monotherapeutikum, sondern in Kombination mit anderen zentral oder auch peripher wirksamen Analgetika und Koanalgetika eingesetzt werden.
Eine alleinige medikamentöse Therapie bei nichttumorbedingten Schmerzen wird in der Schweiz in der Regel nicht durchgeführt. Zu einem tragfähigen integrativen Behandlungskonzept zählen verschiedene Behandlungspfeiler, die in ihrer Kombination eine optimale Schmerztherapie bei minimalen unerwünschten Wirkungen erzielen sollten.
Zu diesen Behandlungspfeilern zählen physiotherapeutische und physikalische Therapien, Patientenedukation und Psychotherapie, gegebenenfalls Lifestyle-Modifikationen, Aufklärung über Möglichkeiten und auch Grenzen der analgetischen Therapie. Von Wichtigkeit ist hier, beim Patienten realistische Erwartungen an die Therapie mit opioidhaltigen Analgetika zu erwecken resp. unrealistischen Erwartungen entgegenzuwirken. Zu erwartende und häufige unerwünschte Arzneimittelwirkungen (Obstipation, Übelkeit, Libidoverlust etc.) sowie potenziell schwerwiegende unerwünschte Arzneimittelwirkungen (Sucht, Sturzgefahr, Atemdepression bei Überdosierung, erhöhte Mortalität bei geriatrischen Patienten) sowie der Einfluss auf die Fahrtüchtigkeit müssen vor Behandlungsbeginn kommuniziert werden. Um unerwünschte Wirkungen frühzeitig zu erkennen sowie Sicherheit und Wirksamkeit der Behandlung zu gewährleisten, müssen regelmässige Kontrolluntersuchungen vereinbart werden. In diesen werden eine regelmässige Indikationsüberprüfung sowie Dosisanpassungen oder Substanzwechsel vorgenommen werden.
In der klinischen Leitlinie zur Langzeitanwendung von Opioiden bei nichttumorbedingten Schmerzen (9) werden u.a. folgende Schlüsselempfehlungen gegeben (zusammengefasst):
1. Differenzialindikation opioidhaltiger Analgetika: Je nach Krankheitsbild und individuellen Bedürfnissen des Patienten wird das Analgetikum nach seinen pharmakodynamischen, kinetischen und galenischen Eigenschaften ausgewählt.
2. Langwirksame Präparate mit retardierter Galenik sollten kurzfristig anflutenden Substanzen vorgezogen werden.
3. Einnahmeschema: Die Einnahme sollte nicht «on demand», sondern nach einem vorher festgelegten Schema erfolgen.
4. Dosierung: Therapiebeginn mit niedrigen Dosen, Erhaltungsdosis nach Erreichen der zuvor formulierten Therapieziele. Die Höchstdosis von >120mg/d orales Morphinäquivalent soll nicht überschritten werden.
5. Therapiedauer: Eine Therapie >3 Monate soll nur bei Therapierespondern durchgeführt werden.
6. Dosisreduktion und Medikamentenpausen sollen nach einem halben Jahr angestrebt werden, um die Wirksamkeit der parallel durchgeführten Therapiemassnahmen zu überprüfen.
7. Eine regelmässige Therapieüberwachung mit den Endpunkten Sicherheit, Verträglichkeit und Fehlgebrauch sollte unter Langzeittherapie mit Opioiden durchgeführt werden.
Mechanismusbasierte Schmerztherapie
Ein brauchbares Werkzeug in der Entscheidungsfindung, wie ein Schmerzsyndrom pharmakologisch optimal behandelt werden kann, bietet eine Mechanismus-basierte Schmerztherapie. Hier erfolgt zunächst eine Identifikation des Schmerzcharakters: Sind Muskel- und Skelettsystem betroffen und liegt ein belastungsabhängiger Schmerz ohne Entzündungszeichen vor, handelt es sich um einen nozizeptiven Schmerz. Beispiele wäre eine Arthrose oder ein myofasziales Schmerzsyndrom. Pharmakologisch kommen hier zunächst periphere Analgetika wie NSAR, Metamizol oder Paracetamol zum Einsatz. Der Einsatz von Opioiden kann in einem zweiten Schritt überlegt werden.
Ist das Muskel- und Skelettsystem betroffen und liegen Entzündungszeichen vor, handelt es sich um einen nozizeptiv / entzündlichen Schmerz mit Nozizeptoraktivierung und -sensibilisierung sowie zentraler Sensitivierung und Ausweitung rezeptiver Felder. Beispielerkrankungen wären eine Arthritis oder aktivierte Arthrose. In diesem Kontext sind eher NSAR, Glucokortikoide und kurzfristig evtl. auch transdermale Opioidsysteme sinnvoll.
Sind nervale Strukturen betroffen, ist der Schmerz einschiessend, ausstrahlend und liegen neurologische Begleitsymptome vor, redet man von neuropathischem Schmerz. Beispiel wäre eine diabetische Neuropathie oder eine Post- Zoster Neuralgie. Hier werden neue, Schmerzintensivierende Kanäle und Rezeptoren an nervalen Strukturen synthetisiert, es kommt zu nervaler Spontanaktivität und zentraler Sensitivierung mit reduzierter endogener Schmerzhemmung. Neuropathischer Schmerz wird lokal (Lidocain, Capsaicin), mit Antidepressiva und Antiepileptika sowie mit Opioiden angegangen.
Zeigt ein Patient schliesslich eine allgemeine Hyperalgesie, vegetative und evtl. psychische Symptome ohne passende radiologische oder laborchemische Befunde handelt es sich am ehesten um multilokulären Schmerz. Beispiele wären somatoforme Schmerzen oder das Fibromyalgiesyndrom. Pathophysiologisch liegen eine reduzierte endogene Schmerzhemmung und veränderte Schmerzverarbeitung zugrunde. Antidepressiva aus der Gruppe der Trizyklika und SNRI sind in diesem Fall indiziert (12).
Abhängigkeit von Opioiden beim chronischen Schmerzpatienten
Wir unterscheiden die physische von der psychischen Abhängigkeit. Eine chronische Verabreichung von Opioiden führt zu Toleranz-
entwicklung – diese tritt im klinischen Kontext jedoch selten auf und kann durch ein entsprechendes Medikamentenmanagement meist verhindert werden (z.B. eine Opioidrotation). Zudem kommt es zu einer physischen Abhängigkeit. Ein plötzliches Sistieren führt zu einer Hyperaktivität des sympathischen Nervensystems (mit z.B. Diarrhoe, Schwitzen, Mydriasis, Blutdruckanstieg), gleichzeitig
Verlangen nach dem Opioid, verstärkten Schmerzen, Magen- und Knochenschmerzen sowie Myalgien. Diese Symptomatik kann durch langsames Ausschleichen der Dosis verhindert werden.
Eine psychische Abhängigkeit ist charakterisiert durch negative Konsequenzen, die mit dem Opioidgebrauch einhergehen, wie Kontrollverlust, Tendenz zu inadäquater Dosissteigerung, Eingrenzung von Denken und Verhalten auf die Beschaffung. Wie hoch bei Schmerzpatienten in Europa das Risiko einer Abhängigkeit ist («Prescription Opioid Use Disorder, POUD»), ist bisher nicht bekannt (13, 14). Geschätzt wird, dass ca. 10-15% chronischer Schmerzpatienten eine Sucht entwickeln.
Für eine psychische Abhängigkeit werden genetische und epigenetische Ursachen postuliert (14). Solange keine spezifischeren pharmakologischen Behandlungsoptionen für einzelne Schmerzsyndrome existieren, an denen aktuell wegen der Opioidkrise rege geforscht wird, muss der Schmerztherapeut sein analgetisches Armamentarium kennen und einsetzen können.
Dr. med. Antje Heck
Fachärztin für Klinische Pharmakologie und Toxikologie FMH
Fachärztin für Anästhesie FMH, Schmerzspezialistin SGSS
Leiterin Sprechstunde Medikamente in Schwangerschaft und Stillzeit
Oberärztin Psychiatrische Klinik Königsfelden
Postfach 432
5201 Brugg
antje.heck@pdag.ch
Prof. Dr. med. Eli Alon
Facharzt für Anästhesiologie FMH, Schmerzspezialist SGSS
Professor für Anästhesiologie und Schmerzmedizin an der
Universität Zürich
Praxis für Schmerztherapie
Arzthaus Zürich City
Lintheschergasse 3
8001 Zürich
eli.alon@arzthaus.ch
Die Autoren haben in Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
Auch wenn in der Schweiz innert 30 Jahren der durchschnittliche
Opioidverbrauch von 18 auf 421 mg/Person/ Jahr angestiegen ist, konnte in den letzten 20 Jahren ein signifikanter Rückgang der an Opioid- Überdosis verstorbenen Menschen registriert werden.
In der multimodalen Schmerztherapie werden Optionen wie physi-
kalische Massnahmen, Physiotherapie, die interventionelle Schmerztherapie sowie ein breites Spektrum an verhaltenstherapeutischen Massnahmen und Coping-Instruktionen berücksichtigt. Ein Abstützen jeder analgetischen Therapie auf mehrere dieser Therapiesäulen hilft, den Analgetikabedarf zu minimieren.
Die Wahl geeigneter Analgetika erfolgt mit Vorteil aufgrund des dem Schmerz zugrunde liegenden Schmerzmechanismus, ja nach dem,
ob ein nozizeptiver Schmerz ohne oder ein nozizeptiv/entzündlicher Schmerz mit Nozizeptoraktivierung und -sensibilisierung vorliegt, ob eine zentrale Sensitivierung mit Ausweitung rezeptiver Felder besteht, oder ein neuropathischer oder multilokulärer Schmerz besteht.
Opioide nehmen heute einen unverzichtbaren Stellenwert in einer modernen, multimodalen Schmerztherapie ein.
1. Uhlmann B: Opioid-Krise in den USA: Ein Land unter Drogen. Süddeutsche Zeitung 26. Oktober 2017.
2. Verhamme KMC, Bohnen AM. The Lancet: Are we facing an opioid crisis in Europe? August 20, 2019.
3. Lyapustina T, Rutkow L, Chang HY, et al. Effect of a »pill mill” law on opioid prescribing and utilization: the case of Texas. Drug Alcohol Depend 2016; 159: 190–97.
4. hhs.gov/opioids. What is the U.S. opioid epidemic? United States Department of Health and Human Services. Jan 22, 2019. https://www.hhs. gov/opioids/about-the-epidemic/index.html.
5. Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region, 2000-2016. Geneva, World Health Organization; 2018.
6. Ruchad D et al: Opioid consumption from 1985 to 2015 : The situation in Switzerland, with an international comparison. Rev med. Suisse 2018 Jun 20;14(612):1262-1266.
7. Heck A, Alon E: Nicht-Opioid-Analgetika in der Geriatrie. Der Informierte Arzt; Sept 2019.
8. World Health Organization. Traitement de la douleur cancéreuse. Geneva, Switz: World Health Organization; 1987.
9. Häuser W eta al: Clinical practice Guideline: Long- term opioid use in non- cancer pain. Dtsch Arztebl. Int 2014; 111: 732-40.
