Aufgrund der raschen Entwicklung im molekular-genetischen Verständnis primärer Hirntumoren wird 2021 eine Revision der WHO Klassifikation publiziert. Dort finden sich wesentliche Neuerungen zum diagnostischen Algorithmus und zur Nomenklatur primärer Hirntumoren. Diese werden unter anderem den Empfehlungen des interdisziplinären Konsortiums «cIMPACT-NOW» (Consortium to Inform Molecular and Practical Approaches to CNS Tumour Taxonomy – Not Officially WHO) entnommen.
En raison de l’ évolution rapide de la compréhension moléculaire et génétique des tumeurs cérébrales primaires, une révision de la classification de l’ OMS sera publiée en 2021. Cela comprendra des modifications importantes de l’ algorithme du diagnostic et de la nomenclature des tumeurs cérébrales primaires. Celles-ci sont tirées des recommandations du consortium interdisciplinaire «cIMPACT-NOW» (Consortium to Inform Molecular and Practical Approaches to CNS Tumour Taxonomy – Pas officiellementde l’ OMS).
Die letzte und vierte Auflage der WHO-Klassifikation von Hirntumoren, die 2016 veröffentlicht wurde, führte zu einer wesentlichen Umstrukturierung des diagnostischen Ansatzes für Hirntumoren. Obwohl streng histologische Kriterien für bestimmte Tumoren informativ sind, wurden zunehmend Bedenken hinsichtlich des subjektiven Einflusses der Neuropathologen auf die histo-pathologische Beurteilung laut. Ein klassisches Beispiel war die geringe Interobserver-Konkordanz bei der Diagnose von diffusen Gliomen mit überlappenden astrozytären und oligodendroglialen Merkmalen. Nach dem Aufkommen der standardmässigen Suche nach IDH 1/2 Mutationen und einer Ko-Deletion von 1p19q im Falle einer IDH Mutation, fielen jedoch die meisten diffusen Gliome in die Kategorien Astrozytom oder Oligodendrogliom, wodurch die mehrdeutige Bezeichnung Oligoastrozytom weitgehend obsolet wurde. Im letzten Jahrzehnt hat die umfassende molekulare Hochdurchsatz-Profilierung in Verbindung mit Fortschritten beim maschinellen Lernen die Diagnose von Hirntumoren weiter verändert, indem sie eine genauere Orientierung der Ursprungszellen eines Tumors erlauben und Hinweise auf das Therapieansprechen liefern. Zum ersten Mal wurden molekulare Parameter mit histologischen Merkmalen in einem komplementären, integrativen Format zusammengeführt, wodurch die WHO-Klassifikation einer zunehmenden Anzahl von primären Hirntumoren verfeinert wurde (1).
Angesichts des rasanten Fortschritts im molekular-genetischen Verständnis, ist es nicht verwunderlich, dass die WHO Klassifikation 2016 zum Zeitpunkt der Veröffentlichung bereits veraltet war. Die fortlaufende Entdeckung von Biomarkern und neuer medikamentöser Targets verstärkte die Notwendigkeit, den Revisionsprozess zu beschleunigen. Deshalb wurde cIMPACT-NOW gegründet, um eine zeitnahe Aktualisierungen zu vermitteln und Empfehlungen für zukünftige WHO-Publikationen zu geben (2).
Bis heute hat das Konsortium 7 Positionspapiere veröffentlicht.
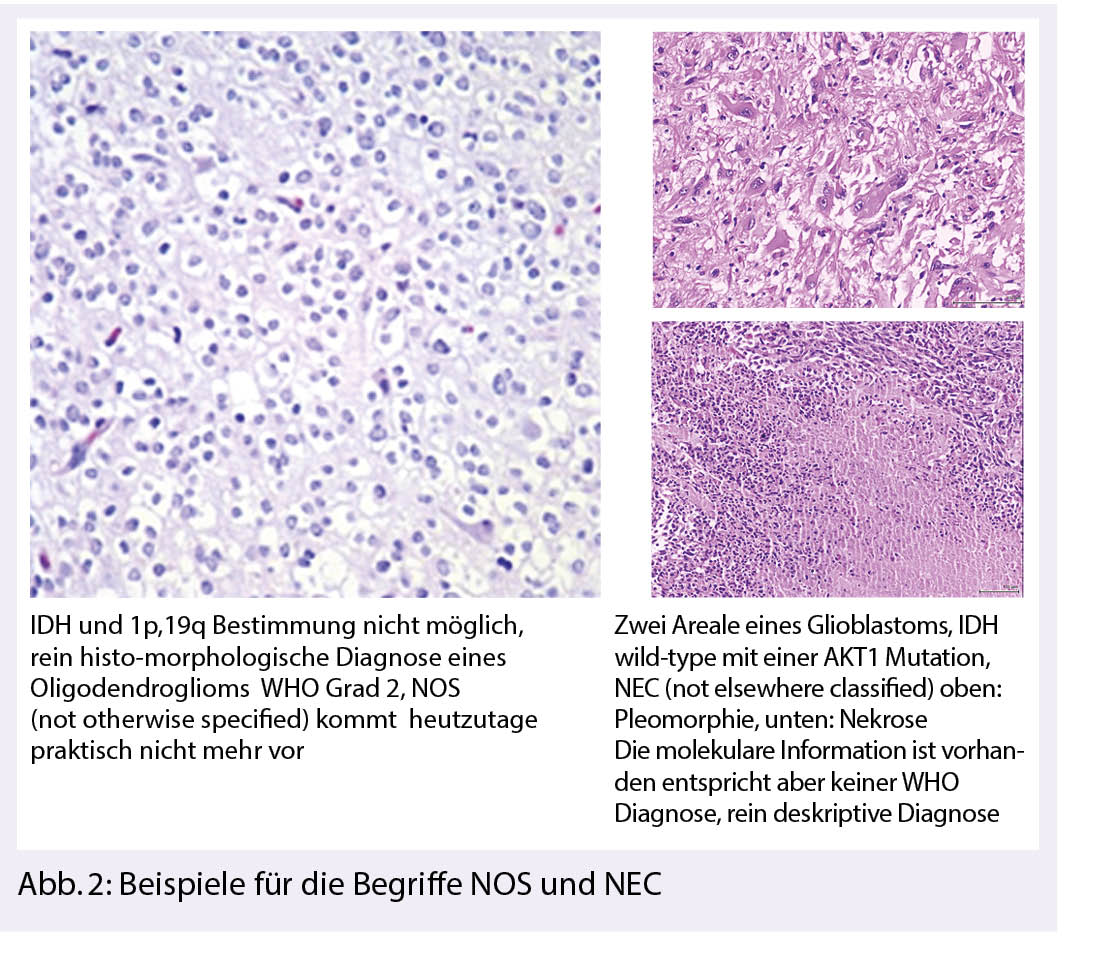
Das erste Update widmet sich der Klärung der Verwendung der Begriffe NOS (Not Otherwise Specified) und NEC (Not Elsewhere Classified). Die NOS-Bezeichnung sollte angewendet werden, wenn für die Diagnose dafür notwendige diagnostische (z. B. molekulare) Informationen für eine spezifischere Klassifizierung fehlen. Der NEC-Qualifikator kann angewendet werden, wenn eine Nichtübereinstimmung zwischen histologischen Merkmalen und molekularen Ergebnissen besteht. Alternativ kann NEC verwendet werden, wenn diagnostische Tests nicht-kanonische Ergebnisse zeigen, die eine Zuordnung zu einer bekannten WHO-Entität ausschliessen und daher auf einen neuen/entstehenden Tumortyp hindeuten (3) (Abb. 2).
Nach mehreren Fallberichten über H3K27-Mutationen bei so unterschiedlichen Tumoren wie Ependymomen der hinteren Schädelgrube (4), pilozytischen Astrozytomen und glioneuronalen Tumoren (5), entschied cIMPACT-NOW, dass hier eine Klärung angebracht ist. Daher wurde in einem zweiten Update empfohlen, den Begriff «diffuses Mittelliniengliom, H3K27m» und die dazugehörige WHO-Grad 4-Bezeichnung auf diffuse Mittelliniengliome zu beschränken und nicht auf andere Tumoren anzuwenden, die lediglich diese Mutation aufweisen. Die prognostische Bedeutung einer H3K27-Mutation in einer nicht-kanonischen Lokalisation oder Tumorentität ist noch unklar. In der gleichen Aktualisierung wurde die Diagnose des diffusen Astrozytoms, IDH-mutiert, durch die Einführung von Diagnosekriterien vereinfacht, welche eine molekulare LOH (loss of heterzygosity)-1p19q-Bestimmung nicht zwingend notwendig machen. Basierend auf immunhistochemischen Ergebnissen allein, können IDH-mutierte Gliome mit Nachweis eines Verlustes der nukleären ATRX-Expression und gleichzeitiger p53-Überexpression sicher den Astrozytomen zugeordnet und von Oligodendrogliomen abgegrenzt werden (6).
Nach dem dritten Update cIMPACT-NOW 3 können IDH-wild type Astrozytome der WHO-Grade 2 und 3 de facto als Glioblastome angesehen werden, wenn sie die folgenden molekularen Kriterien erfüllen: EGFR-Amplifikation und/oder Gewinn des gesamten Chromosoms 7 und Verlust des gesamten Chromosoms 10 (+7/-10) und/oder TERT (Telomerase reverse Transkriptase)-Promotor-Mutation.
Mit anderen Worten, zuverlässige molekulare Marker ermöglichen es, am Beispiel einer Biopsie die Diagnose eines Glioblastoms zu stellen, ohne dass die sonst notwendigen histologischen Kriterien, wie Nekrosen oder mikrovaskuläre Proliferation, vorhanden sein müssen (7).
Das vierte Update konzentriert sich auf Durchbrüche bei der Klassifizierung von IDH-, H3-Wildtyp, meist hemisphärische, pädiatrische diffuse Gliome. Es wurden sechs neue Gliom-Subtypen eingeführt, darunter diffuses Gliom, MYB-verändert, diffuses Gliom, MYBL1-verändert, diffuses Gliom, FGFR1 TKD-dupliziert, diffuses Gliom, FGFR1-mutiert, und diffuses Gliom, BRAFV600E-mutiert (ohne CDKN2A/2B-Deletion) sowie diffuses Gliom mit anderen MAPK-Signalweg-Veränderungen. Das Fazit dieses Updates war, dass diffuse Gliome im Kindesalter trotz histologischer Ähnlichkeiten zu Gliomen Erwachsener eindeutige molekulare Veränderungen aufweisen. Molekulare Tests sind für eine genaue Klassifizierung und die Identifizierung potenzieller therapeutischer Ziele unerlässlich (8).
In der fünften cIMPACT-NOW Publikation werden IDH-mutierte Astrozytome in einer eigenen Kategorie in WHO Grad 2-4 unterteilt. Sie haben generell einen weniger aggressiven klinischen Verlauf im Vergleich zu ihren Wildtyp-Pendants, mit Ausnahme der IDH-mutierten Astrozytome mit einer homogzygoten CDKN2A/B Deletion WHO Grad 4.
Anstelle von römischen Ziffern wurden arabische Ziffern für die Grade 2-4 vergeben, wobei Tumore des Grades 3 eine «signifikante» mitotische Aktivität aufweisen. Aufgrund der prognostischen Auswirkungen wird empfohlen, Astrozytome WHO Grad 2 und 3 auf homozygote CDKN2A/B-Deletionen zu testen. Tumoren mit homozygoten CDKN2A/B-Deletionen oder mikrovaskulärer Proliferation und/oder Nekrose sollen fortan als IDH-mutierte Astrozytome WHO Grad 4 und nicht mehr als Glioblastome bezeichnet werden (9).
Im sechsten Update werden allgemeine Prinzipien für die künftige Einteilung und Klassifizierung der hirneigenen Tumoren skizziert. So wird z. B. die Ersetzung von «Entität» und «Variante» durch «Typ» und «Subtyp» empfohlen. Vier weitere wichtige Empfehlungen werden in der WHO Klassifikation erscheinen: die erste Kategorie umfasst neu anerkannte Typen, Subtypen, Diagnosekriterien oder Tumorfamilien. Ein Beispiel ist das neu anerkannte diffuse Gliom, H3.3 G34-mutant mit einem insgesamt längeren Überleben im Vergleich zum klassischen IDH-Wildtyp-Glioblastom. Ein weiteres Beispiel ist das Astroblastom, MN1-alteriert (10). Zu den Nomenklaturänderungen (Kategorie 2), gehört der Wegfall der Lokalisation für das chordoide Gliom (ehemals «of the third ventricle»). Darüber hinaus sollen Ependymome kombinierte histologisch-molekulare Bezeichnungen tragen, die auf den einzigartigen epigenetischen und genetischen Signaturen an verschiedenen anatomischen Stellen beruhen. Die Kategorie der supratentoriellen Ependymome umfasst die klinisch aggressiven RELA-Fusion (C11orf95)-positiven und YAP1-Fusion-positiven Tumoren, die mit einer günstigeren Prognose assoziiert sind. Ependymome, die in der hinteren Schädelgrube entstehen, werden in den pädiatrischen Typ/PFA und den Erwachsenentyp/PFB unterteilt, wobei letzterer mit einer besseren Prognose assoziiert ist (10).
Einige bestehende Typen wie das extraventrikuläre Neurozytom und das pilozytische Astrozytom (Kategorie 3) werden keine Änderungen erfahren. Die vierte Kategorie umfasst Entitäten mit unzureichender Literatur, um eine Empfehlung auszusprechen. Beispiele sind pilozytäre Astrozytome mit anaplastischen Merkmalen, die durch ihr Methylierungsprofil definiert sind, und verschiedene infantile hemisphärische Gliome, die durch eine spezifische molekulare Signatur charakterisiert sind, z. B. Tyrosinrezeptor-Kinase-Fusionen wie NTRK, MET, ALK oder ROS1 (8).
cIMPACT-NOW 7 konzentriert sich erneut auf Fortschritte in der Klassifikation von Ependymomen, wobei die meisten Änderungen bereits im sechsten Update angedeutet wurden. Angesichts der häufigen Rezidive wurde das myxopapilläre Ependymom auf WHO-Grad 2 hochgestuft. Darüber hinaus wurde eine neue, klinisch aggressive, molekulare Untergruppe des spinalen Ependymoms, das MYCN-amplifizierte Ependymom, anerkannt (10, 11).
Jüngste Publikationen, die seit dem letzten cIMPACT-NOW Update erschienen sind, unterstreichen die unterschiedliche molekulare Landschaft von Hirntumoren bei Kindern. Bis heute wurden vier molekulare Subtypen des diffusen Mittellinienglioms mit H3.1/3.2 K27-, H3.3 K27-Mutationen, EZHIP-Überexpression und EGFR-Mutationen berichtet (13). Die meisten EGFR-mutierten Tumoren sind bithalamisch, während H3.1 K27-Mutationen die Pons bevorzugen (14). Der polymorphe niedriggradige neuroepitheliale Tumor (PLNTY) der Jugend ist ein neu anerkannter, Epilepsie-assoziierter Tumortyp mit MAPK-Veränderungen, der bei Kindern und jungen Erwachsenen gefunden wurde (15). Zwei weitere Entitäten, der diffuse glioneuronale Tumor mit oligodendroglialen Merkmalen und nukleären Clustern (DGONC) und der myxoide glioneuronale Tumor, haben sich der wachsenden Liste glioneuronaler und neuronaler Tumoren angeschlossen (16).
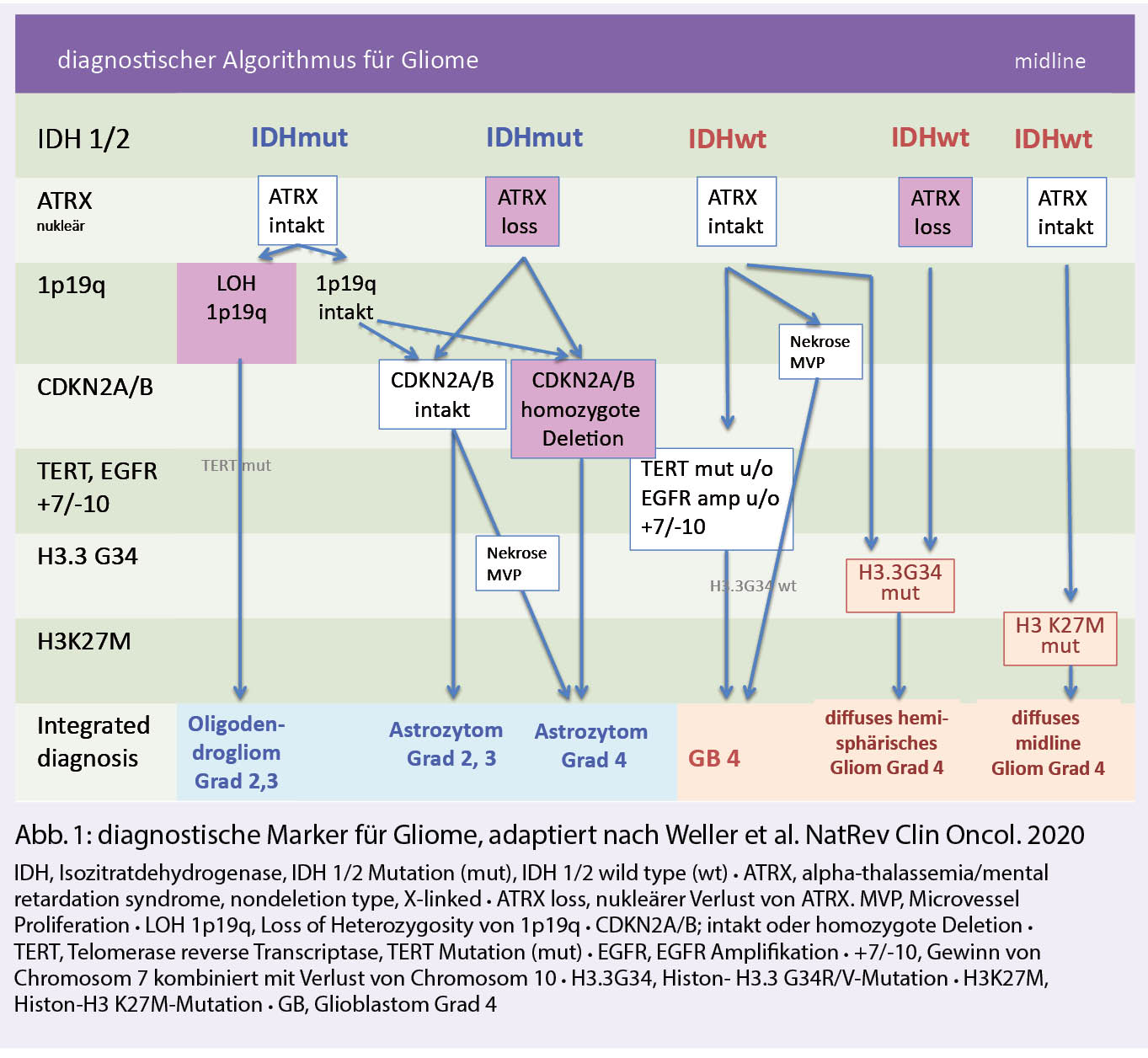
Zusätzlich zu den pädiatrischen Tumoren unterstreicht die Entdeckung aktionsfähiger Zielmoleküle in etwa einem Drittel der Gliome die Bedeutung eines optimalen Gewebemanagements, um der Herausforderungen der personalisierten Medizin zu begegnen. Vor dem Hintergrund begrenzter Gewebeverfügbarkeit besteht der Bedarf an einem optimalen Diagnosealgorithmus (Abb. 1), einschliesslich der Wahl der Analysemethoden sowie der Gewebeanalyse im Rahmen von Studien.
Patientenalter, histologische Diagnose und Tumorlokalisation sind wichtige Faktoren für die Auswahl geeigneter Nukleinsäuretests wie FISH, PCR, DNA- und RNA-Sequenzierung.
Next Generation Sequencing, (NGS) setzt sich als kosteneffiziente Methode zur gleichzeitigen Untersuchung mehrerer Gene durch.
Bei histologisch unklaren Tumoren ist das DNA-Methylom-Profiling ein weiteres Analyseverfahren, das den diagnostischen Prozess ergänzen kann. So lassen sich z. B. infantile hemisphärische Gliome, die histologisch vielfältig sind und eine Vielzahl von Genfusionen beherbergen, tendenziell einer eigenen Methylierungsklasse zuordnen (18). Noch sind nicht alle regulatorischen und methodologischen Fragen geklärt. Im Falle von NTRK-Fusionen z.B. hat sich das Methylierungsprofiling als nicht informativ erwiesen (19).
Zusammenfassend:
Nach den neuen Empfehlungen der cIMPACT-NOW kann bei einem Gliom der histologischen WHO-Grade 2 oder 3, das keine IDH-Mutation aufweist, ein Glioblastom diagnostiziert werden, wenn entweder eine EGFR-Amplifikation oder ein chromosomaler Gewinn auf Chromosom 7 bei komplettem Verlust von einem Chromosom 10 oder aber eine TERT (Telomerase reverse Transkriptase)-Promotor-Mutation vorliegt.
Astrozytäre Tumoren mit IDH-Mutation werden als Astrozytom, IDH-mutiert, WHO-Grad 2, 3 oder 4 klassifiziert. Der Verlust von CDKN2A wird als schlechter prognostischer Marker erkannt, der die Zuordnung zu einem WHO Grad 4 erlaubt.
Frühere IDH-mutierte Glioblastome gelten jetzt als Astrozytome, IDH-mutiert, WHO-Grad 4. Zu den IDH-mutierten Astrozytomen mit einer schlechteren Prognose gehören infratentorielle IDHmut Astrozytme, welche zudem non-IDH1-R132H Varianten aufweisen (21) und primär Mismatch-Repair defiziente IDHmut Astrozytome mit einem medianen Überleben von lediglich 15 Monaten (22).
Neue Kategorien sind auch die Histon-mutierten Gliome, hier speziell das Histon-3 K27M-mutierte diffuse Mittellinien-Gliom mit schlechter Prognose, und das Histon-3.3 G34-mutierte diffuse hemisphärische Gliom. Beide Gliome wurden früher überwiegend als Glioblastome diagnostiziert.
Ein ähnlicher Artikel zu diesem Thema wurde auf Anfrage an Prof. E. J. Rushing in der englischsprachigen Zeitschrift MEMO (Magazine of European Medical Oncology) eingereicht.
Copyright bei Aerzteverlag medinfo AG
Dr. med. Silvia Hofer
Universitätsspital Zürich
Institut für Pathologie und Molekularpathologie
Schmelzbergstrasse 12
8091 Zürich
silvia.hofer@usz.ch
Prof. Dr. med. Elisabeth Jane Rushing
Institut für Neuropathologie
Universitätsspital Zürich
Frauenklinikstr. 38
8091 Zürich
Die Autorinnen haben im Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
◆ Die aktuelle Revision der WHO-Klassifikation von Tumoren des Zentralnervensystems 2021 führt entscheidende Veränderungen in der Diagnostik und Nomenklatur ein, mit Folgen auf die Art, wie wir Patienten mit Gliomen behandeln.
◆ Heute kann die Diagnose eines Glioblastoms nicht nur auf der Grundlage morphologischer Kriterien gestellt werden, sondern zweifelsfreier auf der Basis mehrerer molekularer Marker, was im Falle von kleinen Gewebeproben besonders hilfreich ist.
◆ Für viele der neu definierten Krankheitsentitäten in der neuesten WHO-Klassifikation sind noch keine Daten über spezifische Behandlungsergebnisse verfügbar; die Extrapolation aus Daten früherer klinischer Studien bleibt eine Herausforderung.
Messages à retenir
◆ La révision actuelle de la classification des tumeurs du système nerveux central 2021 de l’ OMS introduit des changements cruciaux dans le diagnostic et la nomenclature, avec des conséquences sur la façon dont nous traitons les patients atteints de gliomes.
◆ Aujourd’ hui, le diagnostic d’ un glioblastome peut être établi non seulement sur la base de critères morphologiques, mais aussi, de manière plus univoque, sur la base de multiples marqueurs moléculaires, ce qui est particulièrement utile dans le cas de petits échantillons de tissu.
◆ Pour de nombreuses entités pathologiques nouvellement définies dans la dernière classification de l’ OMS, les données sur les résultats de traitements spécifiques ne sont pas encore disponibles ; l’ extrapolation à partir de données provenant d’ essais cliniques antérieurs reste un défi.
1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. International Agency for Research on Cancer, World Health Organization Histological Classification of Tumours of the Central Nervous System (Revised 4th edition). Lyon: 2016.
2. Louis DN, Aldape K, Brat DJ, et al. Announcing cIMPACT-NOW: the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy. Acta Neuropathol. 2017;133:1-3
3. Louis DN, Wesseling P, Paulus W, et al. cIMPACT-NOW update 1: Not Otherwise Specified (NOS) and Not Elsewhere Classified (NEC). Acta Neuropathol. 2018;135:481-484.
4. Gessi, Capper D, Sahm F, et al. Evidence of H3 K27M mutations in posterior fossa ependymomas. Acta Neuropathol .2016;132:635-7.
5. Orillac C, Thomas C, Dastagirzada, Y, el al. Pilocytic astrocytoma and glioneuronal tumor with histone H3 K27M mutation. Acta Neuropathol Commun. 2016;4:84
6. Louis DN, Giannini C, Capper D, et al. cIMPACT-NOW update 2: diagnostic clarifications for Diffuse Midline Glioma, H3 K27M–mutant and Diffuse Astrocytoma/Anaplastic Astrocytoma, IDH-mutant. Acta Neuropathol 135:639-642, 2018.
7. Brat DJ, Aldape K, Colman H, et al. cIMPACT-NOW update 3: recommended diagnostic criteria for „Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV“. Acta Neuropathol. 2018;136:805-810.
8. Ellison DW, Hawkins C, Jones DTW, et al. cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF V600E mutation. Acta Neuropathol. 2019;137:683-687.
9. Brat DJ, Aldape K, Colman H, et al. cIMPACT-NOW update 5: recommended grading criteria and terminologies for IDH-mutant astrocytomas Acta Neuropathol. 2020;139:603-608.
10. Louis DN, Wesseling P, Aldape K, et al. cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020;30:844-856.
11. Ghasemi DR, Sill M, Okonechnikov K, et al. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol. 2019;138:1075-1089.
12. Ellison DW, Aldape KD, Capper D, et al. cIMPACT-NOW update 7: advancing the molecular classification of ependymal tumors. Brain Pathol. 2020;30:863-866.
13. Cooney TM, Lubanszky E, Prasad R, et al. Diffuse midline glioma: review of epigenetics. J Neurooncol 2020. Online ahead of print.
14. Antony R, Solomon D, Lee-Way J, et al. HGG-12. Primary bithalamic glioma with EGFR mutation: A rare case report. Neuro Oncol. 2018; 20(Suppl 2): i91.
15. Huse JT, Snuderl M, Jones DTW, et al. Polymorphous low-grade neuroepithelial tumor of the young (PLNTY): an epileptogenic neoplasm with oligodendroglioma-like components, aberrant CD34 expression, and genetic alterations involving the MAP kinase pathway. Acta Neuropathol 2017;133:417-429.
16. Deng MY, Sill M, Sturm D, et al. Diffuse glioneuronal tumour with oligodendroglioma-like features and nuclear clusters (DGONC) – a molecularly defined glioneuronal CNS tumour class displaying recurrent monosomy 14. Neuropathol Appl Neurobiol. 2020;46:422-430.
17. Jonsson P, Lin AL, Young RJ, et al. Genomic Correlates of Disease Progression and Treatment Response in Prospectively Characterized Gliomas. Clin Cancer Res. 2019;25: 5537–5547
18. Capper D, Stichel D, Sahm F, et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol. 2018;136:181-210.
19. Torre, M, Vasudevarja V, Serrano J, et al. Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol Commun. 2020;8:107
20. Weller M, van den Bent M, Preusser M, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. doi: 10.1038/s41571-020-00447-z.
21. Banan R, Stichel D, Beck A, et al. Infratentorial IDH-mutant astrocytoma is a distinct subtype. Acta Neuropathol. 2020; 140: 569–581
22. Suwala A, Stichel D, Schrimpf D et al. Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. Acta Neuropathol 2021; 141:85-100
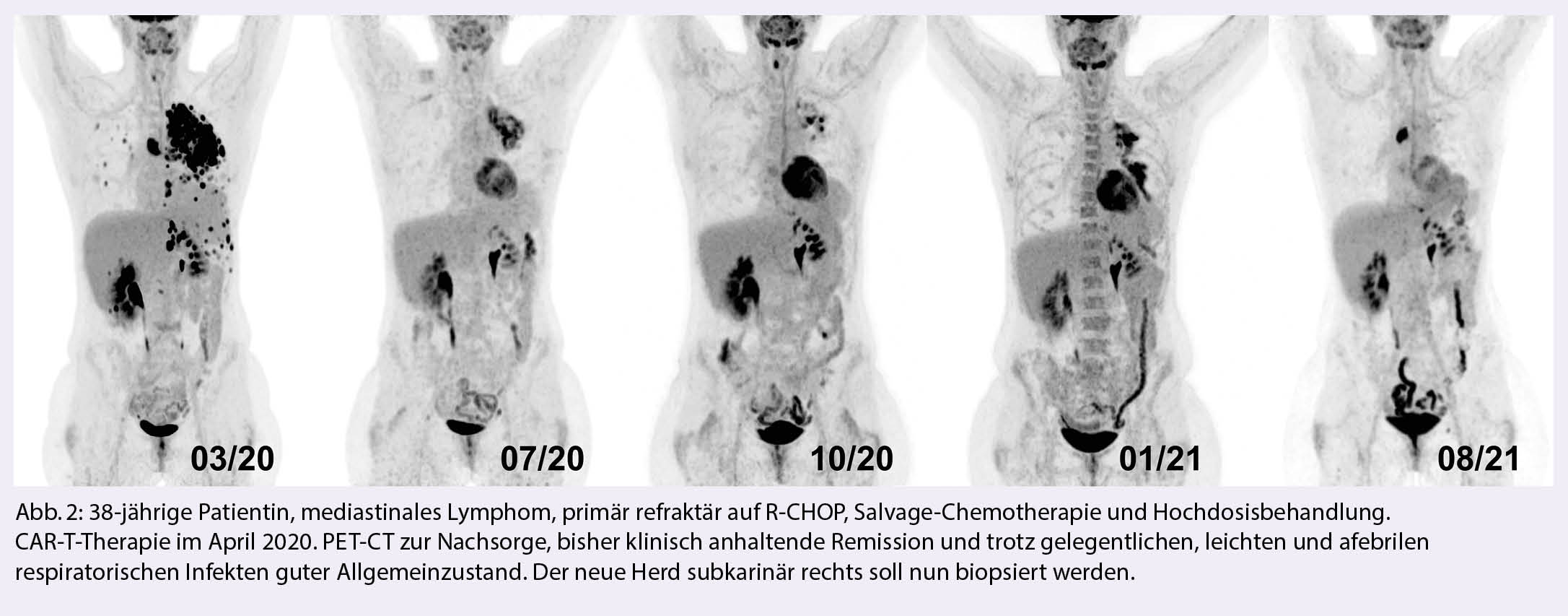
Seit der Zulassung der ersten CAR-T-Therapie in der Schweiz im Oktober 2018 und der schweizweit ersten Behandlung am Inselspital im Januar 2019 wurden bis zum Januar 2021 hierzulande 110 dieser Behandlungen durchgeführt (SBST, Bern Januar 2021). In diesem Übersichtsartikel besprechen wir die aktuelle Datenlage dieser neuen Therapieoption und kommentieren sie mit unseren eigenen Erfahrungen von bisher ca. 80 PatientInnen, welche wir bei aggressivem B-Zell-Lymphom, akuter lymphatischer Leukämie, Mantelzelllymphom und multiplem Myelom behandelt haben.
Depuis l’autorisation de la première thérapie CAR-T en Suisse en octobre 2018 et le premier traitement en Suisse à l’Insel-spital en janvier 2019, 110 de ces traitements ont été réalisés dans notre pays d’ici janvier 2021 (SBST, Berne janvier 2021). Dans cet article de synthèse, nous discutons des données actuelles sur cette nouvelle option thérapeutique et nous la commentons à l’aide de notre propre expérience d’environ 80 patients que nous avons traités jusqu’à présent pour un lymphome agressif à cellules B, une leucémie aiguë lymphoblastique, un lymphome à cellules du manteau et un myélome multiple.