10. Häuser W et al: Untying chronic pain: prevalence and societal burden of chronic pain stages in the general population- a cross- sectional survey. BMC Public Health 2014;14: 352.
11. Kissin I: Long-term opioid treatment of chronic nonmalignant pain: unproven efficacy and neglected safety? Journal of Pain Research 2013:6 513–529.
12. Alon E: Opioide sind nicht 1. Wahl, aber häufig unverzichtbar. Beilage Medical Tribune 49/2018.
13. Kraus M et al: Consensus and Controversies Between Pain and Addiction Experts on the Prevention, Diagnosis, and Management of Prescription Opioid Use Disorder. J Addict Med. 2020 Jan/Feb;14(1):1-11.
14. The Opioid Crisis and the Future of Addiction and Pain Therapeutics. J Pharmacol Exp Ther 371: 396-408; Nov 2019.
Der erste «Corona-Patient» in der Schweiz wurde in Lugano in eurem Spital Moncucco behandelt. Wie seid ihr auf die Diagnose gekommen und wie war der Verlauf?
Dr. med. Daniel Hagara
D. Hagara: Der erste Coronavirus-Patient war ein 70-jähriger Zahnarzt, der sich wahrscheinlich bei einem Ärztesymposium in Mailand angesteckt hat. Die Diagnose wurde am 25.2.2020 gestellt, da bei uns im Spital Moncucco bereits ab 24.2.2020, also 3 Tage nach den alarmierenden Nachrichten der Lombardei, eine Notfalltriage auf Covid errichtet worden war. Dieser Patient wurde regelhaft isoliert und hatte in der Folge einen guten Krankheitsverlauf. Seine durchgemachten sozialen Kontakte wurden in Zusammenarbeit mit dem Kantonsarzt zurückverfolgt.
Prof. Dr. med. Andreas Cerny
A. Cerny: Am 7.2. hatten wir schon einen jungen Patienten auf dem Notfall gesehen, welcher seit kurzem aus China zurückgekehrt war. Gottlob waren seine Grippesymptome nicht auf das SARS CoV-2 zurückzuführen. Diese Erfahrung half uns, das interne Dispositiv zu verbessern. Obschon wir täglich von den Ereignissen in den italienischen Medien informiert worden waren, traf uns die Epidemie mental unvorbereitet. Es ist die enorme Geschwindigkeit der Ausbreitung, welche wir alle unterschätzten.
Was waren die unmittelbaren Konsequenzen aus diesem ersten Fall fürs Personal, Spital und den Kanton?
D. Hagara: Bereits in dieser ersten Woche nach den Ereignissen in der Lombardei wurden in unserem Spital wie auch im Kanton Krisenstäbe täglich abgehalten. Meiner Meinung nach war die Tragweite der Geschehnisse allen Akteuren des Gesundheitswesens wie auch den politischen Kräften klar. Die ersten Entscheidungen waren aber zunächst sehr umstritten: so wurde der Karneval von Bellinzona noch abgehalten und Grossanlässe wie z.B. Sportanlässe wurden durchgeführt. Am 26.2.2020 wurden dann Karneval und Grossanlässe verboten. Die Schulen aber blieben weiterhin geöffnet und wurden dann erst am 11.3.2020 (post-obligatorische Schulen) und 16.3.2020 (alle) geschlossen.
In unserer Klinik wurden bereits nach 2 Tagen Massnahmen auf der Notfallstation ergriffen, die dem Covid-Verdacht Rechnung trugen. Nach einer Woche wurden stationäre Betten errichtet für die Covid-Verdachtsfälle. Es wurde erst nach 10 Tagen eine allgemeine Maskentragpflicht des Personals eingeführt.
Wie konnte der ambulante und stationäre Spitalbetrieb aufrechterhalten werden, auch um Schaden bei Patienten mit anderen Problemen abzuwenden?
D. Hagara: Wie das Tessin stark mit der Lombardei verknüpft ist, zeigt folgende Anekdote: eine 80-jährige Frau wurde am 2.3.2020 (also in einer Zeit, als die Klinik die Sicherheitsmassnahmen hochfuhr) bei uns wegen einer Synkope aufgenommen. Beim Eintritt klagte sie über keinerlei Beschwerden, die auf Covid hingewiesen hätten. Erst nach 2 Tagen stellte sich heraus, dass diese Frau, die ihre Angehörigen im Tessin hat und sich oft im Tessin aufhält und auch in der Schweiz versichert ist, am 22.2.2020 von der roten Zone in Lodi (Lombardei) «geflüchtet» ist, um bei ihren Kindern im Tessin zu verweilen. Unmittelbar auf Covid positiv getestet, zeigte sie einen akut sich verschlechternden Verlauf mit intensivmedizinischer Beatmung. Diese Episode führte in der Folge dazu, dass sich 5 Mitarbeiter und 1 Patientin auf dieser Abteilung mit dem Covid infiziert haben.
Wie hat diese Corona-Pandemie euren beruflichen Alltag verändert? (z.B. wie habt ihr die vielen Coronapatienten betreut, welche nicht, beziehungsweise nicht mehr, intensivmedizinisch behandelt werden mussten? Zusammenarbeit mit niederge-lassenen Kollegen etc.)
D. Hagara: In unserer Gemeinschaftspraxis haben wir sofort täglich einen Krisenstab durchgeführt. Ambulante Patienten wurden ab sofort einen Tag vor der geplanten Visite telefonisch auf eventuelle grippeähnliche Symptome befragt. Wir haben auch sofort hygienische Massnahmen erarbeitet und umgesetzt für Covidverdachtsfälle. Dies trifft ebenfalls auf Niveau der Klinik zu. Mitte März wurde die gesamte Klinik in eine Klinik verwandelt, welche ausschliesslich Covid-Patienten aufnimmt mit max. 180 Spitalbetten und 40 Intensivplätzen.
Die niedergelassenen Ärzte waren am Anfang überfordert, v.a. aufgrund von mangelndem Schutzmaterial. Letzteres wurde dann durch die Tessiner Ärztegesellschaft den niedergelassenen Ärzten zu Verfügung gestellt. Ebenfalls hat die Ärztegesellschaft in der Folge sogenannte Checkpoints errichtet, wo die Covid-Beurteilung und Diagnose mittels Nasenabstrich durchgeführt werden konnte.
Wie hat diese Corona-Pandemie euren persönlichen Alltag verändert?
D. Hagara: Mir erscheint diese relativ kurze Zeit von 2 Monaten wie gefühlte durchgemachte 3 Jahre. Dies aufgrund von einer Vielzahl von neuen Anforderungen, wie unzählige Telefonate, Kommunikation und Kollaboration mit Ärzten, Hygienemassnahmen, etc.
Am 4.4.2020 habe ich mich mit dem Coronavirus angesteckt: Die Krankheit ist bei mir relativ glimpflich verlaufen, vielleicht gerade, weil ich sofort mit Plaquenil 3x200mg, Azithromycin 1x500mg und Clexane 80 U s.c.1×1 angefangen habe (mit prompter EKG-Kontrolle). Nach ein paar Tagen mit deutlichen Grippesymptomen wurde ich von einem trockenen Husten eingeholt, der über 3 Wochen anhielt. Für meine Familie bedeutete diese Krankheit, dass ich währende 4 ganzen Wochen komplett isoliert in einem Zimmer blieb (zum Glück mit einem separaten WC), sodass sich niemand meiner Familie angesteckt hat. A. Cerny: Es ging alles viel schneller, sehr viele Kommunikationen auf allen Kanälen, oft wusste ich gar nicht mehr, welchen Wochentag wir hatten, wenig Schlaf und stressige Träume.
Wie habt ihr euch lokal und international ausgetauscht, um die Diagnostik und Behandlung zu optimieren?
A. Cerny: Ja, wir hatten enge Kontakte mit unseren Kollegen in Mailand, Pavia, Bergamo und Parma und hatten so Zugang zu vielen Informationen wie dem Thromboserisiko, den ersten Erfahrungen mit antiviralen Substanzen und Immunsuppressiva und bekamen regelmässig die letzten Versionen ihrer Guidelines. Wir erfuhren auch vom stark erhöhten Risiko, sich bei der Arbeit mit dem Virus zu infizieren. Wir tauschten diese Erfahrungen regelmässig mit Kollegen in der Deutschschweiz und im Welschland aus, welche initial etwas skeptisch waren.
Was kam von wissenschaftlicher Seite dazu, wie z.B. Studienteilnahme, Compassionate use Programme, Patienten-Register oder Biobanking etc.
A. Cerny: Die neuesten Studienresultate wurden rasch untereinander ausgetauscht und in unserer WhatsApp-Gruppe rege diskutiert. Bei uns und auch im Ente Ospedaliero wurden das Compassionate-use Programm für Remdesivir regelmässig benützt und es wurden verschiedene Studien begonnen. Dank grosszügiger und unkomplizierter Unterstützung von Privaten konnten wir eine Biobank für COVID-19 Patienten aufbauen, welche gekoppelt mit einer klinischen Datenbank helfen wird, diese heimtückische Erkrankung in Zukunft besser zu verstehen.
Wie hat sich die mediale und kommunikative Begleitung angesichts der Bedrohungslage abgespielt?
A. Cerny: Da ich die Landessprachen spreche und meine Infektiologen-Kollegen im Krisenstab eingebunden waren und somit weniger frei waren, über die Ereignisse zu berichten, wurde ich oft von den Medien im Tessin und anderswo in der Schweiz angefragt. Mir war es wichtig, vor allem in der Anfangsphase, als der Rest der Schweiz noch keine konkrete Erfahrung mit der Krankheit und deren Gefährlichkeit hatte, darüber zu berichten.
Wie war die Zusammenarbeit mit den Behörden und Medien? (Haben die Behörden in dieser Notlage zeitgerecht und angemessen gehandelt, sowohl gegenüber der Bevölkerung wie auch gegenüber den Ärzte-Pflegenden etc?)
D. Hagara: Im Nachhinein ist es immer leicht zu kritisieren. Denn es musste schnell gehandelt werden. Für einige wichtige Massnahmen, wie zum Beispiel Contact Tracing, fehlte es sowohl an Zeit als auch Personal. Der grosse Fehler allerdings war in der Anfangsphase die Zulassung des Bellinzoneser Karnevals, welche meiner Meinung nach der breiten Ansteckung der Bevölkerung Vorschub leistete. In der folgenden Phase hat man auch den Altersheimen zu wenig Beachtung geschenkt, was man an der sehr grossen Zahl der Toten in Altersheimen ersehen kann (fast 50% der Toten im Tessin).
Wie seht ihr die Rolle der WHO und der internationalen politischen Zusammenarbeit in dieser und auch kommenden Pandemien?