Einführung, Indikationen
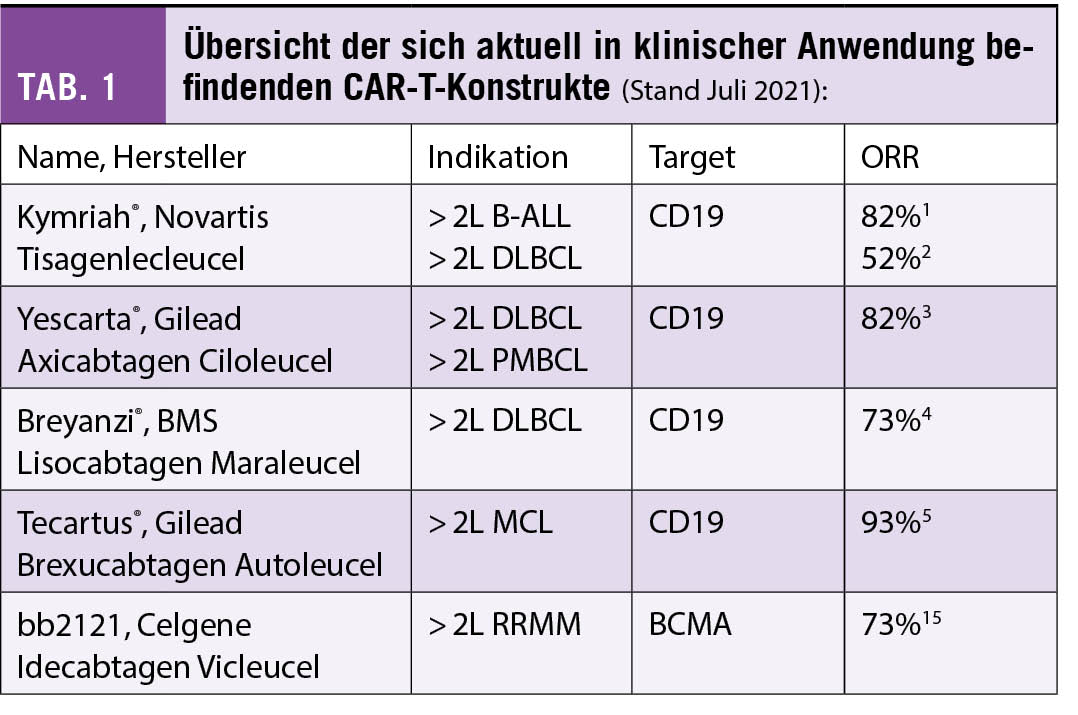
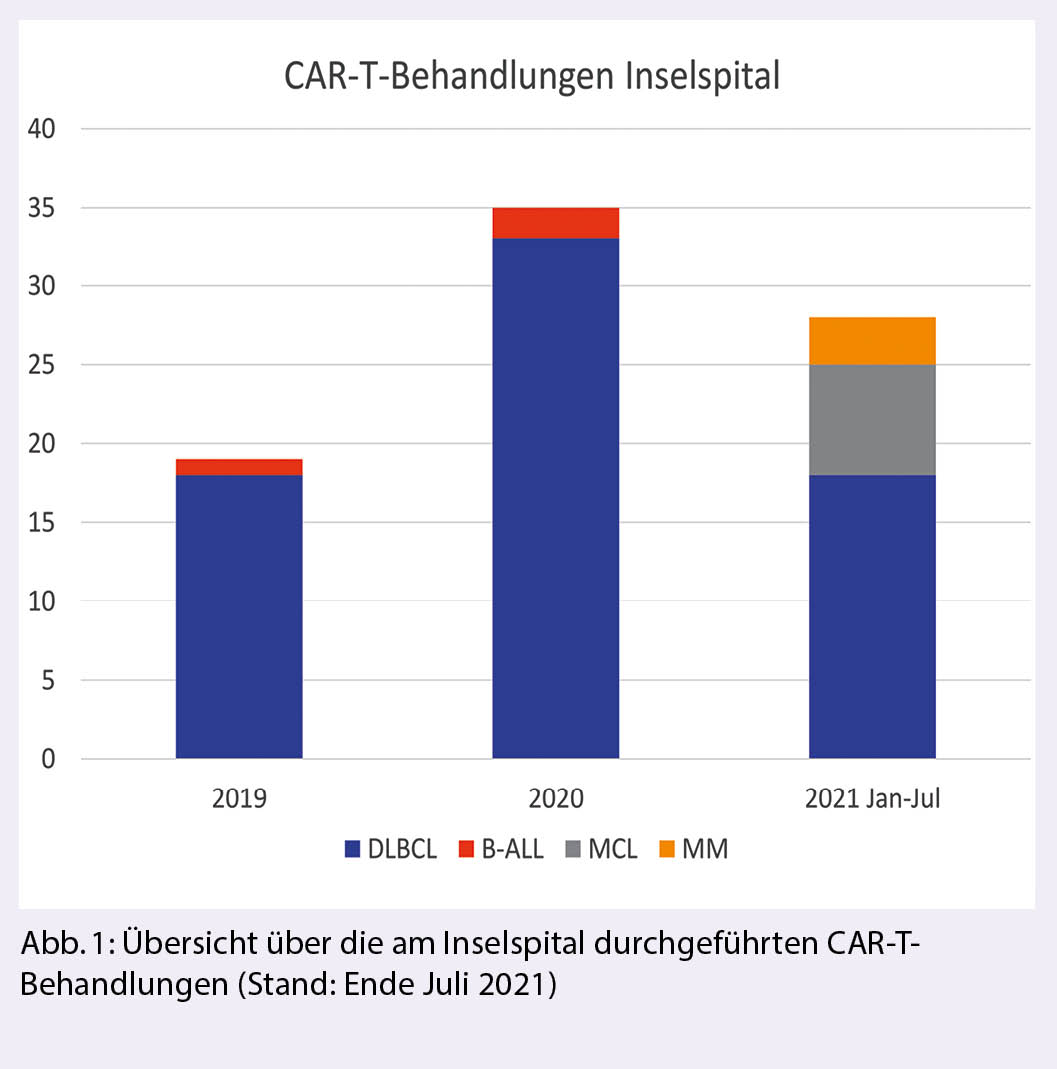
Die Zulassung der ersten CAR-T-Therapie (Kymriah®, Tisagenlecleucel, Novartis) zur Behandlung von rezidivierender oder refraktärer B-ALL (1) und rezidivierendem diffusem grosszelligem B-Zell-Lymphom (DLBCL) (2), in beiden Fällen nach mindestens zwei Vortherapien, erfolgte in der Schweiz im Oktober 2018. Im April 2019 wurde auch Yescarta® (Axicabtagen Ciloleucel, Gilead) zur Behandlung von rezidivierendem DLBCL oder primär mediastinalem grosszelligem B-Zell-Lymphom (PMBCL) nach zwei vorgegangenen Therapielinien zugelassen (3). Breyanzi® (Lisocabtagen Maraleucel, BMS) wurde Anfang 2021 durch die FDA aufgrund der Resultate der Transcend-Studie (4) ebenfalls für die Behandlung von DLBCL nach zwei Therapielinien zugelassen, die Zulassung in der Schweiz steht derzeit noch aus. Das Inselspital hat an einer Phase II und einer Phase-III-Studie mit diesem Produkt teilgenommen. Weiter verabreichten wir PatientInnen mit rezidivierendem Mantelzelllymphom (MCL) nach zwei Therapielinien inklusive Versagen von Ibrutinib das CAR-T-Präparat Tecartus® (Brexucabtagen Autoleucel, Kite) (5) im Rahmen eines Early Access Programmes. Im Frühling/Sommer 2021 wurden zudem die ersten drei PatientInnen mit dem anti-BCMA CAR-T-Konstrukt «bb2121» von BMS/Celgene bei rezidivierendem multiplem Myelom (RRMM) im Rahmen der Phase-III-Studie KarMMa-3 behandelt. Der aktuelle Stand der Zulassungen ist in Tabelle 1 zusammengefasst. Bis zum 2. August 2021 haben wir am Inselspital Bern total 82 CAR-T-Behandlungen durchgeführt. Während aggressive Lymphome weiterhin den grössten Teil der Behandlungen ausmachen, nimmt die Zahl der Behandlungen in den weiteren Indikationen stetig zu (Abb. 1).
Ablauf
Auf Zuweisung hin werden die PatientInnen, die für eine CAR-T-Therapie in Frage kommen, zu einem Vorgespräch gesehen. Die Indikationsstellung zur CAR-T-Behandlung erfolgt interdisziplinär am Lymphom-, Myelom- oder Leukämie-Tumorboard. Neben der Medizinischen Onkologie, der Radiologie und der Pathologie sind auch die SpezialistInnen der Hämatologie und des Hämatologischen Zentrallabors beteiligt, welche sowohl diagnostische (Morphologie, Flowzytometrie, molekulare Diagnostik) Aufgaben übernehmen als auch die Apherese, die initiale Präparation für das Kymriah-Produkt, den Export und Import der Zellen, die Lagerung und die Reinfusion der CAR-T-Zellen durchführen. Im Rahmen der Indikationsstellung zur CAR-T-Zell-Therapie fordern wir eine bioptische Verifizierung des vermuteten Lymphomprogresses oder Rezidivs, da sich hier auch Zweitmalignome gezeigt haben, und damit die Indikation zur CAR-T-Behandlung entfällt. Auch wenn die Voraussetzungen für eine Kostenübernahme der Behandlung klar geregelt sind, machen wir nicht selten die Erfahrung, dass die Erteilung der Kostengutsprache durch die Krankenkassen immer noch mit viel Mühe und Arbeit verbunden ist, und dies die Behandlung weiter verzögern kann. Im Rahmen einer Dissertation haben wir eine durchschnittliche Bearbeitungsdauer auf Seiten der Krankenkasse von 34 Tagen (1-70 Tage) ermittelt. Dieser Zeitrahmen und die verbleibenden Möglichkeiten für die Bridging-Therapie müssen bei der Indikationsstellung für eine CAR-T-Zell-Therapie berücksichtigt werden.
Während für die Behandlung in CAR-T Studien strikte Einschlusskriterien weit über den Zulassungstext hinaus gelten, haben wir die Erfahrung gemacht, dass die CAR-T Therapie auch von älteren PatientInnen und solchen mit gewissen Komorbiditäten, welche für eine Studienbehandlung nicht qualifiziert hätten, gut toleriert wird. Wenn immer möglich und falls noch nicht durchgeführt, sollten die PatientInnen, bei welchen eine CAR-T-Zell Therapie vorgesehen ist, gegen Covid-19 geimpft sein.
Erster Schritt in Richtung Behandlung mit CAR-T-Zellen ist die Lymphapherese, welche in der Regel die Einlage eines Dialysekatheters erfordert. Die Modalitäten der Sammlung sind für alle Produkte unterschiedlich, und die Komplexität zu deren Implementierung im Rahmen der Zertifizierung durch die Firmen sind nicht zu unterschätzen (6). Nach erfolgter Sammlung werden die T-Zellen je nach Produkt bei Raumtemperatur oder, nach einer Aufarbeitung durch das Stammzelllabor, tiefgefroren an den Hersteller verschickt. Der Produktionsprozess umfasst die Isolation der T-Zellen aus dem Lymphapheresat, die Aktivierung der isolierten T-Lymphozyten, die Transfektion der T-Zellen mit dem spezifischen Genkonstrukt, sodass diese den entsprechenden chimären T-Zell-Rezeptor exprimieren, und die abschliessende Vermehrung der transfizierten T-Lymphozyten. Der ganze Prozess ist von den Behörden zugelassen, d.h. Modifikationen können ohne grossen Aufwand der involvierten Firmen nicht vorgenommen werden. Für Aussenstehende ist die Produktion weitgehend eine Blackbox, und detaillierte Informationen zum individuellen Produkt liegen nicht vor. Das gefrorene Endprodukt wird anschliessend zurück in unser Stammzelllabor gesandt, wo es bis zur finalen Verwendung bei -170°C in Stickstoff gelagert wird. Zwischen der Sammlung und der Lieferung vergehen gewöhnlich 3-4 Wochen. Wir haben für beide kommerziellen CARs auch Produktionsversager verzeichnen müssen, was mehrere Lymphapheresen zur Herstellung eines finalen Produktes erforderlich gemacht hat. In einzelnen Fällen haben wir auch «out-of-specification» Produkte verabreichen müssen.
Eine meist erfolgreiche Produktion vorausgesetzt, werden die PatientInnen anschliessend für die eigentliche CAR-T-Therapie hospitalisiert. Dies ist gesetzlich gefordert, da die Vergütung derzeit nur im stationären Setting möglich ist. Gewöhnlich ist mit einer Aufenthaltsdauer von 3 Wochen zu rechnen. Wenn nicht kürzlich erfolgt, wird bei Therapiebeginn eine radiologische Standortbestimmung mittels CT Hals-Thorax-Abdomen und Schädel-MRI durchgeführt. Diese dienen neben dem Therapiemonitoring auch dazu, bei Auftreten von Nebenwirkungen eine initiale Vergleichsuntersuchung zur Verfügung zu haben. Während die Lymphodepletion per se nicht kardiotoxisch ist, gehört eine kardiologische Standortbestimmung inklusive EKG und Echokardiographie, wenn nicht kürzlich erfolgt, zum Workup ebenso dazu wie definierte Laboruntersuchungen. Zudem werden vor der Aufnahme der Therapie wie auch an verschiedenen Zeitpunkten im Verlauf Blut- und ggf. Knochenmarksproben für unsere Biobank abgenommen.
Die lymphodepletierende Chemotherapie mit Fludarabin und Cyclophosphamid wird über drei Tage durchgeführt und meist gut toleriert. Für beide kommerziellen Produkte verwenden wir hierzu Fludarabin und Cyclophosphamid mit den Dosierungen der Zulassungsstudien, lediglich für beide Medikamente etwas höhere Dosierungen vor der Applikation von Yescarta® (30 statt 25 mg/m2 Fludarabin und 500 statt 250 mg/m2 Cyclophosphamid). Die wichtigsten Nebenwirkungen dieser lymphodepletierenden Therapie umfassen Müdigkeit, Übelkeit, Erbrechen, Diarrhoe und eine vorübergehende Myelosuppression. Da die PatientInnen in der Regel umfangreich vorbehandelt sind und es durch die Lymphodepletion längerfristig zu einer relevanten B-Zell-Depletion kommt, verabreichen wir an den Tagen -2 und +7 Immunglobuline. Zur Verkürzung der möglichen Neutropenie erhalten die PatientInnen einmalig peg-Filgrastim, da infektiöse Trigger das Auftreten von Nebenwirkungen begünstigen können.
Nach der CAR-T-Reinfusion, in der Regel 1-2 Tage nach Ende der Lymphodepletion, müssen die PatientInnen mindestens 14 Tagen stationär überwacht werden. Nach dieser Zeit ist das Auftreten von Nebenwirkungen unwahrscheinlich. Eine Betreuung im ambulanten Setting ist schon nur aus regulatorischen Gründen nicht möglich. Wir haben aber auch PatientInnen behandelt, bei welchen die Betreuung aus rein medizinischer Sicht sicherlich im ambulanten Setting möglich gewesen wäre.
Nebenwirkungen und deren Management
Im Allgemeinen ist die CAR-T-Therapie gut verträglich und kann somit auch bei älteren PatientInnen zum Einsatz kommen. Schwerwiegende Nebenwirkungen, vor allem das Cytokine Release Syndrom (CRS) und das Immunoeffektorzell-assoziierte Neurotoxizitätssyndrom (CAR-Related Encephalopathy Syndrome (CRES) oder Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS)), mit potentiell letalem Ausgang sind jedoch möglich; in der Literatur wird dies mit 1-2 % angegeben (7). Prospektiv validierte Screeninginstrumente fehlen, sodass für den Einzelfall nicht zuverlässig vorausgesagt werden kann, bei welchen PatientInnen stärkere Nebenwirkungen zu erwarten sind. Dennoch wurden in den bisherigen Studien Risikofaktoren identifiziert, welche Anhaltspunkte für die Entwicklung höhergradiger CRS geben können. So sind eine hohe Tumorlast, ein reduzierter Allgemeinzustand und die frühe Entwicklung eines CRS in den ersten drei Tagen nach der T-Zell-Reinfusion mit schweren Verläufen assoziiert (8,9). Mit zunehmender klinischer Erfahrung der Zentren und früherer medikamentöser Intervention hat die Inzidenz von schweren CRS über die Zeit abgenommen (10–12).