A. Cerny: In der Anfangsphase war die Mensch-zu-Mensch-Übertragung noch in Frage gestellt und die Gefährlichkeit der Krankheit unterschätzt worden. Wie sehr politische Einflüsse den raschen Austausch vitaler Informationen behinderte, wird sicher Teil der Aufarbeitung dieser katastrophalen globalen Krise sein. Der Ende Februar publizierte Report, der von Experten der WHO und des chinesischen CDC verfasst worden war, war trotzdem sehr hilfreich. Leider hat sich ausser Italien kaum ein Land daran gehalten. Ich denke, wir hätten uns auch auf Nationaler Ebene mehr mit italienischen Experten und Behörden austauschen sollen.
Welches sind für euch die wesentlichsten Erkenntnisse für eine zukünftige Pandemie für unser Land?
D. Hagara: Ich sehe keine einzeln eruierbare Erkenntnis, aber bin der Überzeugung, dass diese Pandemie global äusserst viel verändern wird. Die Menschheit hat vor allem erfahren, dass das, was als selbstverständlich galt, in einem Augenblick nicht mehr sicher sein kann. Und: Händewaschen wird nicht mehr nur eine Frage der Hygiene sein, sondern eine Frage des Überlebens. A. Cerny: Das Pandemiekonzept wird sicher überarbeitet werden müssen, dieses muss raschere Entscheidungsprozesse vorsehen, die Reserven an persönlichem Schutzmaterial und Desinfektionsmaterial müssen angepasst werden. Das Kommunikationskonzept von Bund und Kantonen muss verbessert werden, insbesondere im Hinblick auf Klarheit und Koordination. Die rasche wissenschaftliche Aufarbeitung neuer Erkenntnisse muss verbessert werden, insbesondere sollte die Task Force in eine ständige Beratungseinheit übergeführt werden, welche sich vermehrt mit WHO und ECDC austauscht und auch im Inland offene Kanäle mit den wichtigen Stakeholdern wie z.B. der forschenden Pharmaindustrie, den Universitäten und Swissmedic etabliert.
Nun werden zunehmend Lockerungen wirksam: seid ihr auf eine weitere Welle gefasst und was ist die persönliche Erwartung in die nahe Zukunft (z.B. Stichwort: Neue Normalität, Medikamente und Impfung?)
A. Cerny: Ich erwarte eine erneute Zunahme der Fälle Ende Juni anfangs Juli. Die Lockerungs-Massnahmen betreffen zu rasch gleichzeitig weite Bereiche des öffentlichen Lebens und ich befürchte, dass das «contact tracing» bei einem erneuten Anstieg der Fälle rasch dekompensiert. Die nächsten Wochen im Mai und Juni, wo wir weiterhin wenig neue Fälle sehen werden, könnten für viele als Zeichen missgedeutet werden, dass der «böse Traum» vorbei sei und dass wir unsere normalen Sommeraktivitäten wieder aufnehmen dürfen. Es wird vermehrt Stimmen geben, welche den Lockdown als unnötig bezeichnen und Verantwortlichkeiten fordern. Unser Spital und Ambulatorium werden die Achtsamkeit bestimmt nicht vermindern und sicherstellen, dass wir nicht wieder auf dem linken Fuss erwischt werden.
Zur Bewältigung der zweiten Welle hoffen wir auf neue Medikamente, die Resultate der vielen z.T. schon abgeschlossenen und z.T. noch laufenden Studien sollten uns neue Informationen zur Pathogenese und Impulse für die Behandlungen geben. Ich zweifle, dass wir im Juli schon soweit sind. Die Impfung scheint noch weit weg.
medinfo bedankt sich ganz herzlich bei Prof. Dr. med. Thomas Cerny, der dieses Interview organisiert und geführt hat.