Während bei Hochdosischemotherapie mit autologer Stammzelltransplantation infektiöse Komplikationen während der Aplasiephase im Vordergrund stehen, sind die Nebenwirkungen von CAR-T-Behandlungen vor allem auf Immunphänomene, ausgelöst durch die Zytokinfreisetzung, zurückzuführen. Da aufgrund der lymphodepletierenden Chemotherapie eine relevante Myelosuppression eintritt, gehört eine Infektfokussuche und ggf. –Behandlung aber in jedem Fall zum Workup bei Fieber im Rahmen der CAR-T-Therapie dazu.
Cytokine release syndrome (CRS)
Durch aktivierte oder lysierte Effektorzellen (CAR-T-Zellen, Makrophagen) können grosse Mengen proinflammatorischer Zytokine wie IFN-Gamma, TNF-Alpha, IL-6 und IL-1 freigesetzt werden. Diese Zytokinfreisetzung führt zu einer weiteren Aktivierung des Immunsystems und zu Symptomen wie Fieber, Hypotonie und Hypoxämie. Weiter kann es zu einer Endothelaktivierung mit «capillary leak» und disseminierter intravaskulärer Gerinnung kommen.
Die Häufigkeit von CRS in allen Schweregraden wird in den Zulassungsstudien mit einer Frequenz von 40 bis 80% angegeben (13).Neben Unterschieden im Nebenwirkungsprofil der verschiedenen CAR-Konstrukte erschweren auch verschiedene Methoden der Gradierung des CRS die Vergleichbarkeit zwischen den Studien. Wir verwenden die Gradierung nach Lee et al (14).
Mit der Einführung neuer CAR-T-Konstrukte mit neuen Zielen wandeln sich auch die Häufigkeit und vor allem das zeitliche Auftreten der Nebenwirkungen. Während Zytokin-vermittelte Nebenwirkungen bei mit Anti-CD19-CARs behandelten PatientInnen gewöhnlich um den 3.- 6. Tag nach Infusion der Zellen auftreten (2, 3), entwickeln PatientInnen, die mit Anti-BCMA-CARs therapiert werden tendenziell früher CRS, häufig bereits am 1. und 2. Tag nach der Infusion (15). Auch hier deckt sich unsere bisher beschränkte Erfahrung mit den Daten aus den Zulassungsstudien.
Die Behandlung des CRS umfasst neben Antipyretika zur Fiebersenkung und intravenöser Volumengabe zur Stabilisierung des Kreislaufes auch Medikamente, welche in den Zytokinstoffwechsel eingreifen. Etabliert hat sich einerseits die Anwendung von Tocilizumab, welches mit hoher Affinität an den IL-6-Rezeptor bindet und diesen blockiert. Am Inselspital verabreichen wir Tocilizumab nach erstmaligem Auftreten von Fieber nach der CAR-T-Reinfusion und nach Prämedikation mit Dexamethason. In refraktären Fällen kommt auch der lösliche IL-6-Rezeptor Siltuximab zum Einsatz. Durch frühe Intervention und den niederschwelligen Einsatz von Tocilizumab und Steroiden kann die Entwicklung eines höhergradigen CRS verhindert werden, ohne dass dadurch der Therapieerfolg negativ beeinflusst wird (11, 12, 16). Dennoch kann bei schweren Symptomen eine intensivmedizinische Behandlung nötig werden. In einem Fall haben wir ein letal verlaufendes Makrophagenaktivierungssyndrom am Tag 43 gesehen.
Aufgrund der Immunsuppression müssen auch infektiöse Ursachen von Fieber und Hypotonie ausgeschlossen bzw. proaktiv adäquat behandelt werden, zumal auch infektiöse Trigger schwere CRS-Verläufe auslösen oder begünstigen können (7). Etwa jeder vierte Patient erleidet im Rahmen der CAR-T-Behandlung einen Infekt, meist sind dies Bakteriämien und virale Atemwegsinfekte (17). Die immunsuppressive Wirkung von Tocilizumab scheint hier nicht die primäre Ursache zu sein (18).
Neben der klinischen Überwachung können auch verschiedene Laborparameter Anhaltspunkte zum Verlauf des CRS liefern, so z.B. das im Serum gemessene IL-6 oder das Ferritin. Es muss jedoch beachtet werden, dass die Aussagekraft der gemessenen Werte durch die zytokinhemmende Therapie verfälscht werden kann, insbesondere nach Verabreichung von Siltuximab, welches das freie IL-6 bindet. So kann es unter dieser Behandlung zu relevanten Infekten bei normwertigem CRP kommen.
Die Ursache der Neurotoxizität der CAR-T-Zellen ist nicht vollständig geklärt. Die CAR-T-Zellen scheinen Zytokine auszuschütten, welche das Endothel aktivieren und so eine Störung der Blut-Hirn-Schranke bewirken. In der Folge können einerseits T-Zellen das Hirngewebe infiltrieren und so direkt entzündliche Reaktionen auslösen, andererseits kommt es durch die Störung der Blut-Hirn-Schranke zu einem Ödem (19). PatientInnen mit ZNS-Lymphom waren unter anderem aufgrund der Befürchtung, häufiger Neurotoxizität zu entwickeln, für die erwähnten Zulassungsstudien nicht zugelassen. Fallserien legen jedoch nahe, dass weder die Rate noch die Intensität der Neurotoxizität exzessiv hoch sind (20, 21). Hingegen scheint die Remissionsdauer bei diesen PatientInnen kürzer zu sein (21).
In den meisten Fällen (>90%) geht dem Auftreten eines ICANS ein CRS voran, die Entwicklung von neurologischen Nebenwirkungen ohne CRS ist selten (22). Das Risiko von neurologischen Nebenwirkungen hängt nicht zuletzt auch vom eingesetzten CAR-T-Produkt ab: In den Zulassungsstudien wurden ICANS aller Schweregrade mit Kymriah® während den ersten 8 Wochen nach der Infusion in 39% der PatientInnen beobachtet (2), bei Yescarta® aber in 64% (3). Diese Zahlen decken sich mit unseren Erfahrungen. Das Risiko von Neurotoxizität bei der Behandlung von Myelomen mit dem Anti-BCMA-Produkts bb2121 scheint mit 18% in der Phase-2-Studie etwas kleiner zu sein (15).
Die mediane Zeit bis zum Auftreten von neurologischen Symptomen lag in den Studien bei Anti-BCMA-CARs bei 2 Tagen, bei Yescarta® waren es 5 Tage und bei Kymriah® 6 Tage, gefolgt von einer kompletten Remission der Symptome nach 3, 17 und 14 Tagen nach der Infusion des jeweiligen CAR-T-Produktes (2, 3, 15).
Zum Monitoring der kognitiven und exekutiven Funktionen wird der ICE (Immune Effector Cell Encephalopathy) Score oder CARTOX-Test verwendet, welcher täglich zwei Mal durchgeführt wird. Neben der zeitlichen und örtlichen Orientierung und einer einfachen Rechenübung wird das Schriftbild geprüft, welches oft erste Anzeichen für eine sich entwickelnde Neurotoxizität liefert. Die Symptome des ICANS reichen von Kopfschmerzen, Schriftbildveränderungen und Desorientiertheit über Vigilanzstörungen und epileptische Anfälle bis hin zu motorischen Ausfällen wie Hemi- und Paraparesen. Schwere derartige Verläufe haben wir bisher glücklicherweise nur ganz vereinzelt gesehen.
Bei klinischen Hinweisen für eine Neurotoxizität erfolgen weitere Abklärungen, einerseits die Wiederholung der Bildgebung, um Blutungen oder Ischämien als Ursache auszuschliessen. In unserer bisherigen Kohorte sind spezifische radiologische Befunde selten. Ein EEG führen wir bei Verdacht auf ein ICANS regelmässig durch, um iktale Ereignisse auszuschliessen, allerdings behandeln wir alle PatientInnen bei Auftreten eines ICANS prophylaktisch anti-epileptisch.
Die Behandlung von PatientInnen mit ICANS erfordert eine interdisziplinäre Zusammenarbeit. Bei schweren Verläufen ist eine intensivmedizinische Überwachung zwingend und ggf. eine Schutzintubation nötig. Bei erhöhtem Hirndruck erfolgt die Anlage einer externen Ventrikeldrainage zum Monitoring und zur Therapie. Die medikamentöse Therapie umfasst neben der zytokinhemmenden Behandlung und der Gabe von Antiepileptika auch den Einsatz hochdosierter Steroide. Während in den Zulassungsstudien der Anti-CD19-CARs vor allem Methylprednisolon zum Einsatz kam, findet in unserem klinischen Alltag aufgrund der guten ZNS-Gängigkeit vor allem Dexamethason Anwendung. Bei klinischer Besserung können die Steroide schrittweise ausgeschlichen, und in der Folge auf eine perorale Verabreichung umgestellt werden. In seltenen Fällen ist eine längerdauernde Steroidbehandlung, d.h. auch nach Austritt notwendig. Gerade bei älteren PatientInnen ist die Beurteilung einer neurologischen Verschlechterung häufig erschwert, da die Abgrenzung zwischen ICANS und Delir schwierig sein kann. Dies ist insbesondere auch bei PatientInnen zu berücksichtigen, welche unter Steroidtherapie erneut neurologische Symptome zeigen.
Prolongierte Zytopenien und Infektionen:
Bei bis zu einem Drittel der behandelten PatientInnen verläuft die hämatologische Erholung nach der CAR-T-Therapie verzögert, bei vielen über mehrere Monate, wobei insbesondere Neutro- und Thrombopenien auftreten (23, 24). Diesbezüglich sei auf die kürzlich erschienene Zusammenstellung zur beobachteten Hämatotoxizität der Deutschen Kollegen hingewiesen (25). Während die frühe Hämatotoxizität durch die lymphodepletierende Chemotherapie gut erklärt ist, sind die Ursachen der protrahierten Erholung (definiert als persistierende Zytopenien >28 Tage nach CAR-T-Therapie) weniger gut verstanden. Eine mögliche Erklärung ist, dass die PatientInnen in der Regel umfangreich vorbehandelt und zu einem Teil bereits stammzelltransplantiert sind, sodass die Knochenmarkreserven eingeschränkt sind. Weiter scheinen aber auch durch die CAR-T-Zellen ausgelöste Immun- und Entzündungsphänomene eine Rolle zu spielen. Hierfür spricht auch, dass prolongierte Zytopenien bei PatientInnen mit höhergradigem CRS häufiger auftreten (26). Diesbezüglich zu beachten ist, dass der Einsatz von Tocilizumab bei rheumatologischen PatientInnen ebenfalls zu Zytopenien geführt hat, sodass eine medikamentöse Genese nicht gänzlich ausgeschlossen werden kann (27).
Neben der Neutropenie erhöhen auch eine prolongierte B-Zell-Aplasie und Hypogammaglobulinämie das Risiko infektiöser Komplikationen, sodass eine adäquate Prophylaxe gegen Herpesviren, Pneumozystis und Pilzinfektionen nötig ist. Valaciclovir, Trimpetoprim/Sulfometoxazol und Fluconazol oder Posaconazol haben ihren festen Platz in der Infektprophylaxe bei aplasierenden Chemotherapien, und sind auch im Rahmen der CAR-T-Behandlung essentiell. Die supportive Therapie ist derzeit individuell und in Analogie zum Usus im autologen und allogenen Setting. Wir würden uns wünschen, dass die Evidenz hierzu mit akademischen Studien erarbeitet würde.
Einfluss von Nebenwirkungen und deren Behandlung auf das Outcome:
Aus der klinischen Erfahrung mit Checkpoint-Inhibitoren gibt es immer mehr Evidenz dafür, dass das Auftreten von immunvermittelten Nebenwirkungen ein Therapieansprechen anzeigt und mit einer besseren Prognose assoziiert ist (28). Ob dies auch für die CAR-T-Therapie gilt, ist derzeit noch unklar. Die aktuellen Daten weisen eher darauf hin, dass die Entwicklung von CRS und ICANS ein Surrogatmarker für eine hohe Tumorlast und so eher mit einem schlechten Outcome assoziiert sind (29). Frühe Studien zeigten einen negativen prognostischen Effekt der Behandlung von CRS und ICANS mit Steroiden9,30, sodass aktuell primär Tocilizumab zum Einsatz kommt. In neueren Untersuchungen konnte aber gezeigt werden, dass auch der Gebrauch von hochdosierten Steroiden primär einen Einfluss auf die Nebenwirkungen hat, ohne dabei die Aktivität der CAR-T-Zellen zu beeinträchtigen (11, 31). Es ist also möglich, Steroide zur Behandlung von Nebenwirkungen früher einzusetzen.
Zusammenfassung und Ausblick:
Unsere Erfahrung aus der Behandlung von bisher ca. 80 CAR-T-PatientInnen ist grösstenteils positiv. Die Anzahl an PatientInnen ist für Schweizer Verhältnisse gross, aber international immer noch vergleichsweise klein, und wir lernen mit jeder Behandlung dazu. Wie in den Zulassungsstudien zeigen sich bei diesem, teils intensiv vorbehandelten Kollektiv sehr gute Ansprechraten und auch langanhaltende Remissionen. Da die CAR-T-Zell-Therapie in kurativer Absicht durchgeführt wird, haben wir unser Programm auch während der Covid-19 Pandemie ohne wesentliche Einschränkung fortgesetzt.
Wie bereits aus anderen Ländern berichtet, haben auch wir einige PatientInnen mit aggressiven Lymphomen erfolgreich mit der CAR-T-Zelltherapie behandelt, welche die Einschlusskriterien der Studien z. B. aufgrund von Komorbiditäten nicht erfüllt hätten (10). Trotzdem scheinen sich die Ergebnisse mit den Zulassungsstudien mindestens zu decken und die Toleranz – wahrscheinlich durch die frühe, niederschwellige Behandlung von CRS und ICANS – sogar besser zu sein (12, 32–35). Die enge Patientenselektion für die Studienbehandlung ist insbesondere bei der KarMMa-3-Studie (für das Multiple Myelom) problematisch. Die Optionen für eine Bridging-Therapie sind durch die Studie sehr limitiert, sodass bei raschem Myelomprogress die Wartezeit bis zur eigentlichen CAR-T-Therapie zu lang sein kann. Hier werden die kommerziellen Produkte andere Möglichkeiten eröffnen. Wir haben auch Erfahrungen mit Hepatitis-B-positiven PatientInnen und solchen mit Lymphommanifestionen im ZNS sammeln können, allesamt Ausschlusskriterien für eine Studienbehandlung. Über diese Fälle sowie die klinischen Resultate an unserem Zentrum werden wir in wissenschaftlichen Publikationen ebenso berichten wie über den Prozess zur Erteilung der Kostengutsprachen. Die CAR-T-Produkte für LymphompatientInnen unterscheiden sich von der Lymphapherese bis zum Nebenwirkungsprofil. Wir sind daher angehalten, die Produkte in unsere Algorithmen einzubauen, und haben hierzu eine Initiative unter Einbezug der EMBT gestartet.
Die CAR-T-Therapie hat die Behandlung von fortgeschrittenen lymphoiden Neoplasien revolutioniert. Auch die bisherigen Studienergebnisse bei der Behandlung des multiplen Myeloms und anderen indolenten Lymphomen sind vielversprechend (15, 36).Die Voraussetzungen wären also gegeben, die CAR-T-Therapie für andere Krankheiten, insbesondere auch solide Tumoren, weiterzuentwickeln. Die Hürde hierfür ist jedoch gross: Während die malignen Zellen bei lymphoiden Neoplasien in der Regel in grösserer Zahl im Blutstrom vorliegen und die CAR-T-Zellen so aktiviert werden können, ist bei soliden Tumoren zuerst eine Migration ins Gewebe nötig. Zusätzlich besitzt das Tumormikromilieu meist eine immunsuppressive Wirkung, sodass die Aktivität von CAR-T-Zellen hier abgeschwächt werden kann. Aktuell läuft eine Vielzahl von Phase I/II-Studien mit verschiedenen CAR-T-Konstrukten in soliden Tumoren, deren Ergebnisse mit Spannung erwartet werden.