In den letzten 40 Jahren hat sich die Anzahl der Mehrlingsschwangerschaften in der Schweiz praktisch verdoppelt. Während 1979 insgesamt 20 Mehrlingsgeburten pro 1000 Lebendgeburten registriert wurden, waren es 2018 35.5 Zwillings- und 1.1 Drillingsgeburten pro 1000 Lebendgeburten (1). Diese Entwicklung erklärt sich durch das gestiegene mütterliche Alter und die Verbreitung der Fortpflanzungsmedizin (ART). Da Mehrlingsschwangerschaften mit einem deutlich erhöhten Risiko für mütterliche und fetale Komplikationen einhergehen, ist deren rechtzeitige Diagnose essentiell für die weitere Schwangerschaftsbetreuung (2). Oft ist eine Mehrlingsbetreuung in Zusammenarbeit mit oder an einem Perinatalzentrum sinnvoll.
En Suisse, depuis 40 ans, le nombre de grossesses multiples a quasiment doublé. En 1979 on enregistrait 20 grossesses multiples par 1000 naissances avec enfant vivant, en 2018 on comptait 35,5 grossesses gémellaires et 1,1 triplés par 1000 naissances avec enfant vivant (1). Cette évolution s’explique par l’âge maternel croissant et le recours de plus en plus fréquent à la procréation médicalement assistée (PMA). Vu que les grossesses multiples ont un risque maternel et fœtal nettement augmenté, le diagnostic précoce est essentiel pour la prise en charge ultérieure (2). Un suivi en collaboration avec ou primairement dans un centre de périnatologie est conseillé.
Ultraschall im ersten Trimester
Die Ersttrimester-Sonographie ist ein integraler Bestandteil der Schwangerschaftsbetreuung, nicht zuletzt, um Mehrlingsschwangerschaften frühzeitig zu erkennen. Die sorgfältige Diagnose der Mehrlingsschwangerschaft, die Bestimmung des Schwangerschaftsalters sowie das Festlegen der Chorionizität/Amnionizität sind für die weitere Schwangerschaftsbetreuung von zentraler Bedeutung (3).
Diagnose
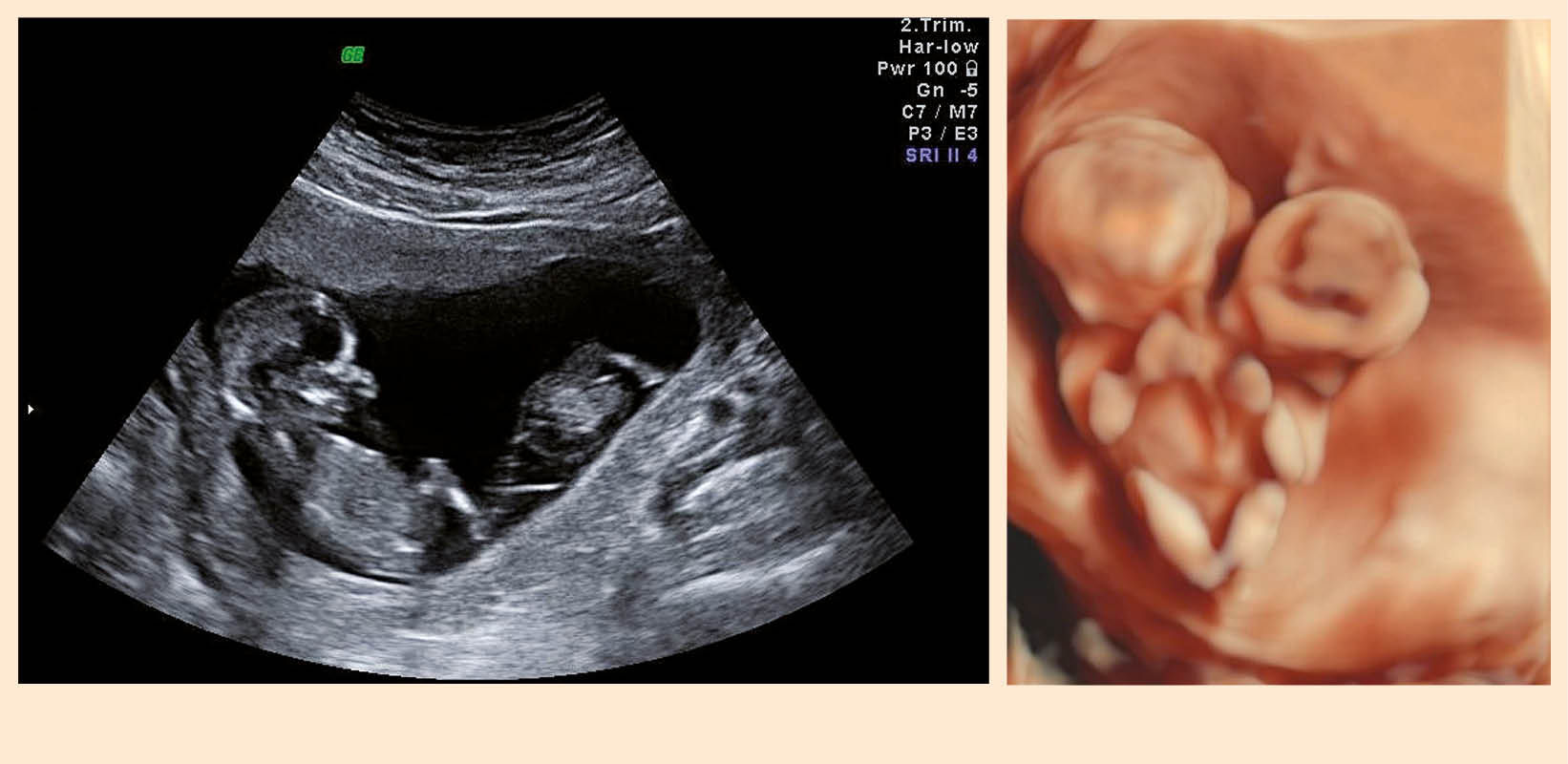
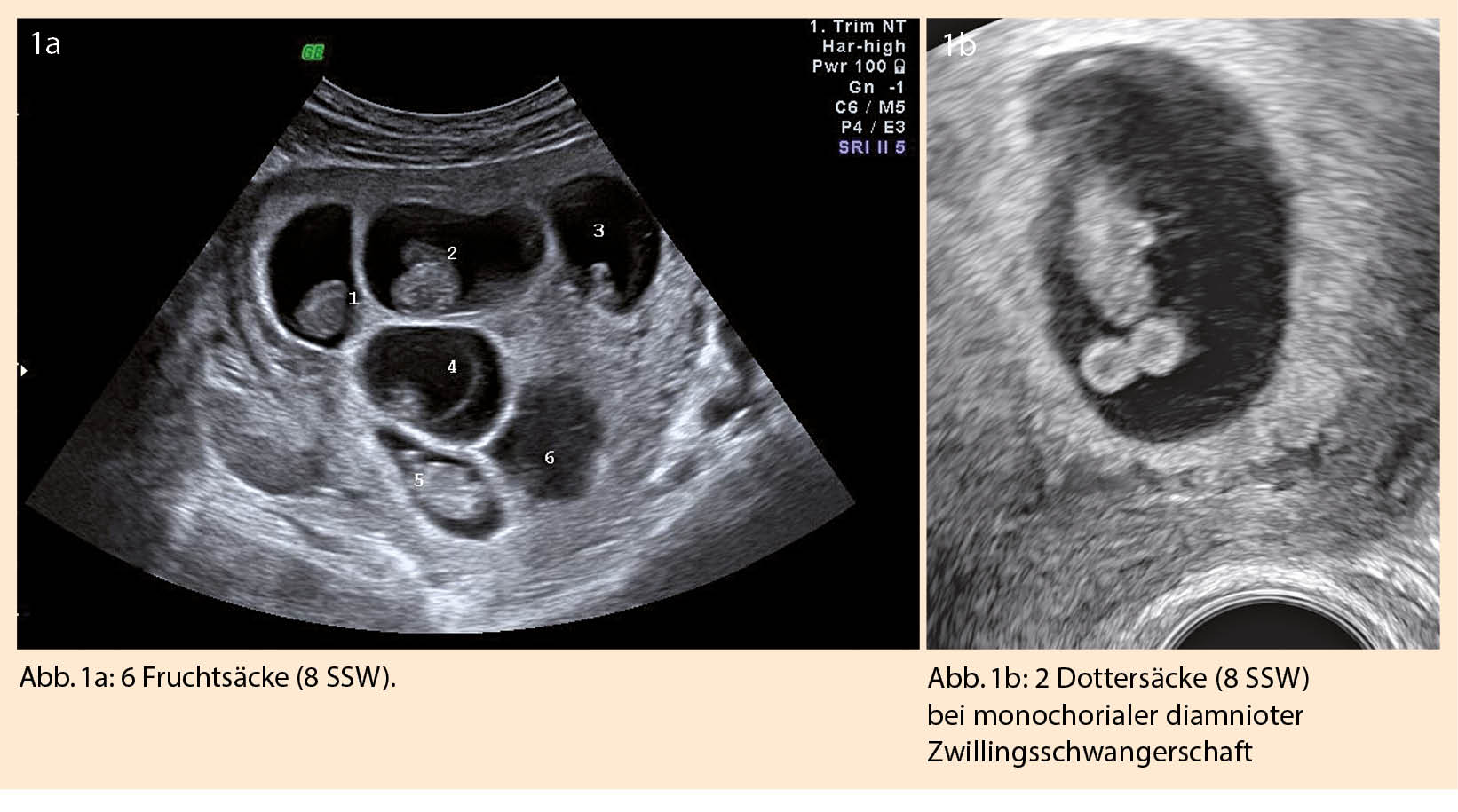
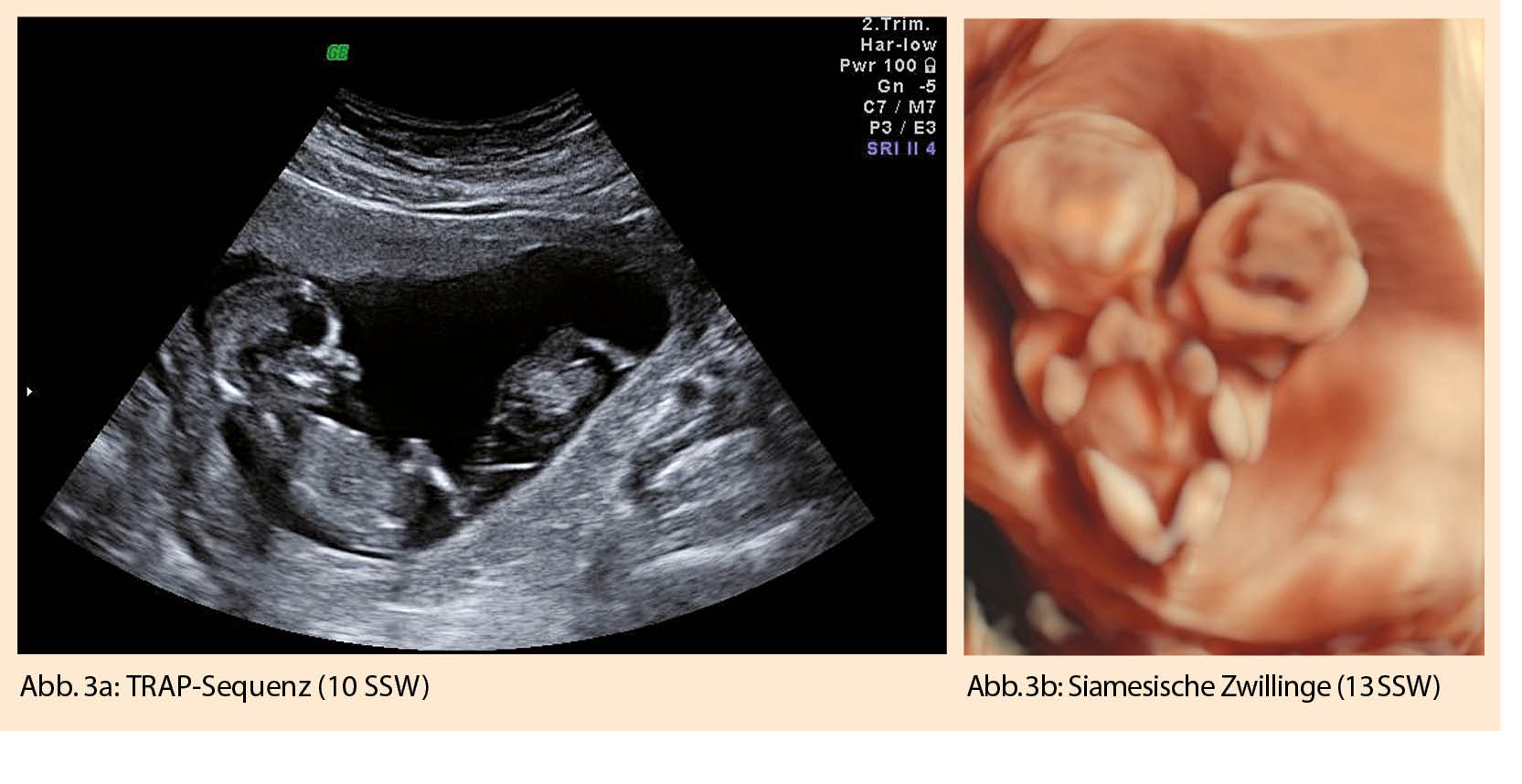
Die Anzahl der Feten lässt sich sonographisch ab der 7. SSW darstellen, indem die Fruchtsäcke gezählt werden (Abb. 1a). Ist mehr als 1 Dottersack zu sehen, muss immer an eine Mehrlingsschwangerschaft gedacht werden (Abb. 1b).
Bestimmung des Schwangerschaftsalters
Wenn keine durch ART entstandene Schwangerschaft vorliegt, wird auch bei Mehrlingen das Schwangerschaftsalter mittels Messung der Scheitelsteisslänge (SSL) zwischen 11 und 14 Schwangerschaftswochen (SSW) bestimmt (4, 5). Eine ungleiche plazentare Verteilung, Anomalien oder ein auffälliger Karyotyp eines Feten, können schon im ersten Trimester zu diskordantem Wachstum führen (6). Zur Bestimmung des Schwangerschaftsalters soll bei Mehrlingen die SSL des grösseren Fetus verwendet werden (7).
Chorionizität/Amnionizität
Die Chorionizität lässt sich schon ab der 7. SSW bestimmen, muss aber spätestens bis 14 SSW festgelegt, dokumentiert und mit Bild in der Patientenakte hinterlegt worden sein. Hintergrund ist, dass mit jeder weiteren SSW das Risiko der Fehlklassifikation um 10% ansteigt (8). Beim geringsten Zweifel, sollte deshalb rechtzeitig eine Zuweisung an ein Perinatalzentrum erfolgen. Dadurch kann beispielsweise die richtige Klassifikation einer Zwilllingsschwangerschaft um über 50% auf 95% gesteigert werden (9).
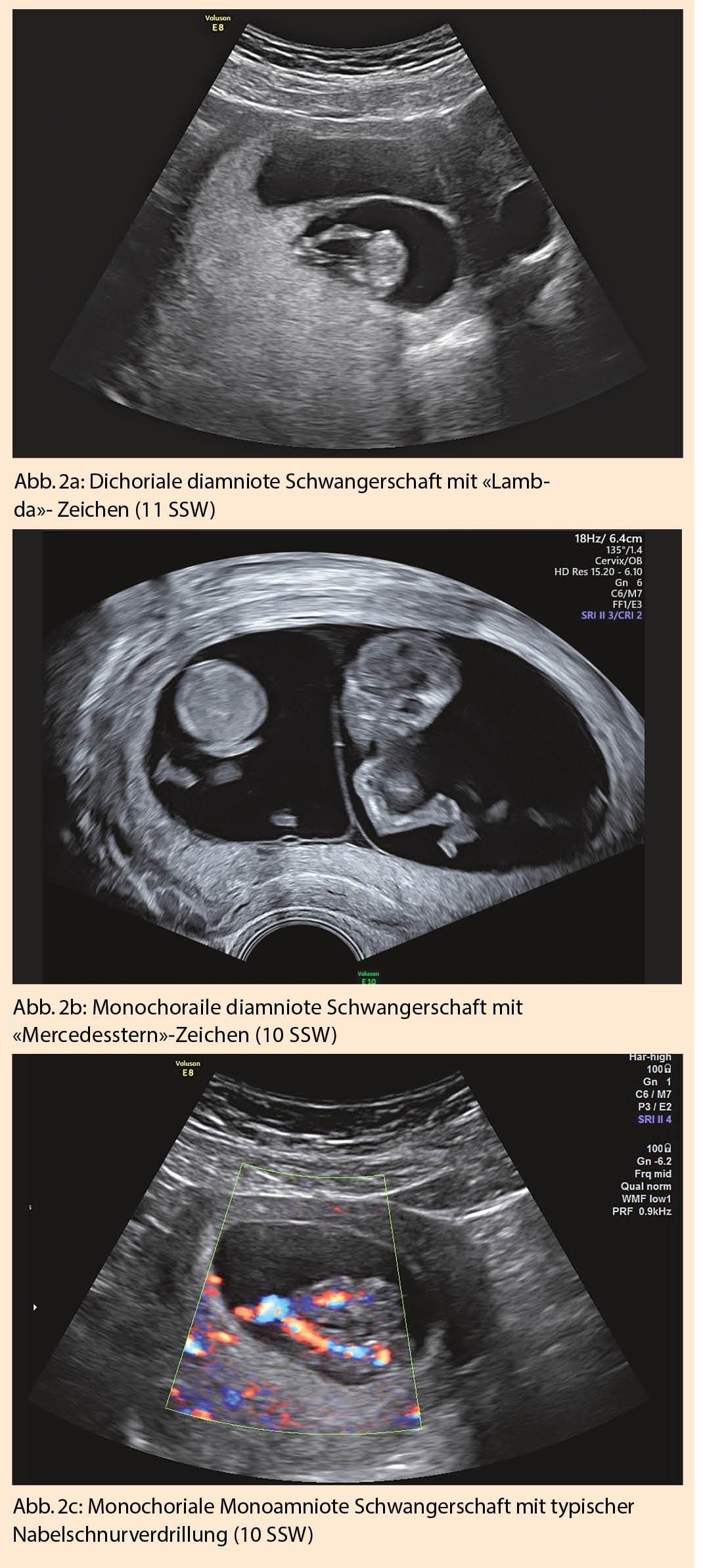
Sieht man sonographisch zwei klar getrennte, von Throphoblast umgebene Fruchthöhlen, liegt eine dichoriale diamniote Schwangerschaft vor. Um 12-14 SSW entwickelt sich daraus das typische «Lambda»-Zeichen (Abb. 2a), da nur noch plazentaseitig ein Choriondreieck zwischen die beiden Amnionblätter zieht. In 7% verschwindet das Lambda-Zeichen nach 20 SSW (10).