Dr. med. Alexander D. Heini1, Dr. med. Marie-Noëlle Kronig/1, Dr. med. Barbara Jeker1, Dr. med. Myriam Legros/2, PD Dr. med. Michael Daskalakis/3,4, Prof. Dr. med. Ulrike Bacher/3,
Prof. Dr. med. Thomas Pabst/1, Prof. Dr. med. Dr. phil. nat. Sacha Zeerleder/3, Prof. Dr. med. Urban Novak/1, urban.novak@insel.ch
1/Universitätsklinik für Medizinische Onkologie, Inselspital, Universitätsspital Bern, 3010 Bern
2/Zentrum für Labormedizin, Inselspital, Universitätsspital Bern, 3010 Bern
3/Universitätsklinik für Hämatologie und Hämatologisches Zentrallabor, Inselspital, Universitätsspital Bern, 3010 Bern
4/Department of BioMedicalResearch (DMBR), Universität Bern, Murtenstrasse 40, 3008 Bern
Copyright bei Aerzteverlag medinfo AG
Dr. med. Alexander D. Heini
Universitätsklinik für Medizinische Onkologie
Inselspital, Universitätsspital Bern
3010 Bern
PD Dr. med. Urban Novak
Inselspital
Universitätsspital Bern
Universitätsklinik für Medizinische Onkologie
Loryhaus
3010 Bern
urban.novak@insel.ch
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
◆ Am Inselspital sind die Erfahrungen aus der Behandlung von bisher
ca. 80 CAR-T-PatientInnen grösstenteils positiv.
◆ Einige PatientInnen mit aggressiven Lymphomen, welche die
Einschlusskriterien der Studien z. B. aufgrund von Komorbiditäten
nicht erfüllt hätten, wurden mit der CAR-T-Zelltherapie erfolgreich behandelt.
◆ Die CAR-T-Therapie hat die Behandlung von fortgeschrittenen
lymphoiden Neoplasien revolutioniert. Auch die bisherigen Studien-ergebnisse bei der Behandlung des multiplen Myeloms und anderen indolenten Lymphomen sind vielversprechend.
◆ Die Voraussetzungen wären gegeben, die CAR-T-Therapie auch
für andere Krankheiten, insbesondere solide Tumoren, weiterzu-
entwickeln, auch wenn die Hürden hierfür gross sind.
◆ Aktuell läuft eine Vielzahl von Phase I/II-Studien mit verschiedenen CAR-T-Konstrukten in soliden Tumoren.
Messages à retenir
◆ À l’Inselspital, l’expérience tirée du traitement d’environ 80 patients CAR-T jusqu’à présent a été largement positive.
◆ Certains patients atteints de lymphomes agressifs qui n’auraient pas satisfait aux critères d’inclusion des études, par exemple en raison de comorbidités, ont été traités avec succès grâce à la thérapie par cellules CAR-T.
◆ La thérapie CAR-T a révolutionné le traitement des néoplasmes lymphoïdes avancés. Les résultats des études menées à ce jour dans le traitement du myélome multiple et d’autres lymphomes indolents sont également prometteurs.
◆ Les conditions seraient réunies pour poursuivre le développement de la thérapie CAR-T pour d’autres maladies, notamment les tumeurs solides, même si les obstacles à surmonter pour y parvenir sont importants.
◆ Un grand nombre d’essais de phase I/II sont actuellement en cours avec différentes constructions CAR-T dans les tumeurs solides.
1. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018. doi:10.1056/nejmoa1709866
2. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med. 2019. doi:10.1056/nejmoa1804980
3. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017. doi:10.1056/nejmoa1707447
4. Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020. doi:10.1016/S0140-6736(20)31366-0
5. Wang M, Munoz J, Goy A, et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med. 2020. doi:10.1056/nejmoa1914347
6. Korell F, Laier S, Sauer S, et al. Current Challenges in Providing Good Leukapheresis Products for Manufacturing of CAR-T Cells for Patients with Relapsed/Refractory NHL or ALL. Cells. 2020. doi:10.3390/cells9051225
7. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med. 2020. doi:10.1056/nejmra2026131
8. Maude SL, Frey N, Shaw PA, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med. 2014;371(16). doi:10.1056/nejmoa1407222
9. Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25-224ra25. doi:10.1126/scitranslmed.3008226
10. Nastoupil LJ, Jain MD, Feng L, et al. Standard-of-care axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma: Results from the US lymphoma CAR T consortium. J Clin Oncol. 2020. doi:10.1200/JCO.19.02104
11. Topp M, Van Meerten T, Houot R, et al. Earlier Steroid Use with Axicabtagene Ciloleucel (Axi-Cel) in Patients with Relapsed/Refractory Large B Cell Lymphoma. Blood. 2019. doi:10.1182/blood-2019-126081
12. Riedell PA, Walling C, Nastoupil LJ, et al. A Multicenter Retrospective Analysis of Clinical Outcomes, Toxicities, and Patterns of Use in Institutions Utilizing Commercial Axicabtagene Ciloleucel and Tisagenlecleucel for Relapsed/Refractory Aggressive B-Cell Lymphomas. Blood. 2019. doi:10.1182/blood-2019-127490
13. Garcia Borrega J, Gödel P, Rüger MA, et al. In the eye of the storm: Immune-mediated toxicities associated with car-t cell therapy. HemaSphere. 2019;3(2). doi:10.1097/HS9.0000000000000191
14. Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014. doi:10.1182/blood-2014-05-552729
15. Munshi NC, Anderson LD, Shah N, et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Engl J Med. 2021. doi:10.1056/nejmoa2024850
16. Gardner R, Leger KJ, Annesley CE, et al. Decreased Rates of Severe CRS Seen with Early Intervention Strategies for CD19 CAR-T Cell Toxicity Management. Blood. 2016. doi:10.1182/blood.v128.22.586.586
17. Hill JA, Li D, Hay KA, et al. Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood. 2018. doi:10.1182/blood-2017-07-793760
18. Frigault MJ, Nikiforow S, Mansour MK, et al. Tocilizumab not associated with increased infection risk after CAR T-cell therapy: Implications for COVID-19? Blood. 2020. doi:10.1182/BLOOD.2020006216
19. Titov A, Petukhov A, Staliarova A, et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis. 2018. doi:10.1038/s41419-018-0918-x
20. Frigault MJ, Maus M V., Dietrich J, et al. Tisagenlecleucel CAR T-cell therapy in secondary CNS lymphoma. Blood. 2019. doi:10.1182/blood.2019001694
21. Ghafouri S, Timmerman J, Larson S, Mead MD. Axicabtagene Ciloleucel CAR T-cell therapy for relapsed/refractory secondary CNS non-Hodgkin lymphoma: comparable outcomes and toxicities, but shorter remissions may warrant alternative consolidative strategies? Bone Marrow Transplant. 2021. doi:10.1038/s41409-020-01099-4
22. Yáñez L, Sánchez-Escamilla M, Perales MA. CAR T cell toxicity: Current management and future directions. HemaSphere. 2019. doi:10.1097/HS9.0000000000000186
23. Fried S, Avigdor A, Bielorai B, et al. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplant. 2019. doi:10.1038/s41409-019-0487-3
24. Jain T, Knezevic A, Pennisi M, et al. Hematopoietic recovery in patients receiving chimeric antigen receptor T-cell therapy for hematologic malignancies. Blood Adv. 2020. doi:10.1182/bloodadvances.2020002509
25. Rejeski K, Perez A, Sesques P, et al. CAR-HEMATOTOX: A model for CAR T-cell related hematological toxicity in relapsed/refractory large B-cell lymphoma. Hematol Oncol. 2021. doi:10.1002/hon.82_2879
26. Penack O, Koenecke C. Complications after cd19+ car t-cell therapy. Cancers (Basel). 2020. doi:10.3390/cancers12113445
27. Shetty A, Hanson R, Korsten P, et al. Tocilizumab in the treatment of rheumatoid arthritis and beyond. Drug Des Devel Ther. 2014. doi:10.2147/DDDT.S41437
28. Fiala O, Sorejs O, Sustr J, Kucera R, Topolcan O, Finek J. Immune-related adverse effects and outcome of patients with cancer treated with immune checkpoint inhibitors. Anticancer Res. 2020. doi:10.21873/anticanres.14063
29. Westin JR, Tam CS, Borchmann P, et al. Correlative Analyses of Patient and Clinical Characteristics Associated with Efficacy in Tisagenlecleucel-Treated Relapsed/Refractory Diffuse Large B-Cell Lymphoma Patients in the Juliet Trial. Blood. 2019. doi:10.1182/blood-2019-129107
30. Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013. doi:10.1126/scitranslmed.3005930
31. Liu S, Deng B, Yin Z, et al. Corticosteroids do not influence the efficacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer J. 2020. doi:10.1038/s41408-020-0280-y
32. Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012. doi:10.1182/blood-2011-10-384388
33. Faramand R, Jain M, Staedtke V, et al. Tumor Microenvironment Composition and Severe Cytokine Release Syndrome (CRS) Influence Toxicity in Patients with Large B-Cell Lymphoma Treated with Axicabtagene Ciloleucel. Clin Cancer Res. 2020. doi:10.1158/1078-0432.CCR-20-1434
34. Pasquini MC, Locke FL, Herrera AF, et al. Post-Marketing Use Outcomes of an Anti-CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy, Axicabtagene Ciloleucel (Axi-Cel), for the Treatment of Large B Cell Lymphoma (LBCL) in the United States (US). Blood. 2019. doi:10.1182/blood-2019-124750
35. Jaglowski S, Hu Z-H, Zhang Y, et al. Tisagenlecleucel Chimeric Antigen Receptor (CAR) T-Cell Therapy for Adults with Diffuse Large B-Cell Lymphoma (DLBCL): Real World Experience from the Center for International Blood & Marrow Transplant Research (CIBMTR) Cellular Therapy (CT) Registry. Blood. 2019. doi:10.1182/blood-2019-130983
36. Jacobson C, Chavez JC, Sehgal AR, et al. Primary Analysis of Zuma-5: A Phase 2 Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients with Relapsed/Refractory (R/R) Indolent Non-Hodgkin Lymphoma (iNHL). Blood. 2020. doi:10.1182/blood-2020-136834
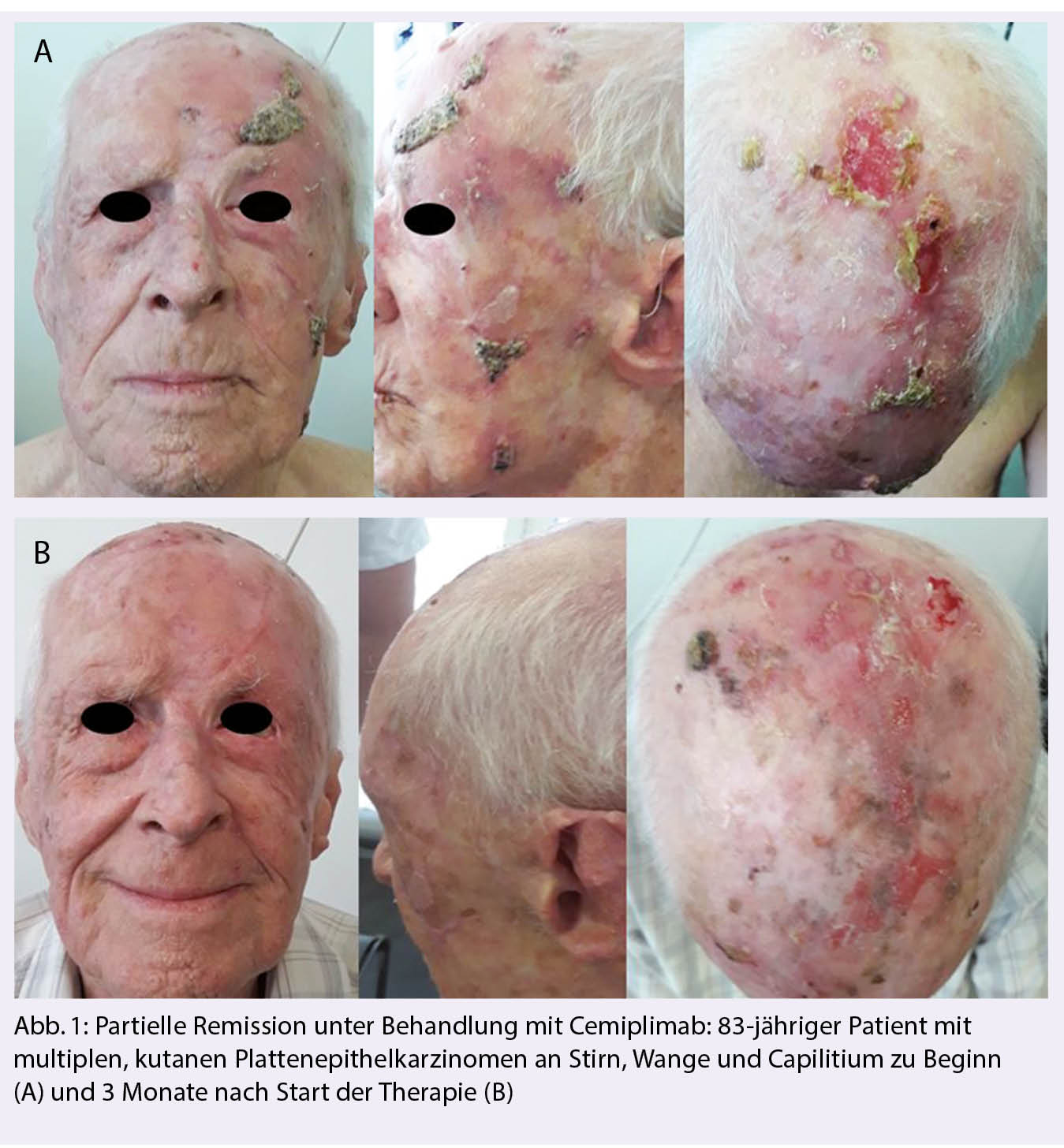
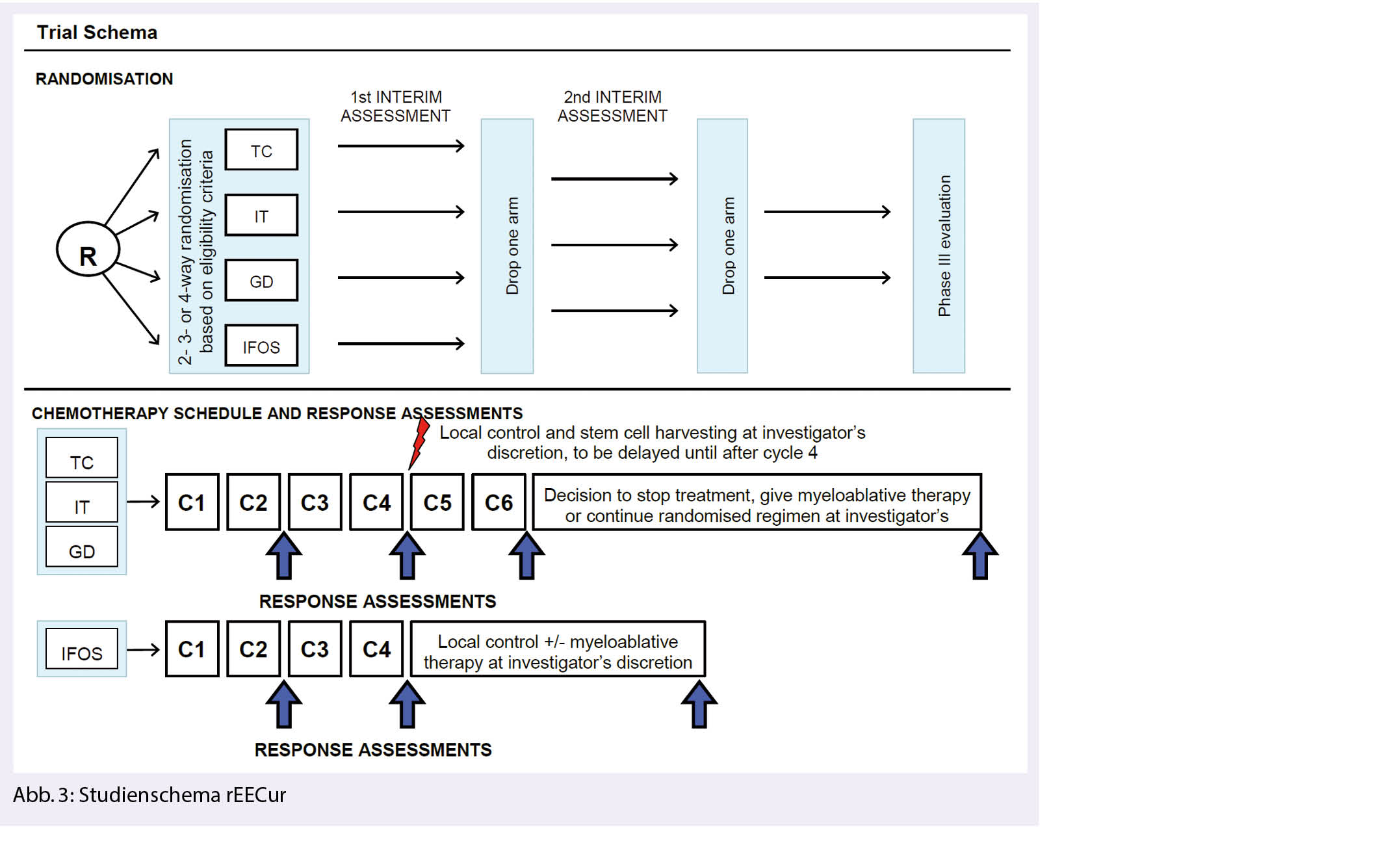
Aufgrund der raschen Entwicklung im molekular-genetischen Verständnis primärer Hirntumoren wird 2021 eine Revision der WHO Klassifikation publiziert. Dort finden sich wesentliche Neuerungen zum diagnostischen Algorithmus und zur Nomenklatur primärer Hirntumoren. Wir haben in der Ausgabe 03-21 der «info@onkologie« darüber berichtet. Therapeutische Anpassungen an die neue Klassifikation sind Thema des folgenden Beitrags.
En raison de l’évolution rapide de la compréhension moléculaire-génétique des tumeurs cérébrales primaires, une révision de la classification de l’OMS sera publiée en 2021. Il s’agira notamment de modifier de manière significative l’algorithme de diagnostic et la nomenclature des tumeurs cérébrales primaires. Nous en avons parlé dans le numéro 03-21 de l’«info@oncologie». Les adaptations thérapeutiques à la nouvelle classification font l’objet de l’article suivant.
IDH1– und viel seltener IDH2-Mutationen sind frühe Ereignisse in der Gliomentwicklung. Biologisch und prognostisch unterscheiden sich Gliome dahingehend, ob sie mit oder ohne diese Mutationen auftreten und auch die Therapiestruktur richtet sich danach. Wir gliedern deshalb im Folgenden die Gliome in die beiden Gruppen «IDH-mutiert» und «IDH-wild-type».
Therapie IDH-mutierter diffuser Gliome
Hintergrund:
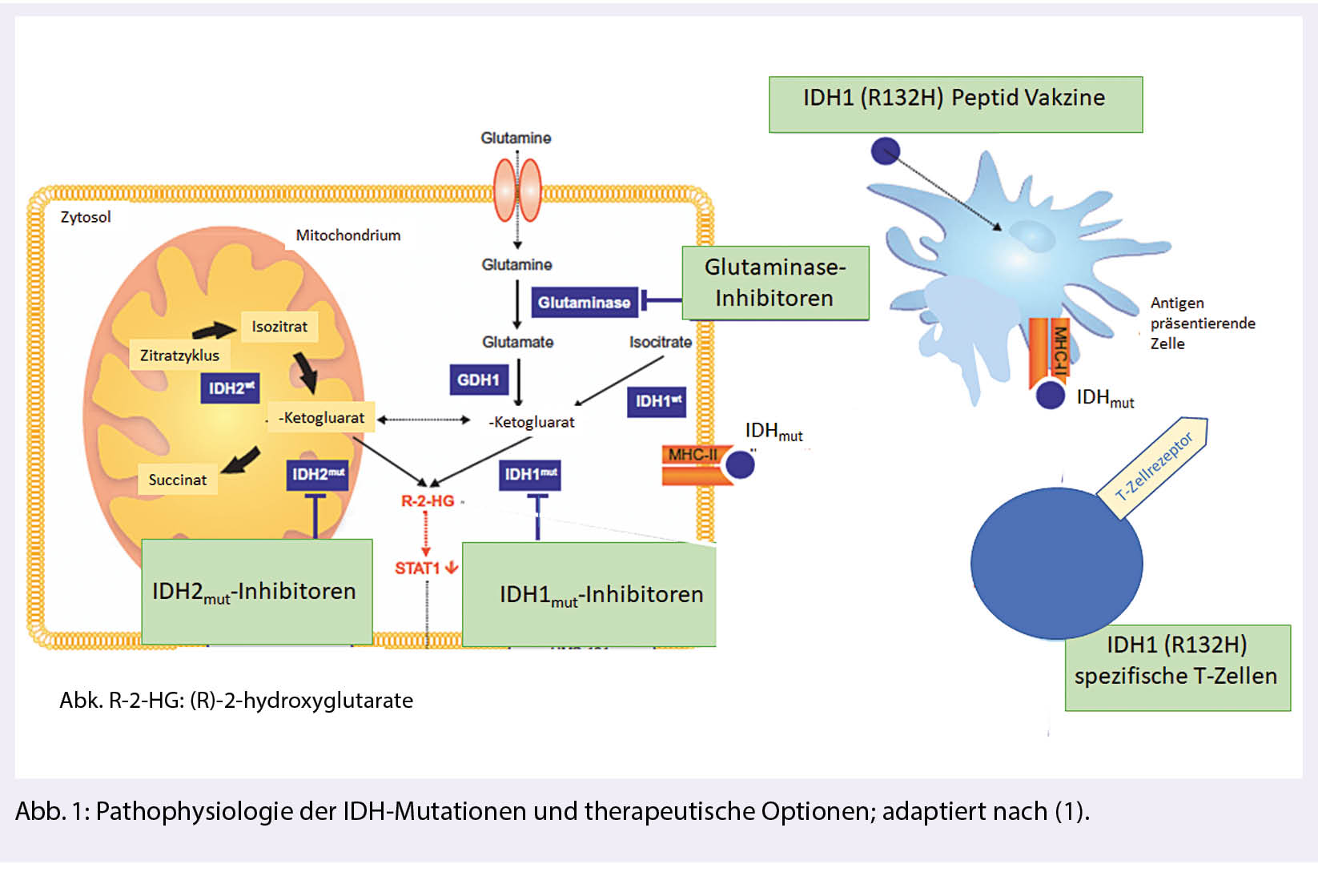
Mutationen im Gen für Isozitrat-Dehydrogenase (IDH) sind mit bis zu 80 % die weitaus häufigste genetische Veränderung diffuser Gliome der WHO-Grade 2-4. Sie unterscheiden sich in Bezug auf den natürlichen Verlauf und das Therapieansprechen von IDH-wild-type Gliomen. In klinischen Studien werden Therapien gegen mutiertes IDH in Form von spezifischen Inhibitoren oder Impfungen mit dem Peptid, welches die charakteristische Mutation enthält, geprüft (Abb. 1).
IDH-Mutationen treten in Gliomen überwiegend in Form einer IDH1R132H-Mutation auf. Sie führen zu einer neomorphen, d.h. Tumor definierenden enzymatischen Funktion des IDH-assoziierten Proteins mit Konversion von α-Ketoglutarat (α-KG) zu 2-Hydroxyglutarat (2-HG).
Der Onkometabolit 2-HG akkumuliert in Tumorzellen und führt über verschiedene epigenetische Mechanismen zu genomischer Instabilität, Akkumulation weiterer Mutationen und maligner Transformation. IDH-Mutationen werden in der klinischen Routine anhand eines mutationsspezifischen Antikörpers, welcher die häufige IDH1R132H-Mutation erkennt, immunhistochemisch diagnostiziert. Bei fehlender Reaktivität und Vorliegen eines diffusen Glioms ist eine Sequenzierung erforderlich, um das Vorliegen seltener IDH1– oder IDH2-Mutationen auszuschliessen. Viele Institutionen machen dies bis zu einem Patientenalter von 60 Jahren, da darüber hinaus kaum IDH-mutierte Gliome zu erwarten sind. Indirekte Hinweise für das Vorliegen einer IDH-Mutation können Methylierungs-Arrays liefern, die eine charakteristische, durch die Akkumulation von 2-HG bedingte globale DNA-Hypermethylierung anzeigen. Mit der 2-HG-Magnetresonanzspektroskopie ist ein Verfahren in Entwicklung, das eine nicht-invasive bildgebende Diagnose von IDH-mutierten Gliomen ermöglichen soll.
In der grossen Gruppe IDH-mutierter Gliome wird eine zunehmende Zahl molekular definierter Subtypen mit Bedeutung für Prognose und Therapiewahl zusammengefasst. IDH-mutierte Astrozytome weisen einen nukleären Verlust des Transkriptionsregulators ATRX auf und unterscheiden sich damit von den IDH-mutierten Oligodendrogliomen, die ihrerseits einen kombinierten Allelverlust auf den Chromosomen 1p und 19q aufweisen. Innerhalb der IDH-mutierten Astrozytome ist das Vorliegen von mikrovaskulären Proliferaten, Nekrosen und insbesondere der homozygote Verlust von CDKN2A/B mit einer ungünstigen Prognose assoziiert und wird dem WHO-Grad 4 zugeordnet. Diese Tumoren werden somit nicht mehr als Glioblastome bezeichnet, sondern als «Astroyztom, IDH-mutiert, WHO-Grad 4».
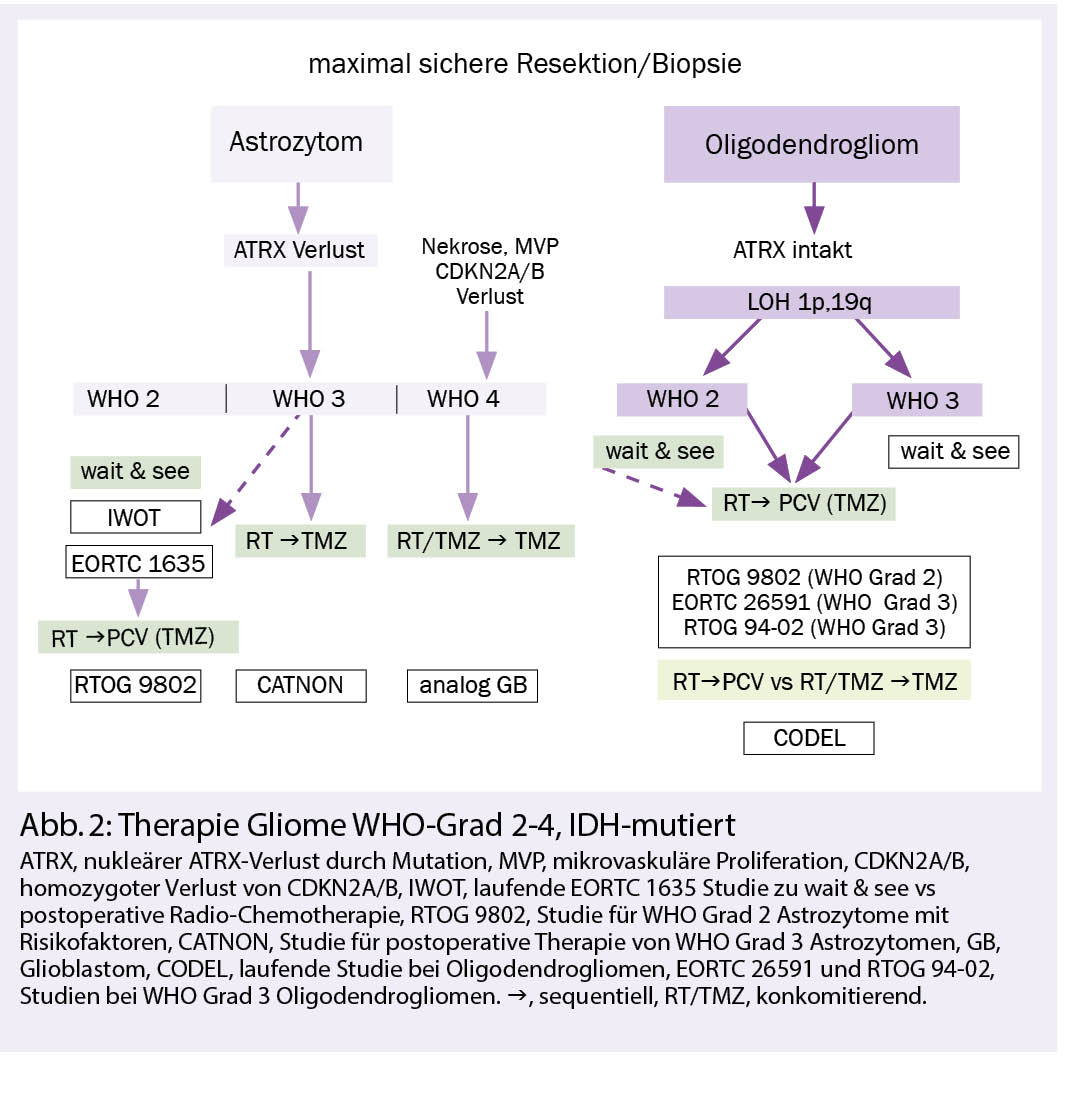
Therapie WHO Grad 2 und 3, IDH-mutierte Gliome:
Nach bildgebender Verdachtsdiagnose erfolgt eine möglichst vollständige und neurologisch schonende Resektion und eine integrierte neuropathologische Diagnose. Diese beinhaltet die histomorphologische und molekulargenetische Gewebeanalyse, welche auch anhand einer stereotaktischen Biopsie gestellt werden kann, wenn eine umfangreichere Resektion nicht indiziert ist. Zunehmend wird bei den Oligodendrogliomen die klinische Bedeutung des WHO-Grades 2 versus 3 in Frage gestellt. Dabei ist allerdings nicht geklärt, was dies für die zu wählende Radiotherapiedosis bedeutet, da es hierfür keine Studiendaten gibt.
Astrozytome und Oligodendrogliome WHO-Grad 2 können bei Fehlen der zwei wichtigsten Prognose bestimmenden Risikofaktoren (RF) – nämlich unvollständige Resektion und Alter über 40 Jahre – zunächst beobachtet werden. Basierend auf den Daten grosser randomisierter Studien wird bei Progress oder Vorhandensein der genannten RF eine Radiotherapie mit 50.4-54 Gy, gefolgt von Procarbazin, CCNU und Vincristin (PCV-Schema) für 6 Zyklen empfohlen oder alternativ, jedoch nicht evidenzbasiert, mit Temozolomid (2, 3).
Die Strahlentherapie bleibt nach wie vor Teil der Standardtherapie bei WHO-Grad-2-Gliomen. In einer randomisierten Phase-III-Studie (EORTC 22033-26033) war die Radiotherapie im Vergleich zur alleinigen Chemotherapie in der molekularen Subgruppenanalyse bei IDH-mutierten Astrozytomen ohne 1p,19q Ko-Deletion der alleinigen Chemotherapie mit Temozolomid über ein Jahr hinsichtlich des progressionsfreien Überlebens überlegen (4).
Die Frage nach dem optimalen Zeitpunkt einer Radio-Chemotherapie bei Grad 2-(und 3-) Astrozytomen ohne LOH 1p,19q ist nicht abschliessend geklärt. Das Abwägen einer postoperativen Tumorkontrolle gegenüber dem Auftreten von Toxizität nach Radiotherapie soll in der laufenden EORTC 1635 IWOT-Studie geklärt werden (NCT03763422). Die Studie vergleicht die postoperative Radio-Chemotherapie mit einer aktiven Nachsorge.
Bei Oligodendrogliomen WHO-Grad 3 belegen Langzeitdaten aus zwei randomisierten Phase-III-Studien (RTOG 9402 und EORTC 26951) den Benefit einer postoperativen Radiotherapie mit 59.4 Gy üblicherweise gefolgt von 6 Zyklen PCV oder wie in der RTOG-Studie mit 4 Zyklen PVC vor der Radiotherapie (5, 6.). Die noch nicht abgeschlossene, internationale CODEL-Studie (NCT00887146) vergleicht bei Oligodendrogliomen in einem Nichtunterlegenheits-Design eine sequenzielle Radiochemotherapie mit PCV (bis 6 Zyklen) mit einer kombinierten (konkomitanten und adjuvanten) Radiochemotherapie mit 6-12 Zyklen Temozolomid.
Bei IDH-mutierten Astrozytomen WHO Grad 3 unterstützen die Ergebnisse der EORTC 26053-22054 CATNON-Studie den Einsatz einer sequenziellen Radiochemotherapie mit 59.4 Gy gefolgt von Temozolomid über 12 Zyklen. Hier ist der Nutzen einer konkomitanten Temozolomid-Therapie parallel zur Radiotherapie noch nicht abschliessend geklärt (7, 8).
Astrozytome WHO-Grad 4, IDH-mutiert sollten wie Glioblastome behandelt werden (Abb. 2). Aufgrund der IDH-Mutation kann von einer Empfindlichkeit gegenüber alkylierenden Substanzen ausgegangen werden.