Findet sich nur eine Fruchthöhle mit zwei Embryonen, liegt eine monochoriale Zwillingsschwangerschaft vor. Bei monochorialen diamnioten Zwillingen liegen die beiden Amnionblätter glatt aufeinander und bilden so eine feine Trennwand zwischen den Feten. Bei 8-10 SSW sind die beiden Amnionblätter meist noch etwas vom Chorionblatt abgelöst, wodurch sonographisch der Eindruck eines leeren «Lambda»-Zeichens oder auch das typische Bild eines «Mercedesstern»-Zeichens erscheint (Abb. 2b). Sobald die Amnion- und Chorionblätter vollständig verschmelzen bildet sich das «T»-Zeichen.
Vor 8-10 SSW ist die Amniozität oft noch schwierig zu beurteilen. Die Dottersackdiagnostik hilft hierbei auch nicht weiter. Bei einer Monoamnionizität, ca. 4% aller monochorialen Schwangerschaften, lässt sich auch bei 10-14 SSW keine Trennwand zwischen den Embryonen darstellen. Typischerweise zeigt sich eine Amnionhöhle mit zwei eng zusammenliegenden Nabelschnuransätzen oder einer Nabelschnurverdrillung (Abb. 2c). Die Messung der Membrandicke und das Zählen der Membranschichten zwischen den Feten sind keine zuverlässigen Diagnosekriterien (10).
Siamesische Zwillinge und TRAP (twin reversed arterial perfusion) -Sequenz
Teilt sich die Zygote erst nach dem 13. Tag post conceptionem, resultieren siamesische Zwillinge (Abb. 3a). Art und Ausmass der Verbindung kann sehr variieren und geht entsprechend mit unterschiedlichsten Prognosen einher. Wichtig sind eine frühzeitige Diagnose und eine entsprechende Beratung des Paares.
Eine weitere für monochoriale Zwillingsschwangerschaften spezifische Diagnose des ersten Trimesters ist die TRAP (twin reversed arterial perfusion)-Sequenz. Typischerweise fehlt hier bei einem Zwilling das Herz und die obere Extremität (=Akardius acranius). Eine arterio-arterielle Anastomose, vom zweiten, ‘pumpenden Zwilling’ kommend, gewährleistet die Durchblutung des Akardius acranius. Mit dem Farbdoppler lässt sich dieser umgekehrte Blutfluss gut darstellen (Abb.3 b). Die Mortalität für den pumpenden Zwilling liegt bei >50 %. Eine rechtzeitige Trennung der beiden Kreisläufe, meist mittels Lasertherapie, kann rettend sein (11).
Höhergradige Mehrlingsschwangerschaften
Wird eine höhergradige Mehrlingsschwangerschaft diagnostiziert, gelten dieselben Regeln zur Bestimmung der Chorionizität/Amnionizität wie bei Zwillingsschwangerschaften (Abb. 1a). Da diese Schwangerschaften mit noch höheren Komplikationsraten und grosser psychosozialen Belastungen einhergehen können, kann sich die Frage nach einer fetalen Reduktion stellen. Die Reduktion einer Drillingsschwangerschaft auf Zwillinge, kann zu einer Schwangerschaftsverlängerung von ca. 3 Wochen führen (12), geht gleichzeitig aber mit einer Abortrate von 5-7% einher (13, 14). Es empfiehlt sich vorgängig ein umfassendes Aneuploidie-Screening durchzuführen. Die Entscheidungsfindung des Paares kann selbstverständlich sehr unterschiedlich sein und hängt vom sozialen Hintergrund und deren Glaubenseinstellungen ab (15).
Aneuploidie-Screening
Fürs Aneuploidie-Screening stehen heutzutage verschiedene Optionen zur Verfügung, welche mit der Patientin umfassend besprochen werden müssen. Bei dizygoten Zwillingen ist das Risiko einer Chromosomenstörung für jeden Fetus gleich gross wie bei einer Einlingsschwangerschaft, für die Schwangerschaft ergibt sich somit ein zweifach erhöhtes Risiko. Monozygote Zwillingen haben, bis auf wenige Ausnahmen, den gleichen Karyotyp (16). Das Risiko einer Chromosomenstörung entspricht somit dem von Einlingsschwangerschaften.
Ersttrimestertest (ETT)
Seit dem Aufkommen des ETT‘s in den 1980-90er Jahren basierte die Aneuploidie-Risikoberechnung bei Mehrlingschwangerschaften, neben dem mütterlichen Alter, auf der Messung der Nackentransparenz (NT) bei einer SSL zwischen 45-84mm. Hierbei soll bei monochorialer Zwillingsschwangerschaft der Durchschnittswert der NT-Messung beider Zwillinge herangezogen werden (17). Eine diskordant erhöhte NT (>20%) oder eine SSL-Differenz >10% bei monochorialen werden als Frühmarker für das Entwickeln eines feto-fetalen Transfusionssyndroms (FFTS) oder einer schweren Wachstumsretardierung im weiteren Schwangerschaftsverlauf diskutiert (17-19).
Nicht-invasiver pränataler Test (NIPT)
Eine weitere Aneuploidie-Screeningmethode für Zwillinge ist der NIPT. Hierbei werden freie ‚fetale‘ DNA-Bruchstücke (ffDNA) aus dem mütterlichem Blut vervielfältigt und für die häufigsten numerischen Chromosomenanomalien getestet. Entscheidend für die Durchführbarkeit des NIPT‘s, analog wie bei den Einlingsschwangerschaften, ist eine ffDNA Fraktion > 4% pro Kind. Bei Zwillingen sollte diese idealerweise über 8% liegen (20). Eine 2017 publizierte Metaanalyse zu ffDNA-Tests bei Zwillingsschwangerschaften zeigte Entdeckungsraten für die Trisomie 21 (T21) von 100% (95% CI, 95.2-100%) bei einer falsch-positiven Rate von 0.0% (95% CI, 0.00-0.003%) (21) . Die T21-Detektionsrate des NIPT’s ist heute für Zwillings- und Einlingsschwangerschaften vergleichbar. Bei höhergradigen Mehrlingen kann der NIPT bisher nicht standardmässig angeboten werden.
In der Schweiz sind die Bedingungen zur Kostenübernahme eines NIPT’s für T21, 18 und 13 durch die obligatorische Krankenversicherung nach einer Risikoberechnung für T21, 18, und 13 ≥ 1:1000 gegeben (22). Bei einem NIPT-Resultat mit Hinweis auf eine Chromosomenanomalie gilt es, diesen mittels invasiver Diagnostik zu bestätigen, um den Ursprung (fetal, plazentar oder mütterlich) zu identifizieren (22).
Invasive Diagnostik
Obwohl in den vergangenen Jahren etwas in den Hintergrund gerückt, gilt die invasive Diagnostik bis heute als ‚Goldstandard‘ des pränatalen, genetischen Screenings. Wünschen die Eltern eine maximale Abklärung kann nach entsprechender Aufklärung über die Risiken und Kosten eine Chorionzottenbiopsie (CVS) oder Amniozentese (AC) angeboten werden. Bei sonographischen Auffälligkeiten (Fehlbildungen oder NT >95. Perzentile) soll über die medizinische Indikation einer invasiven Abklärung zur fetalen Chromosomenuntersuchung inklusive einer Microarray-Analyse aufgeklärt werden.
Dr. med. Ladina Vonzun
Klinik für Geburtshilfe
UniversitätsSpital Zürich
Frauenklinikstrasse 10
8091 Zürich
ladina.vonzun@usz.ch
Prof. Dr. med. Nicole Ochsenbein-Kölble
Klinik für Geburtshilfe
UniversitätsSpital Zürich
Frauenklinikstrasse 10
8091 Zürich
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Beitrag deklariert.
Mehrlingsschwangerschaften lassen sich zuverlässig ab der 7. SSW darstellen
Die sorgfältige Bestimmung des Schwangerschaftsalters, die Festlegung der Anzahl Feten und der Chorionizität/Amnionizität vor 14 SSW ist essentiell für ein adäquates Management der Mehrlingsschwangerschaft.
Als pränatales Aneuploidie-Screening kann Zwillingseltern neben der NT-Messung auch ein NIPT angeboten werden, wobei auffällige Resultate durch eine invasive Diagnostik abgeklärt werden sollen.
Auch bei Mehrlingsschwangerschaften soll bei sonographischen Auffälligkeiten direkt eine invasive Diagnostik angeboten werden.
Die Mehrlingsbetreuung sollte in Zusammenarbeit mit oder am Perinatalzentrum geschehen.
Messages à retenir
L’échographie permet de découvrir de manière fiable une grossesse multiple dès 7 semaines de grossesse (SA, semaines d’aménorrhée corrigée).
La détermination précise de l’âge gestationnel, du nombre de foetus et de la chorionicité/amnionicité avant 14 SA est essentiel pour une prise en charge adéquate d’une grossesse multiple.
Comme dépistage prénatal d’une aneuploïdie on peut offrir d’une part la mesure de la clarté nucale (NT), mais aussi le DPNI (dépistage prénatal non-invasif) avec recours au diagnostic invasif en cas de résultat suspect.
Comme lors de toute grossesse, en cas d’images ultrasonores suspectes dans une grossesse multiple, un diagnostic invasif devrait être envisagé d’emblée.
Le suivi d’une grossesse multiple devrait se faire en collaboration étroite avec ou primairement dans un centre de périnatologie.
1. https://www.bfs.admin.ch/bfs/de/home/statistiken/gesundheit/gesundheitszustand/gesundheit-neugeborenen.html.
2. Lewi L, Gucciardo L, Van Mieghem T, de Koninck P, Beck V, Medek H, et al. Monochorionic diamniotic twin pregnancies: natural history and risk stratification. Fetal Diagn Ther. 2010;27(3):121-33.
3. Oepkes D, Sueters M. Antenatal fetal surveillance in multiple pregnancies. Best Pract Res Clin Obstet Gynaecol. 2017;38:59-70.
4. Dias T, Mahsud-Dornan S, Thilaganathan B, Papageorghiou A, Bhide A. First-trimester ultrasound dating of twin pregnancy: are singleton charts reliable? BJOG. 2010;117(8):979-84.