Neue Therapieansätze für IDH-mutierte Gliome
Neben der Verbesserung des Therapiealgorithmus konventioneller Therapien zielen neue Ansätze auf die IDH-Mutation selbst ab. In den letzten Jahren wurde eine ganze Reihe von IDH-Inhibitoren entwickelt.
Sicherheit, gute Verträglichkeit und biologische Wirkung gemessen an der Reduktion der 2-HG-Produktion im Tumorgewebe konnte in Phase-I-Studien nachgewiesen werden (9). Die bisherigen Effektivitätsdaten mit Tumorvolumenreduktion v. a. bei Patienten mit nicht-kontrastmittelaufnehmenden Tumoren sind ermutigend. Mit der laufenden INDIGO-Studie ist eine internationale randomisierte Phase-III-Studie initiiert, welche die Effektivität (gemessen am PFS) des IDH-Inhibitors AG-881 bei Patienten mit postoperativem Resttumor oder bei progredientem IDH-mutiertem WHO-Grad-2-Gliom untersucht (NCT04164901). Diese Studie rekrutiert Patienten an mehreren Zentren in der Schweiz. Neben spezifischen IDH-Inhibitoren eröffnen spezifische metabolische Abhängigkeiten Möglichkeiten für weitere zielgerichtete Therapien wie beispielsweise DNA-Demethylierungsstrategien oder Glutaminase-Inhibitoren, die bereits in frühen klinischen Studien getestet werden.
Spezifische Impfungen zielen auf die IDH1R132H-Mutation. Die mutierte Sequenz induziert spezifische T-Helferzellen, die das Wachstum IDH-mutierter Tumoren in Tiermodellen kontrollieren können. Die multizentrische NOA-16-Studie (NCT02454634) hat bei Patienten mit neu diagnostizierten IDH1R132H-mutierten Grad 3- und 4-Astrozytomen die Sicherheit und Immunogenität einer IDH1R132H-Peptidvakzine nachweisen können. Die hohe Frequenz an Pseudoprogressionen als Hinweis auf eine intratumorale Immunantwort sowie der Nachweis IDH1R132H-spezifischer T-Helferzellen im Tumorgewebe nach Vakzinierung legen eine biologische Effektivität nahe. Basierend auf präklinischen Hinweisen untersucht die aktuell rekrutierende multizentrische NOA-21-Studie in einer Phase-I-Studie die Sicherheit einer Kombination der IDH1R132H-Vakzine mit dem Immuncheckpoint-Inhibitor Avelumab (NCT03893903). Dabei werden auch mögliche Resistenzmechanismen am Tumorgewebe unter Therapie untersucht (Abb. 1).
Hintergrund: Mutationen im MAPK/ERK Signalweg wurden in ungefähr 95% pilozytischer Astrozytome (PA) gefunden und gelten mittlerweile als krankheitsdefinierend. Die häufigste molekulare Alteration ist die KIAA1549–BRAF Fusion, welche in 60-70% der Fälle auftritt und mit einer besseren Prognose einhergeht, verglichen mit PA ohne diese Fusion. Die BRAF-V600E-Mutation wird in ca. 10% der PA nachgewiesen.
Therapie: Die initiale Therapie der PA besteht in der maximal möglichen Resektion. Erst bei Tumorprogress oder nicht möglicher Operabilität wird eine Radiotherapie empfohlen und es können auf experimenteller Basis zielgerichtete Therapien eingesetzt werden.
Diffuse Astrozytome WHO-Grad 2 und 3, IDH-wild-type
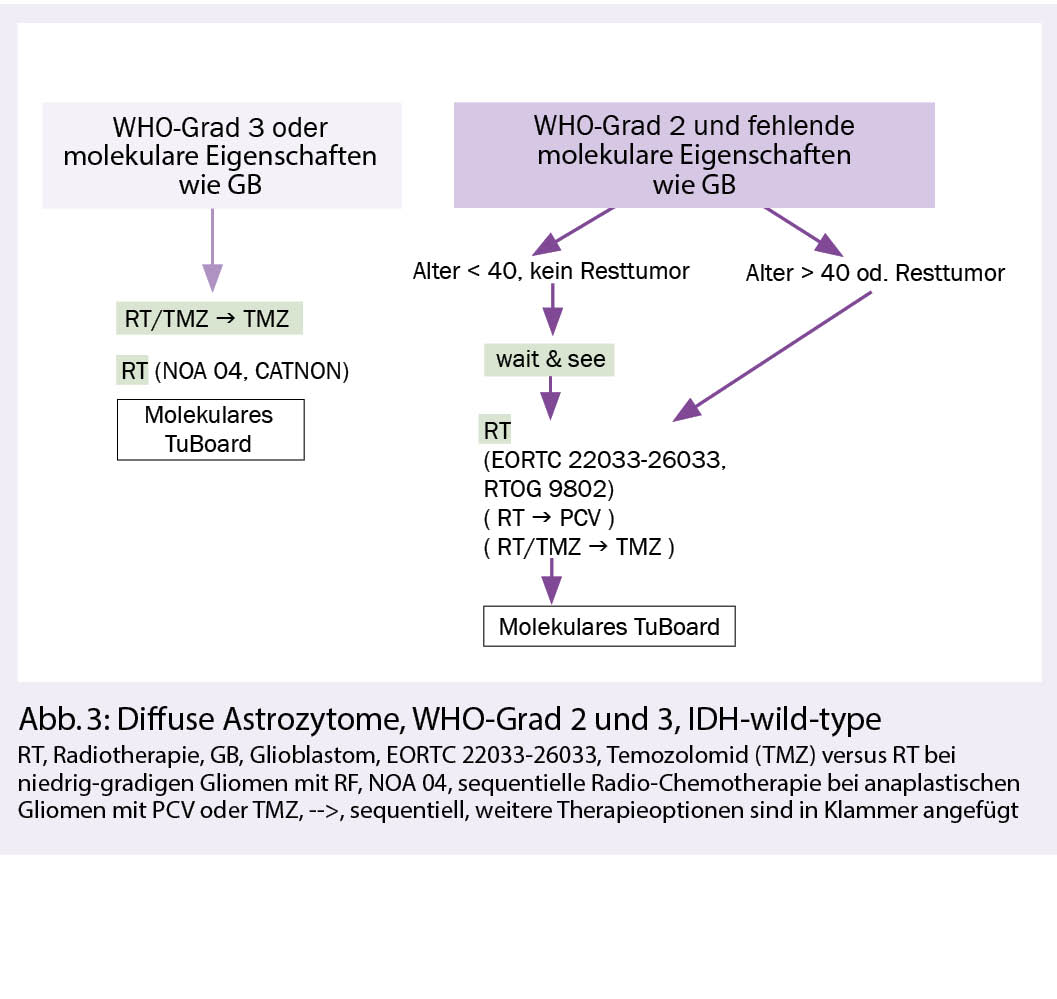
Hintergrund: Diffuse Astozytome IDH-wild-type sind eine heterogene Tumorgruppe, meist mit molekularen Eigenschaften wie bei Glioblastomen und seltener mit einem eher indolentem Verlauf. Bei Gliomen WHO-Grad 2 oder 3, die keine IDH-Mutation aufweisen, kann molekular ein Glioblastom diagnostiziert werden, wenn entweder eine EGFR-Amplifikation oder ein chromosomaler Gewinn auf Chromosom 7 bei komplettem Verlust von einem Chromosom 10 oder aber eine TERT (Telomerase reverse Transkriptase)-Promotor-Mutation vorliegt.
Die postoperative Radiotherapie ist vorläufig die Standardtherapie für Tumoren, die nicht die Kriterien eines Glioblastoms erfüllen. Ein Überlebensvorteil einer zusätzlichen Chemotherapie zur Radiotherapie bei WHO-Grad-2-Astrozytomen war in der RTOG-9802-Studie nur bei Patienten mit IDH-mutierten Tumoren nachweisbar, nicht aber bei IDH-wild-type-Tumoren (3). Es bleibt zu erforschen, weshalb Glioblastome von einer konkomitierenden und adjuvanten Chemotherapie profitieren und Astrozytome vom IDH-wild-type nicht. Die Rolle des MGMT-Promoter-Methylierungsstatus ist bei diesen Gliomen im Gegensatz zu den Glioblastomen auch weniger gut definiert. Nach Versagen der Primärtherapie ist eine molekulare Charakterisierung bei IDH-wild-type-Gliomen mit der Suche therapierbarer Targets eine Option. Therapie: Nach maximal und neurologisch sicherer Resektion kann in Ausnahmefällen und bei fehlenden klinisch-radiologischen Risikofaktoren (d.h. Alter < 40, kein Resttumor) und WHO-Grad 2 eine initiale Wait-and-see-Strategie verfolgt werden. Bei allen anderen WHO-Grad-2-Astrozytomen empfiehlt sich eine postoperative Radiotherapie mit 50.4-54 Gy optional gefolgt von PCV oder Temozolomid. Bei WHO-Grad 3, IDH-wild-type-Astrozytomen ist aufgrund fehlender Alternativen eine postoperative Therapie analog den Glioblastomen empfohlen, d.h. eine kombinierte Radio-Chemotherapie (59.4-60 Gy) und Temozolomid, gefolgt von einer Temozolomid-Erhaltungstherapie über 6 Zyklen. Bei Kontraindikationen für eine Chemotherapie kann auch eine alleinige RT mit einem Aequivalent von 60 Gy durchgeführt werden (Abb. 3).
Die Gruppe der Glioblastome enthält auch histologische Sub-typen wie das Riesenzell-Glioblastom, das Gliosarkom und epitheloide Glioblastome (welche in bis zu 50% eine BRAF-Mutation aufweisen). Therapie: Die Therapierichtlinien berücksichtigen das Alter, den Karnofsky-Performance-Status (KPS) und den MGMT-Promoter-Methylierungsstatus, wie im Algorithmus in Abb. 4 dargestellt.
Die postoperative Therapie bei Glioblastomen wurde 2005 weltweit standardisiert mit konkomitierender Radio-Chemotherapie mit 60 Gy und Temozolomid, gefolgt von einer Erhaltungstherapie mit Temozolomid über 6 Zyklen (10). 2019 wurde die NOA-09-Studie publiziert, welche zusätzlich zur Standardtherapie bei methyliertem MGMT-Promoter Lomustin (CCNU) ergänzt (11). Die Kombination beider Alkylantien, Temozolomid und Lomustin, hat bei insgesamt kleiner Patientenzahl zwar zu einer Verlängerung des OS, aber auch zu erhöhter Knochenmarkstoxizität geführt, weshalb diese Chemotherapie-Kombination eher bei jungen Patienten und nicht breit Anwendung findet. Für neu diagnostizierte Glioblastome ist optional die Tumor-Treating-Field-Therapie (TTFields) in der Schweiz und in anderen europäischen Ländern zugelassen. Es handelt sich dabei um elektrische Wechselfelder mit niedriger Intensität und mittlerer Frequenz (200 kHz), die über der Tumorregion mit speziellen Transducern angelegt werden. Diese Therapie, welche der Patient über mindestens 18 Stunden pro Tag tragen soll, hat in einer randomisierten Phase-III-Studie zu einer Verlängerung des PFS und des OS geführt verglichen mit der Standardtherapie bei Glioblastom. TTFields werden, bis auf mögliche Hautirritationen, gut vertragen und sind in ihrer Wirkung unabhängig von Glioblastom-Subgruppen (12). Bei hohem Alter und schlechtem KPS ist im Gespräch mit Patient und seinen Angehörigen auch «best supportive care» eine mögliche gemeinsame Entscheidung.
Bevacizumab, als hauptsächlich Ödem-reduzierende, antiangiogene Substanz, hat bei neu diagnostizierten Glioblastomen in zwei grossen randomisierten Studien keinen Überlebensvorteil gebracht und wird deshalb nur im Rezidiv oder bei Radionekrose zur Symptombeeinflussung eingesetzt.
Es gibt bislang noch keine positiven Studiendaten, welche einen Immuncheckpoint-Inhibitor in der postoperativen Phase zusätzlich zur Standardtherapie stützen. Die Suche nach Subgruppen, für welche eine Immuntherapie einen Vorteil bringen könnte, geht weiter. Studien hierzu sind in der Schweiz offen und können bei den Verfassern dieses Artikels angefordert werden.
Histon-mutierte Gliome, IDH-wild-type
Hintergrund: Histon H3.3 Missense-Mutationen beeinflussen die epigenetische Regulation der Genexpression. Therapie: Diffuse Mittelliniengliome mit der Mutation H3K27M, WHO-Grad 4 werden mit 54-60Gy radiotherapiert, alternativ chemo-radiotherapiert mit Temozolomid analog den Glioblastomen, aber noch mit ungenügender Evidenzlage.
Diffuse Hemisphären-Gliome mit der Mutation H3.3.G34, WHO-Grad 4 sind zu einem hohen Prozentsatz MGMT methyliert und werden chemo-radiotherapiert mit Temozolomid analog den Glioblastomen. Bisher liegen für diese vergleichsweise neuen Tumorentitäten nur limitierte Daten und Erfahrungswerte vor.
Das diagnostische und therapeutische Management bei Patienten mit neu diagnostizierten Gliomen sollte den Empfehlungen eines interdisziplinären neuro-onkologischen Tumorboards folgen, welche sich auf die neue WHO-Klassifikation, die aktuelle Datenlage abgeschlossener Studien, den Zugang zu laufenden Studien oder auf einen internationalen Konsensus abstützen.
Dabei sind der Allgemeinzustand und die Bedürfnisse der Betroffenen zu berücksichtigen. Neurorehabilitation, Psychoonkologie und soziale Beratung gehören zur ganzheitlichen Betreuung. Für weiterführende Informationen verweisen wir auf die aktuelle EANO (Europiean Association of Neuro-Oncology) Guideline (13).
Copyright bei Aerzteverlag medinfo AG
Dr. med. Silvia Hofer
Universitätsspital Zürich
Institut für Pathologie und Molekularpathologie
Schmelzbergstrasse 12
8091 Zürich
silvia.hofer@usz.ch
Dr. med. Brigitta Baumert, PhD
Institut für Radio-Onkologie
Kantonsspital Graubünden
Loëstrasse 170
7000 Chur
Die Autorinnen und der Autor haben in Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
◆ Die aktuelle Revision der WHO-Klassifikation 2021 von Tumoren des Zentralnervensystems führt entscheidende Veränderungen in der Diagnostik und Nomenklatur ein, mit Folgen in Bezug auf die Art, wie wir Patienten mit Gliomen behandeln.
◆ Für viele der neu definierten Krankheitsentitäten in der neuen WHO-Klassifikation sind noch keine Daten über spezifische Behandlungsergebnisse verfügbar; die Extrapolation aus Daten früherer klinischer Studien bleibt eine Herausforderung.
Messages à retenir
◆ La révision actuelle de la classification OMS 2021 des tumeurs du système nerveux central introduit des changements cruciaux dans le diagnostic et la nomenclature, avec des implications en termes de traitement des patients atteints de gliomes.
◆ Les données ne sont pas encore disponibles pour un grand nombre des entités pathologiques nouvellement définies dans la nouvelle classification de l’OMS, on ne dispose pas encore de données sur les résultats spécifiques des traitements; l’extrapolation à partir des données d’essais cliniques antérieurs reste un défi.