5. Chaudhuri K, Su LL, Wong PC, Chan YH, Choolani MA, Chia D, et al. Determination of gestational age in twin pregnancy: Which fetal crown-rump length should be used? J Obstet Gynaecol Res. 2013;39(4):761-5.
6. Salomon LJ, Cavicchioni O, Bernard JP, Duyme M, Ville Y. Growth discrepancy in twins in the first trimester of pregnancy. Ultrasound Obstet Gynecol. 2005;26(5):512-6.
7. D’Antonio F, Khalil A, Mantovani E, Thilaganathan B, Southwest Thames Obstetric Research C. Embryonic growth discordance and early fetal loss: the STORK multiple pregnancy cohort and systematic review. Hum Reprod. 2013;28(10):2621-7.
8. Blumenfeld YJ, Momirova V, Rouse DJ, Caritis SN, Sciscione A, Peaceman AM, et al. Accuracy of sonographic chorionicity classification in twin gestations. J Ultrasound Med. 2014;33(12):2187-92.
9. Wan JJ, Schrimmer D, Tache V, Quinn K, Lacoursiere DY, James G, et al. Current practices in determining amnionicity and chorionicity in multiple gestations. Prenat Diagn. 2011;31(1):125-30.
10. Lu J, Cheng YKY, Ting YH, Law KM, Leung TY. Pitfalls in assessing chorioamnionicity: novel observations and literature review. Am J Obstet Gynecol. 2018.
11. Lewi L, Valencia C, Gonzalez E, Deprest J, Nicolaides KH. The outcome of twin reversed arterial perfusion sequence diagnosed in the first trimester. Am J Obstet Gynecol. 2010;203(3):213 e1-4.
12. Zipori Y, Haas J, Berger H, Barzilay E. Multifetal pregnancy reduction of triplets to twins compared with non-reduced triplets: a meta-analysis. Reprod Biomed Online. 2017;35(3):296-304.
13. Evans MI, Andriole S, Britt DW. Fetal reduction: 25 years‘ experience. Fetal Diagn Ther. 2014;35(2):69-82.
14. van de Mheen L, Everwijn SM, Knapen MF, Oepkes D, Engels M, Manten GT, et al. The effectiveness of multifetal pregnancy reduction in trichorionic triplet gestation. Am J Obstet Gynecol. 2014;211(5):536.e1-6.
15. Dodd JM, Crowther CA. Reduction of the number of fetuses for women with a multiple pregnancy. Cochrane Database Syst Rev. 2012;10:Cd003932.
16. Lewi L, Blickstein I, Van Schoubroeck D, Gloning KP, Casteels M, Brandenburg H, et al. Diagnosis and management of heterokaryotypic monochorionic twins. Am J Med Genet A. 2006;140(3):272-5.
17. Memmo A, Dias T, Mahsud-Dornan S, Papageorghiou AT, Bhide A, Thilaganathan B. Prediction of selective fetal growth restriction and twin-to-twin transfusion syndrome in monochorionic twins. BJOG. 2012;119(4):417-21.
18. Sebire NJ, Souka A, Skentou H, Geerts L, Nicolaides KH. Early prediction of severe twin-to-twin transfusion syndrome. Hum Reprod. 2000;15(9):2008-10.
19. Kagan KO, Gazzoni A, Sepulveda-Gonzalez G, Sotiriadis A, Nicolaides KH. Discordance in nuchal translucency thickness in the prediction of severe twin-to-twin transfusion syndrome. Ultrasound Obstet Gynecol. 2007;29(5):527-32.
20. Struble CA, Syngelaki A, Oliphant A, Song K, Nicolaides KH. Fetal Fraction Estimate in Twin Pregnancies Using Directed Cell-Free DNA Analysis. Fetal Diagn Ther. 2013.
21. Gil MM, Quezada MS, Revello R, Akolekar R, Nicolaides KH. Analysis of cell-free DNA in maternal blood in screening for fetal aneuploidies: updated meta-analysis. Ultrasound Obstet Gynecol. 2015;45(3):249-66.
22. N. Ochsenbein TB, L. Raio, Y. Vial, D. Surbek, S. Tercanli, A. Rauch, I. Filges, S. Fokstuen. Pränatale nicht-invasive Risikoabschätzung fetaler Aneupoidien. SGGG Expertenbrief No 52. Update vom März 2018:https://www.sggg.ch.
Der nachfolgende Beitrag stellt eine aktuelle Reaktion auf eine Publikation von PRAC/EMA und BfArM zum Thema vaginale Therapie mit Estrogenen vom 17. Januar 2020 dar. Obschon die EMA nicht direkt Anordnungen für die Schweiz geben kann, haben ihre Stellungsnahmen doch über die Medien einen Einfluss auf unsere Patientinnen und Ärzte.
Cet article est une prise de position d’actualité sur une publication du 17 janvier 2020 de l’autorité allemande (BfArM) – qui reprend un document de l’agence européenne des médicaments (PRAC/EMA) – concernant le traitement hormonal aux oestrogènes par voie vaginale. Quoique l’EMA ne soit pas habilitée à légiférer en Suisse, cette recommandation diffusée par les media exerce une influence notable sur les patientes et le corps médical dans notre pays.
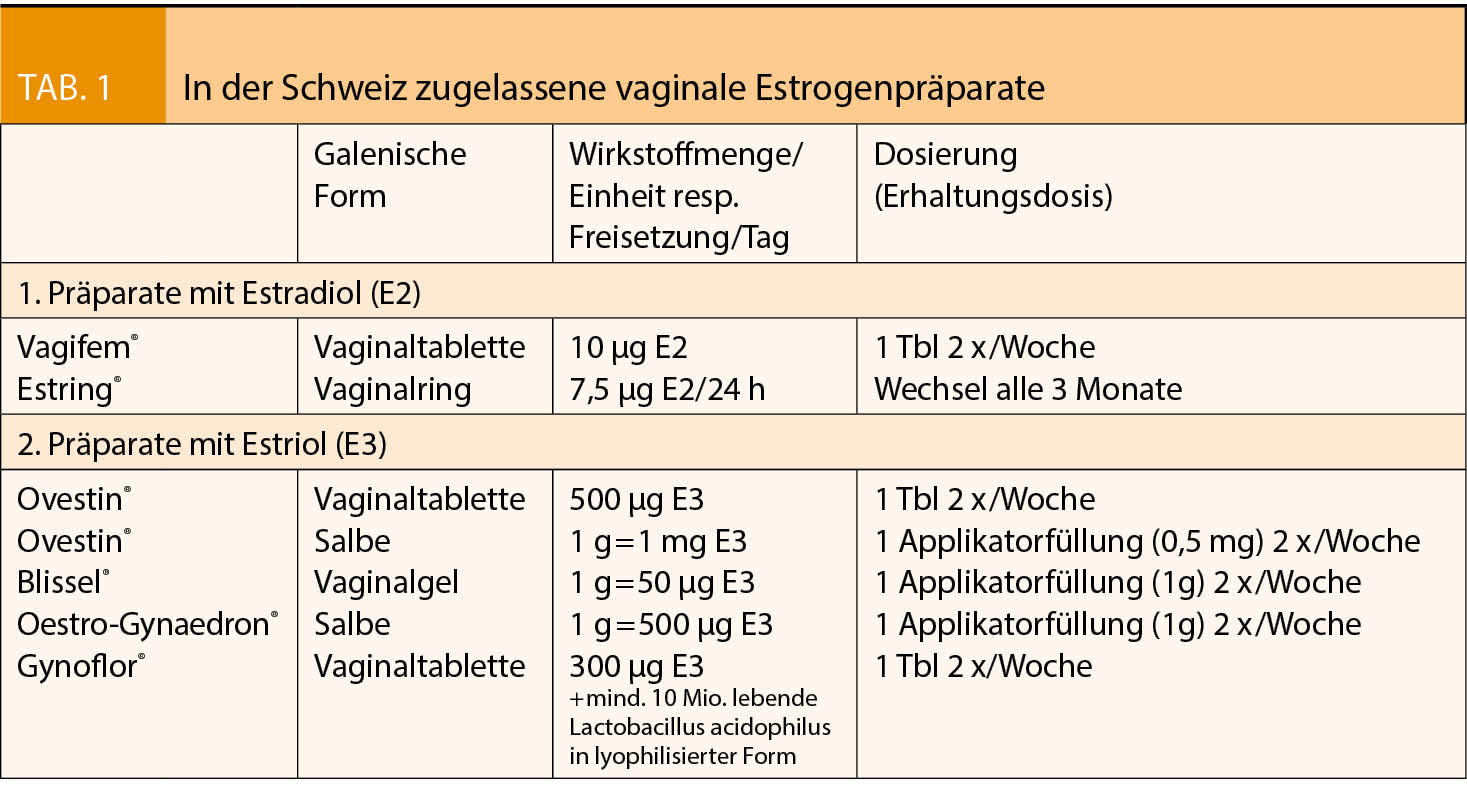
Ein vom Deutschen Bundesamt für Arzneimittel und Medizinprodukte (BfArM) vom PRAC (Ausschuss für Risikobewertung im Bereich der Pharmakovigilanz der Europäischen Arzneimittel-Agentur (EMA)) übernommene Empfehlung vom 17.01.2020 (1) enthält eine neue Risikobewertung für hochdosierte estradiolhaltige Vaginal-Crèmen mit 100µg Estradiol/g Crème (0,01%). Die in der Schweiz zugelassenen niedrig- und ultraniedrig dosierten vaginalen Estradiol-Präparate (Tabelle 1) sind von den neuen Empfehlungen nicht betroffen.
Die Kernbotschaft der BfArM-Empfehlung vom 17.01.2020 lautet:
Hochdosierte estradiolhaltige Crèmes sollten wegen der Risiken, die mit einer systemischen Wirkung von Estradiol assoziiert sind, nicht länger als für einen einzigen Behandlungszeitraum von 4 Wochen verschrieben werden.
Pharmakokinetische Daten über hochdosierte estradiol-haltige Crèmes (100µg/g) für den intravaginalen Gebrauch weisen auf eine erhebliche systemische Estradiolresorption hin, deren Werte über dem normalen postmenopausalen Bereich liegen. Die systemische Exposition des Estradiols könnte mit Nebenwirkungen verbunden sein, die denen von oralen und transdermalen HRT-Arzneimitteln ähnlich sind, wie z.B. endometrialem Hyperplasie/Karzinom, Brust- und Eierstockkrebs und thromboembolische Ereignisse.
Hochdosierte estradiol-haltige Crèmes sollten nicht mit anderen HRT-Medikamenten verschrieben werden.
Obwohl in der Schweiz keines der in Deutschland erhältlichen hochdosierten Präparate (Linoladiol®, Linoladiol N®, Linoladiol Estradiol® Estradiol Wolff®, Montadiol®) zugelassen zugelassen ist und die neue empfehlung von EMA und BfArM die Schweiz rechtlich nicht betrifft, sorgt die Publikation auch hier für Verunsicherung. Dies vor allem, weil auch in den Schweizer Beipackzetteln niedrig-dosierter vaginaler Estradiol-Präparate Warnungen vor möglichen Nebenwirkungen wie erhöhtem kardiovaskulärem, cerebrovaskulärem und thrombo-embolischem Risiko, erhöhtem Brustkrebsrisiko etc. aufgeführt sind, die nicht evidenz-basiert sind. Solche Warnungen können unsere Patientinnen ohne Grund verängstigen, so dass sie ihre korrekt indizierte vaginale Therapie absetzen.
Dieser Beitrag will die Grundlagen für die geltenden Richtlinien zur vaginalen Estrogen-Therapie und die geltenden Empfehlungen von BfArM und Swissmedic evaluieren. Dazu gehören auch die alten Beipackzettel zu niederdosierten Präparaten.
Grundlagen
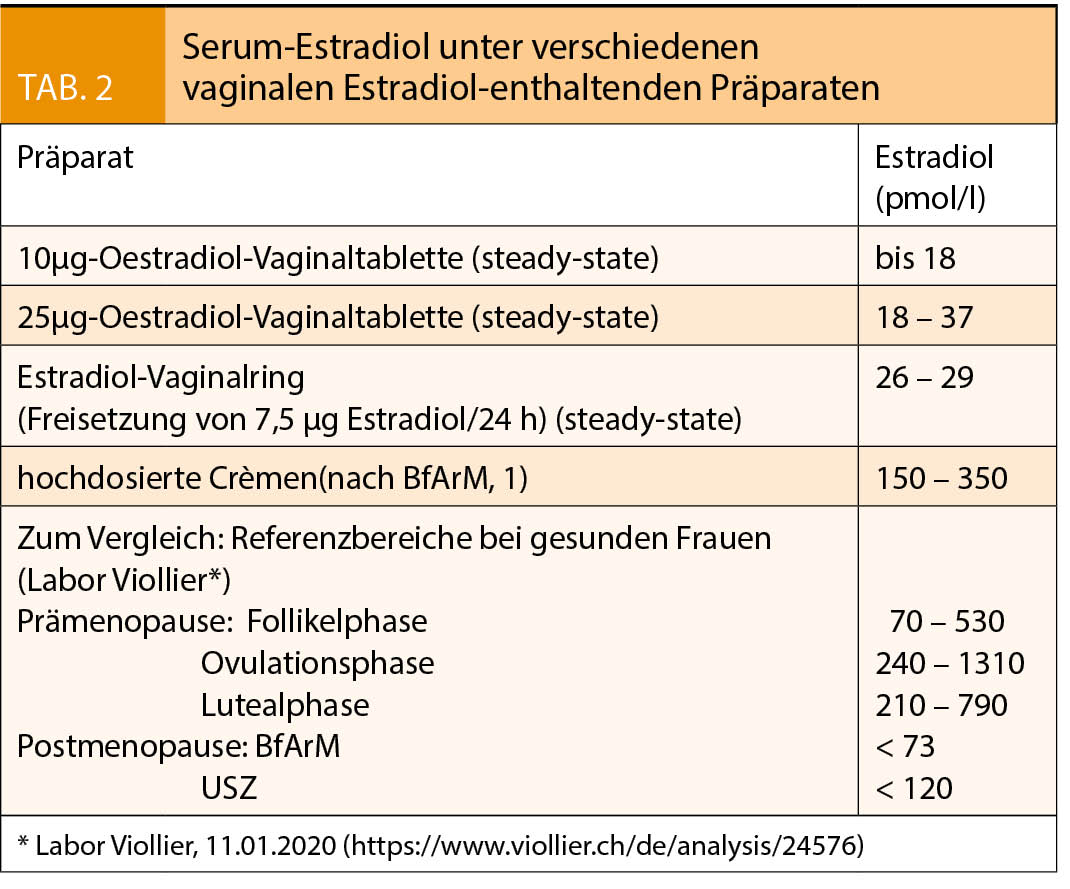
Die Serum-Estradiolspiegel liegen nach BfArM in der Prämenopause zwischen 147 und 1468 pmol/l (40–400 pg/ml) und sinken nach der Menopause auf < 73 pmol/l (20 pg/ml) ab (siehe auch Tab. 2). Diese Abnahme ist mit einer progressiven urogenitalen Atrophie assoziiert, die klinisch im Mittel 4–5 Jahre nach der Menopause manifest wird und zu Beschwerden führen kann, die unter dem Begriff genitourethrales menopausales Syndrom (GMS) zusammengefasst sind.
Objektive Veränderungen und subjektive Beschwerden werden von 25–50% aller postmenopausalen Frauen angegeben (2, 3, 4). Die tatsächliche Inzidenz ist sehr wahrscheinlich höher. Die optimale Therapie eines GMS ist für die urogenitale und sexuelle Gesundheit und damit auch die Sexualität und die Lebensqualität nach der Menopause entscheidend. Für weitere Informationen zum GMS und dessen Behandlung sei auf zwei neuere Übersichtsarbeiten (2, 3) verwiesen.
Risiken einer Behandlung
Niedrig-dosiertes vaginales Estradiol bei leichtem und mittelstarkem GMS
Sind bei einem GMS die nicht-hormonalen Behandlungsmöglichkeiten (2, 3) ausgeschöpft, so strebt eine rationale weitere Behandlung den Ersatz der fehlenden Estrogene und die Wiederherstellung der normalen urogenitalen Physiologie an, um dadurch eine Linderung der Symptome zu erreichen. Wenn eine systemische menopausale Hormontherapie (MHT) nicht aus anderen Gründen indiziert ist, soll die niedrig-dosierte vaginale Estrogengabe einer MHT vorgezogen werden. Sie ist zur Behandlung der vaginalen Atrophie wirksamer als die systemische MHT. Wenn nötig, kann eine vaginale Estrogengabe auch zusätzlich zu einer systemischen MHT angewandt werden.
Lokal-vaginale Estrogene können als Tabletten, Vaginalzäpfchen/Ovula, Crèmen oder als Vaginalring verabreicht werden. In der Cochrane-Review von 2016 (5) fanden sich für die verschiedenen vaginalen Estrogen-Präparate keine unterschiedliche Wirksamkeit. In der Schweiz sind nur Vaginalpräparate mit Estradiol und dem systemisch schwach wirksamen Estriol verfügbar (Tab 1). Hier wird nur auf Estradiol eingegangen, auf das sich die BfArM-Publikation bezieht.
Zur lokalen Behandlung eines leichten bis mittelschweren GMS werden im Vergleich zur systemischen Therapie deutlich kleinere Dosierungen benötigt. Die dabei erreichten Steady-State-Plasmaspiegel bleiben für Vaginalringe (Freisetzung von 7,5μg Estradiol/24h) und Estradioltabletten (25µg und 10µg) alle im normalen postmenopausalen Bereich (6, Tab. 2) und sind damit tiefer als die bei einer transdermalen systemischen Therapie erreichten Serumspiegel. Der Rückzug der vaginalen 25-µg-Estradioltablette vom Schweizer Markt aus Sicherheitsgründen war somit nicht gerechtfertigt. Er benachteiligt Frauen mit stärkeren Symptomen, da nach einem RCT die ultraniedrige 10µg-Estradiol-Tablette nicht in allen Kriterien einer modernen nicht-hormonalen Behandlung überlegen ist (13).
Die allfällige Notwendigkeit einer gleichzeitigen Gestagengabe bei Frauen unter niedrig-dosierten vaginalen Estrogenpräparaten wurde in der Cochrane-Review von 2016, in der Women’s Health Initiative (WHI) Observational Study (medianer Follow-Up 7,2 Jahre) und in zwei Reviews von 2019 und 2020 untersucht (5, 7, 8). Danach erhöht sich weder das Risiko für eine Veränderung der Endometriumhöhe noch dasjenige für Hyperplasien oder Endometriumkarzinome (5, 7, 8, 12). Die Schlussfolgerung, dass bei niedrigdosierter vaginaler Estrogentherapie keine Notwendigkeit für die gleichzeitige Gabe eines Gestagens zum Endometriumschutz besteht, ist auch in den Empfehlungen der IMS (International Menopause Society) (4), der SGGG (9) und der NAMS (North American Menopause Society) (10, 11) festgehalten und wird in den Beipackzetteln respektiert.
Wie die transdermale besitzt auch die vaginale Estradiol-Gabe keinen hepatischen First-Pass-Effekt. Deshalb und wegen der normal-postmenopausalen Estradiolspiegel unter Therapie (Tab. 2) ist es bei der niedrig-dosierten vaginalen Estradiolgabe nicht zu erwarten, dass es zu einer Risikoerhöhung von kardiovaskulären Erkrankungen, Schlaganfällen, thrombo-embolischen Ereignissen, gynäkologischen Karzinomen inkl. Mamma-Ca oder von Demenz kommt. Die WHI Observational Study belegt diese Annahme bei 45 663 nicht-hysterektomierten Studienteilnehmerinnen ohne systemische MHT (12). In diesem Kollektiv waren über die mediane Beobachtungsdauer von 7,2 Jahren die Risiken für CHD, Frakturen und Gesamtmortalität bei den Anwenderinnen von vaginalem Estrogen sogar signifikant niedriger als bei den Nichtanwenderinnen (Global Index, korrigierte Hazard Ratio 0.68; 95% Vertrauensintervall 0.55-0.86). Die übrigen Risiken waren nicht erhöht. Damit ist die in den meisten Beipackzetteln immer noch behauptete Risikozunahme für die in der WHI-Studie untersuchten Erkrankungen formell widerlegt. Diese Fehlinformation zu den Risiken sollte dringend entfernt werden.
Bei Patientinnen mit undiagnostizierten vaginalen Blutungen und solchen mit bekanntem oder vermutetem Endometriumkarzinom ist eine vaginale Estrogengabe kontraindiziert. Bei unklarer vaginaler Blutung unter Estrogen-Therapie muss eine endometriale Pathologie ausgeschlossen werden.
Mit Estrogenen bei schwerem GMS
Ein schweres GMS sollte mit höher bis hoch dosierten lokal-vaginalen Estradiol-Crèmen (0.01%) behandelt werden können (3), weil unter inadäquat niedrig-dosierter Therapie wie auch unter einer hochdosierten Behandlung von nur 4 Wochen keine dauerhafte Besserung eintreten kann. Da in der Schweiz keine entsprechenden Präparate erhältlich sind, muss zu Magistralrezepten oder zu im Ausland erhältlichen Präparaten gegriffen werden.
Die Anordnung von EMA/PRAC für die EU-Länder, dass eine Behandlung mit solchen Präparaten nicht länger als 4 Wochen dauern darf, widerspricht den Grundprinzipien zur vaginalen Gabe von Estrogenen beim GMS (Tab. 3) und ist nicht evidenzbasiert. Jedes deswegen nicht korrekt behandelte schwere GMS kann eine verheerende Wirkung auf die vulvovaginale, urogenitale und sexuelle Gesundheit der betroffenen Frauen haben. Sie kann Partnerschaft, Sexualleben, Selbstsicherheit und Lebensqualität zerstören.
Eine langdauernde höherdosierte vaginale Estradioltherapie ist bei richtiger Durchführung sicher. Unter hochdosierten Vaginalcrèmen fand sich bisher einzig eine mögliche dosisabhängige Zunahme der Endometriumdicke (5). Die WHI Observational Study, in der alle (auch hochdosierten) vaginalen Estrogenpräparate zusammen analysiert wurden, fand bei 45’663 Studienteilnehmerinnen ohne systemische MHT keine Zunahme der bereits oben erwähnten Risiken, auch nicht für Endometriumkarzinome (12).
Zur vom BfArM aus meiner Sicht weit überschätzten Gefahr hochdosierter vaginaler Präparate ist Folgendes festzuhalten: Gemäss BfArM werden unter hochdosierten vaginalen Estrogenen Serumwerte erreicht, die bis fünfmal über dem postmenopausalen Referenzbereich des BfArM liegen (1). Dies entspricht der frühen bis mittleren Follikelphase und damit auch den Estradiolwerten unter transdermaler systemischer MHT. Für diese wurde eine analoge Beschränkung der Behandlungsdauer nie in Betracht gezogen. Warum also bei den vaginalen Präparaten? Werden bei hoch-dosierter vaginaler Estradiol-Gabe Vorsichtsmassnahmen erwogen, so sollten diese aus pharmakodynamischen und metabolischen Gründen mit den Regeln übereinstimmen, die für eine transdermale systemische Estradiol-Gabe (Patches, Gels; ≤ 50µg/Tag) gelten. Ein Vergleich mit peroralen Studien (Million Women Study und WHI-Studie (RCT-Arm)) ist falsch und missachtet die entscheidenden Unterschiede zwischen oraler und vaginaler Estrogengabe. Zudem sollten in den Beipackzetteln, auf die aktuellesten neuen Datenanalysen der WHI-Studien Bezug genommen werden (16), und nicht auf alte, durch neue Daten überholte und deshalb unrichtige Auswertungen. Bestehen bei hochdosiertem vaginalen Estradiol begründete Bedenken wegen eines möglicherweise erhöhten Risikos für Endometrium-Karzinome, wäre die Empfehlung einer Gestagenbeigabe zu hochdosierten Präparaten ausreichend. Für darüber hinausgehende Massnahmen – wie einer Limitierung der Anwendungsdauer auf 4 Wochen – fehlt die erforderliche Evidenz.
Mögliche Alternative: Vaginale Gabe von DHEA
Ospemifen (2) wird vermutlich in der Schweiz nicht eingeführt. Dafür wird noch dieses Jahr ein vaginales Präparat zur Behandlung des GMS mit DHEA (Prasteron) registriert. Klinische Studien mit täglicher intravaginaler Administration von 6.5 mg DHEA (Prasteron) zeigen eine statistisch signifikante günstige Wirkung auf die Symptome einer vulvo-vaginalen Atrophie (GMS) (14). DHEA dringt in die Zelle ein und entfaltet dort durch die intrazelluläre Umwandlung von DHEA zu Estradiol und Testosteron eine intrakrine Wirkung. Die aus DHEA gebildeten intrazellulären Steroide Estradiol und Testosteron werden nicht nach aussen abgegeben: die Serum-Spiegel beider Steroide bleiben unter Therapie unverändert (15). Somit ist auch nicht mit einer Endometriumstimulation oder mit systemischen Nebenwirkungen zu rechnen. Dies bestätigen die klinischen Daten (14).
Prof. em. Dr. med. Martin Birkhäuser
Gartenstrasse 67
4052 Basel
martin.birkhaeuser@bluewin.ch
Der Autor hat keine Interessenskonflikte im Zusammenhang mit diesem Beitrag deklariert.
Für niedrig-dosierte vaginale Estrogenpräparate gilt weiterhin, dass auch bei einer Therapiedauer von > 1 Jahr eine Gestagenbeigabe nicht notwendig ist.
In Beipackzetteln von niedrigdosierten vaginalen Estrogenpräparaten wird fälschlicherweise auf ein mögliches erhöhtes Risiko für Herz-Kreislauf-Erkrankungen, Thromboembolien, Schlaganfälle und Brustkrebs hingewiesen. Diese Warnung ist nicht evidenzbasiert, widerspricht einergrossen prospektiven langdauernden Beobachtungsstudie und verunsichert einzig die Patienti . Sie sollte entfernt werden.
Bei hochdosierten vaginalen Präparaten wären höchstens Risiken zu erwarten, wie sie für eine normaldosierte transdermale systemische Gabe bekannt sind (≤ 50 μg Estradiol/Tag). Allfällige Empfehlungen zur Therapie sollten daher für beide Behandlungsprinzipien die gleichen sein. Eine zusätzliche Gestagengabe ist hier zum Endometriumschutz ausreichend.
Die in der EU geltende Limitierung einer hochdosierten vaginalen Estrogen-Therapie auf < 4 Wochen steht im Gegensatz zum Prinzip, dass eine vaginale Behandlung lange (oft über Jahre!) fortgeführt werden muss, um den Nutzen aufrecht zu erhalten, und ist nicht evidenzbasiert. Sie führt einzig dazu, dass Frauen mit schwerem genito-urethralem menopausalen Syndrom nicht mehr korrekt behandelt werden können.
Die europäische Arzneimittelbehörde (EMA) sollte nicht ohne sichere Grundlage und gegen solide Evidenz die alternativlose Behandlung eines schweren GMS mit einem hochdosierten vaginalen Estradiolpräparat durch Empfehlungen verunmöglichen, sondern sie im Rahmen einer verantwortungsbewussten Nutzen-Risiko-Abwägung sicherer machen.
Messages à retenir
Pour les préparations oestrogéniques faiblement dosées, l’adjonction d’un progestatif n’est-pas nécessaire, même en cas de traitement prolongé (> 1 année).
Les notices d’emballage de préparations hormonales vaginales faiblement dosées mentionnent à tort un risque potentiel augmenté pour les maladies cardiovasculaires, thrombo-emboliques et cérébrovasculaires ainsi que les cancers du sein. Cette mise en garde est contredite par les résultats d’une grande étude observationnelle prospective de longue durée, elle n’est pas fondée sur des preuves et devrait être éliminée des textes. Elle inquiète inutilement les patientes.
Un traitement vaginal hautement dosé provoquerait au maximum des effets secondaires comparables à un traitement hormonal systémique percutané normalement dosé (≤ 50µg estradiol / jour). Les recommandations pour les deux formes de traitement devraient donc être les mêmes. L’adjonction d’un progestatif garantit la protection de l’endomètre.
La limitation dans le temps (< 4 semaines) pour un traitement vaginal fortement dosé telle qu’elle figure dans la publication de l’UE, contredit le principe que, pour être efficace et durable, un traitement vaginal doit être maintenu pendant longtemps (souvent pendant des années). De plus, elle n’est pas fondée sur des preuves. Elle a comme seule conséquence de rendre impossible le traitement correct des femmes souffrant d’un syndrome génito-urinaire de la ménopause sévère (GMS).
L’Agence Européenne des Médicaments (EMA) ne devrait pas empêcher un traitement vaginal à l’estradiol hautement dosé, pour lequel des alternatives manquent, sans fondements sûrs et contre toute évidence. Son rôle serait bien plus de rendre plus sûr un traitement du GMS, en l’occurrence le traitement vaginal à haute dose, par une analyse soigneuse des risques et bénéfices.
1. BfArM. Risikobewertungsverfahren. Hochdosierte, estradiolhaltige Cremes: Ueberprüfung der Risiken. Release-Datum: 17.01.2020. https://www.bfarm.de/SharedDocs/Risikoinformationen/Pharmakovigilanz/DE/RV_STP/a-f/estradiol-creme.html
2. Wüest A, Stute P. Vulvovaginale Beschwerden in der Menopause, Update 2018. GYNÄKOLOGIE 3: 12-17, 2018.
3. Crandall CJ, Treatment of Vulvovaginal Atrophy. JAMA Published online September 26, 2019. doi:10.1001/jama.2019.15100
4. D. W. Sturdee and N. Panay, on behalf of the International Menopause Society Writing Group. Recommendations for the management of postmenopausal vaginal atrophy. CLIMACTERIC; Early Online, 1–14, 2010.
5. Lethaby A, Ayeleke RO, Roberts H. Local estrogen for vaginal atrophy in postmenopausal women. Cochrane Database of Systematic Reviews 2016, Issue 8. Art. No.: CD001500. DOI: 10.1002/14651858.CD001500.pub3.2016.
6. aus United States Pharmacopeia (downloaded 10. Nov. 2010)
7. 32. Constantine GD, Graham S, Lapane K et al.Endometrial safety of low-dose vaginal estrogens in menopausal women: a systematic evidence review. Menopause, 26: 800-807, 2019.
8. Crandall CJ, Diamant A, Santoro N. Safety of vaginal estrogens, a systematic review. Menoopause: January 6, 2020. doi: 10.1097/GME.0000000000001468. [Epub ahead of print]
9. Birkhäuser M, Bürki R, De Geyter C, Imthurn B et al. Aktuelle Empfehlungen zur Menopausalen Hormon-Therapie (MHT). Expertenbrief No 42 der SGGG, 2015.https://www.sggg.ch/fileadmin/user_upload/Formulardaten/42_Menopausale_Hormon-Therapie 2015.pdf
10. NAMS. Management of symptomatic vulvovaginal atrophy: 2013 position statement of The North American Menopause Society. Menopause, Vol. 20, No. 9: 888-902, 2013.
11. Faubion AA, Larkin LC, Stuenkel CA et al. Management of genitourinary syndrome of menopause in women with or at high risk for breast cancer: consensus recommendations from The North American Menopause Society and The International Society for the Study of Women’s Sexual Health Menopause: Vol. 25, No. 6: 1-13, 2018.
12. Crandall CJ, Hovey KM, Andrews CA et al. Breast cancer, endometrial cancer, and cardiovascular events in participants who used vaginal estrogen in the Women’s Health Initiative Observational Study. Menopause; Jan;25(1):11-20, 2018
13. Mitchell CM, Reed SD, Diem S et al. Efficacy of Vaginal Estradiol or Vaginal Moisturizer vs Placebo for Treating Postmenopausal Vulvovaginal Symptoms. A Randomized Clinical Trial. JAMA Intern Med; 178(5): 681–690, 2018
14. Labrie F, Archer DF, Koltun W et al Efficacy of intravaginal dehydroepiandrosterone (DHEA) on moderate to severe dyspareunia and vaginal dryness, symptoms of vulvovaginal atrophy, and of the genitourinary syndrome of menopause. Menopause; 23: 243-256, 2016.
15. Labrie F, Bélanger A, Pelletier et al. Science of intracrinology in postmenopausal women- Menopause 24: 702-712, 2017.
16. JoAnn E. Manson Andrew M. Kaunitz. Menopause Management — Getting Clinical Care Back on Track. N Engl J Med 374;9: 803-806, 2016.