1. Friedrich M, Bunse L, Wick W et al.: Perspectives of Immunotherapy in isocitrate dehydrogenase-mutant gliomas. Curr Opin Oncol 30(6): 368-374, 2018. DOI: 10.1097/CCO.0000000000000478
2. Buckner JC, Chakravarti A, Curran WJ Jr et al.: Radiation plus procarbazine, CCNU, and vincristine in Low-Grade Glioma. NEJM 374:1344-1355, 2016. DOI:10.1056/NEJMc1605897
3. Bell EH , Zhang P, Shaw EG et al.: Comprehenisve Genomic Analysis in NRG
Oncology/RTOG 9802: A Phase III Trial of Radiation versus Radiation plus Procarbazine, Lomustine (CCNU), and Vincristine in High-Risk Low Grade Glioma. J Clin Oncol 38:3407-3417, 2020. DOI: 10.1200/JCO.19.02983
4. Baumert BG, Hegi ME, van den Bent MJ et al.: Temozolomide chemotherapy
versus radio- therapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 17(11):1521-1532, 2016. DOI:10.1016/ S1470-2045(16)30313-8
5. Cairncross G, Wang M, Shaw E et al.: Phase III trial of chemo- radiotherapy for anaplastic oligodendroglioma: long-term re- sults of RTOG 9402. J Clin Oncol 31: 337–343, 2013. DOI:10.1200/JCO.2012.43.2674
6. van den Bent MJ, Brandes AA, Taphoorn MJ, et al.: Adjuvant procarbazine,
lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 31: 344–350, 2013. DOI: 10.1200/JCO.2012.43.2229
7. van den Bent MJ, Baumert B, Erridge SC et al.: Interim results from the CATNON trial (EORTC study 26053-22054) of treatment with concurrent and adjuvant temozolomide for 1p/19q non-co-deleted anaplastic glioma: a phase 3, randomised, open-label intergroup study. Lancet 390(10103):1645-1653, 2017. DOI:10.1016/S0140-6736(17)31442-3. Erratum in: Lancet. 390(10103):1644, 2017
8. van den Bent MJ, Tesileanu CMS, Wick W et al.: 2nd interim analysis and IDH status of the EORTC randomized phase III CATNON trial on adjuvant and concurrent temozolomide in anaplastic astrocytoma. Lancet Oncology 22 (6) 813-823, 2021. DOI:https://doi.org/10.1016/S1470-2045(21)00090-5
9. Mellinghoff I, Ellingson BM, Touat M et al.: Ivosidenib in Isocitrate Dehydrogenase 1 Mutated Advanced Glioma. J Clin Oncol 38(29): 3398-3406, 2020. DOI: 10.1200/JCO.19.03327
10. Stupp R, Mason WP, van den Bent MJ et al.: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352: 987–996, 2005. DOI:10.1056/ NEJMoa043330
11. Herrlinger U, Tzaridis T, Mack F, et al.: Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): a randomised, open-label, phase 3 trial. Lancet 393(10172):678-688, 2019. DOI:10.1016/S0140-6736(18)31791-4
12. Stupp R, Tailllibert S, Kanner A et al.: Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma.A Randomized Clinical Trial. JAMA 318 (23): 2306-2316, 2017. DOI:10.1001/jama.2017.18718
13. Weller M, van den Bent M, Preusser M et al.: EANO Guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nature Reviews | Clinical Oncology 2020. doi.org/10.1038/s41571-020-00447-z
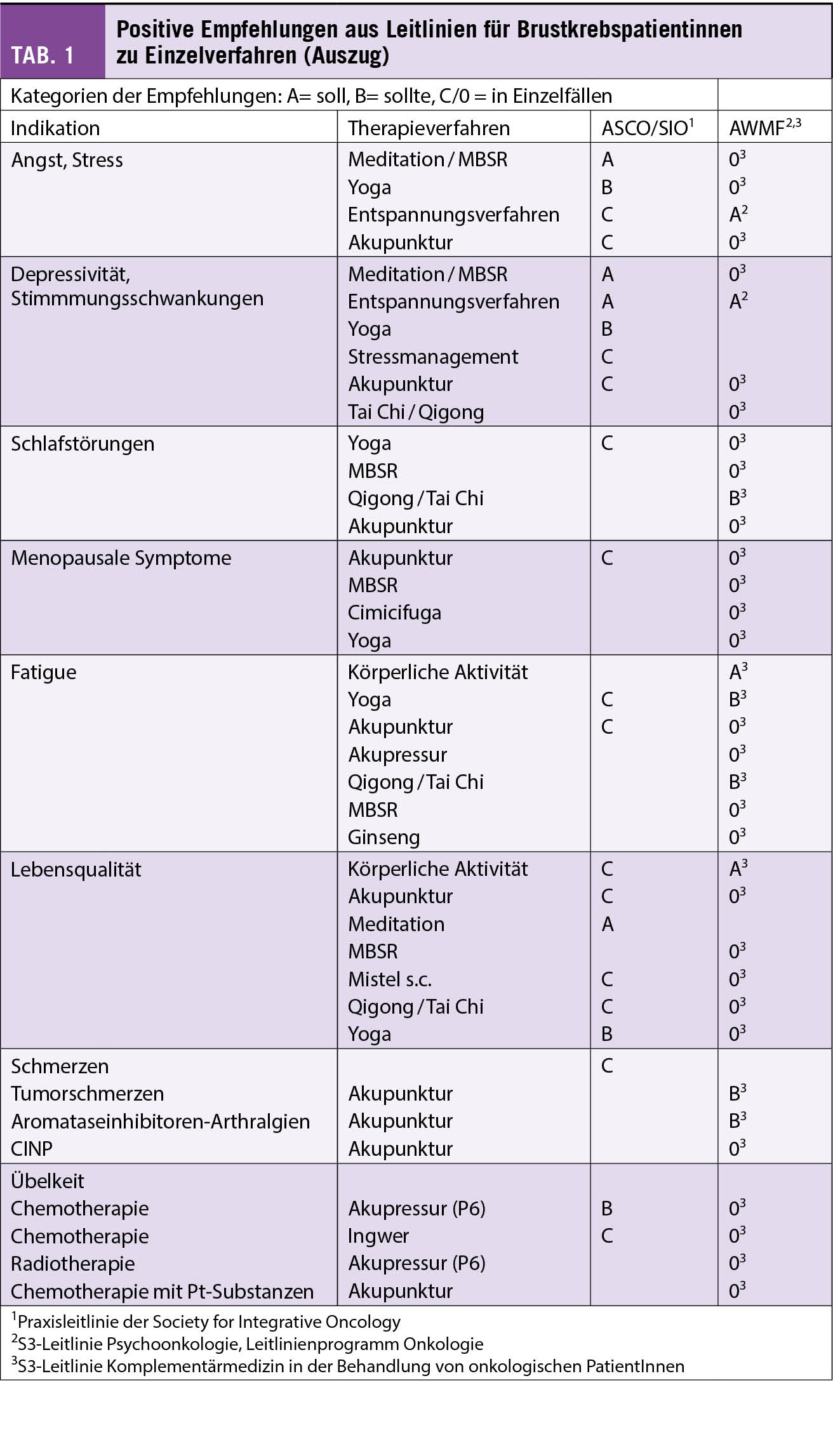
Brustkrebspatientinnen wünschen sich eine Integration von Komplementärmedizin in die ärztliche Versorgung. Dieser Artikel gibt Antworten auf eine häufig gestellte Frage: «Was kann ich noch tun, um meinen Krankheitsverlauf positiv zu beeinflussen oder Nebenwirkungen der Therapie zu reduzieren?» Es gibt evidenzbasierte supportive Massnahmen aus der Komplementärmedizin, die laut Praxis-Leitlinien für bestimmte Symptome oder zur Besserung der Lebensqualität empfohlen werden können.
Les patientes atteintes d’un cancer du sein souhaitent intégrer la médecine complémentaire dans leur suivi thérapeutique. Cet article répond à la question fréquente «Que puis-je faire moi-même pour influencer positivement le décours de ma maladie et/ou minimiser les effets adverses de la thérapie ?» Des mesures de soutien basées sur des preuves provenant de la médecine complémentaire existent. Et selon les lignes directrices pour la pratique elles peuvent être recommandées pour bien des symptômes et/ou pour améliorer la qualité de vie.
Die Situation
Im Juli 2021 wurde die erste S3-Leitlinie zu komplementären Therapien im deutschen Leitlinienprogramm Onkologie publiziert. Ein grosser Teil der dort zusammengestellten Studien wurde mit Brustkrebspatientinnen durchgeführt und je nach Umfrage ist mindestens jede zweite Patientin eine Nutzerin (1). Die Beweggründe der Patientinnen für das Interesse an Komplementärmedizin sind vielfältig. Es ist wichtig, diese zu erfragen, um auch passende Informationen und Empfehlungen geben zu können. Wenn die Erwartungen der Patientin und der Ärztin, des Arztes an die Inhalte des Gesprächs und an die Frage, was Komplementärmedizin leisten kann, nicht abgeglichen werden, kann dies zu Missverständnissen oder gar zu Unzufriedenheit führen.
Der Fokus dieses Artikels liegt auf der supportiven Anwendung von komplementären Therapien im Sinne einer integrativen Onkologie. Diese wurde von der internationalen Society for Integrative Oncology definiert (2) (Definition s. Box 1).
Drei Aspekte aus dieser Definition – Patientenzentrierung, Evidenz und Patientinnen als aktive Teilnehmerinnen in der Krebsbehandlung – sollen in diesem Artikel näher ausgeführt werden.
Ziel ist es auch, das Thema Komplementärmedizin in den Kontext der alltäglichen Praxis zu setzen.
Evidenz aus Leitlinien
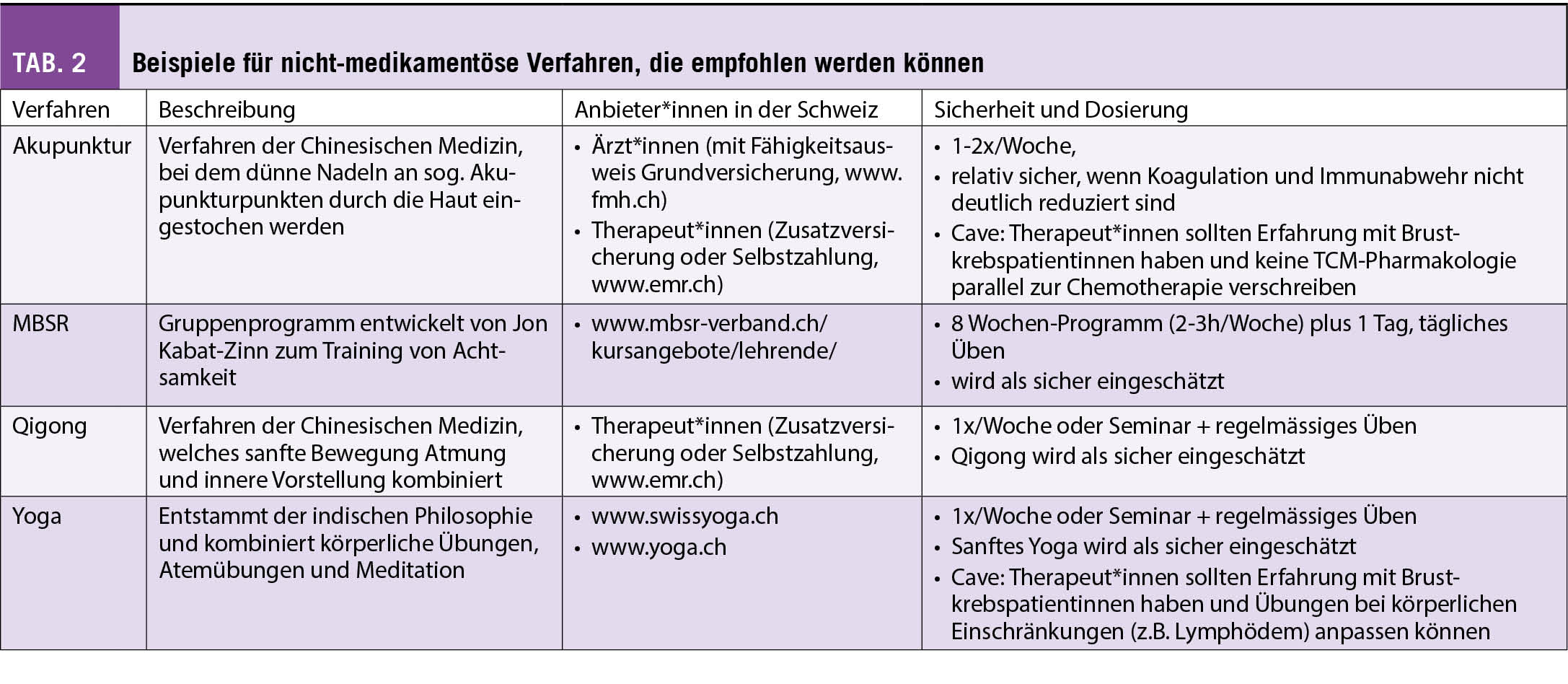
Seit einiger Zeit liegt bereits die Praxisleitlinie der Society for Integrative Oncology (SIO) zur Komplementärmedizin bei Brustkrebs vor, die von der American Society for Clinical Oncology (ASCO) (3) anerkannt wurde. Diese wird nun durch die deutlich umfangreichere S3-Leitlinie zu Komplementärmedizin in der Behandlung von onkologischen PatientInnen der AWMF ergänzt (4). Ein Auszug wichtiger Empfehlungen aus beiden Leitlinien ist in Tab. 1 dargestellt. Bei den beiden Leitlinien wird deutlich, dass hauptsächlich die nicht-pharmakologischen Therapien, wie z.B. Yoga, Achtsamkeitstraining oder Akupunktur/Akupressur empfohlen werden. Diese werden in Tab. 2 etwas näher ausgeführt. Diese Verfahren berücksichtigen zumeist den Wunsch vieler Frauen mit Brustkrebs, selber etwas aktiv tun zu können. Neben der positiven Evidenz aus wissenschaftlichen Studien haben sie auch den Vorteil, dass sie sich zumeist gut mit der antitumoralen Therapie kombinieren lassen, ohne dass man auf Interaktionen achten muss.
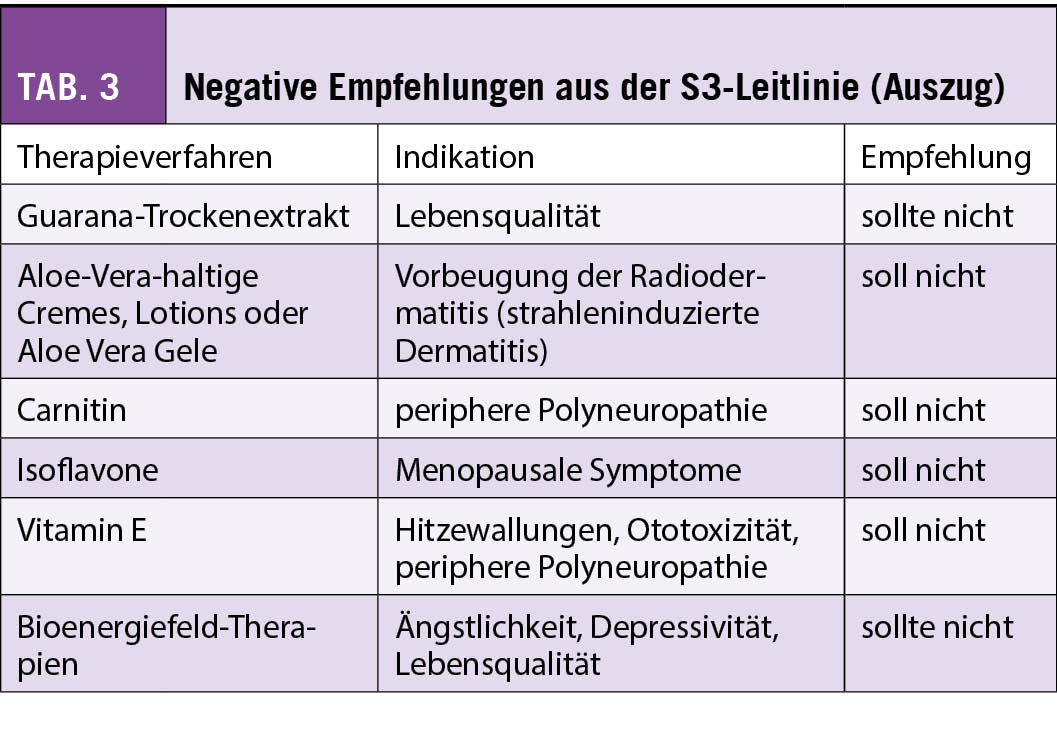
Hingegen liegt für Phytotherapeutika und Nahrungsergänzungsmittel weniger Evidenz vor, und diese sind eher mit einem Risiko der Interaktion mit der antitumoralen Behandlung assoziiert. In der Schweiz wird aber auch die Misteltherapie häufig von Patientinnen mit Brustkrebs nachgefragt. Nach der S3-Leitlinie kann die subkutan verabreichte Misteltherapie im Einzelfall zur Verbesserung der Lebensqualität empfohlen werden, während die Leitlinie der SIO sich aufgrund der unterschiedlichen Präparate und wenig Erfahrung mit Misteltherapie in den USA eher zurückhaltend äussert. In der S3-Leitlinie gibt es jedoch auch Therapien, von denen abgeraten wird. Diese sind in Tab. 3 zusammengefasst.
Die Patientin im Zentrum
Es ist wichtig die Empfehlungen zur Komplementärmedizin und das Gespräch dazu an die Patientin anzupassen, denn «one size fits all» widerspricht dem Prinzip einer Integrativen Onkologie. Es ist gut, die Werte und Wünsche der Patientin sowie mögliche kulturelle Einflüsse zu kennen oder zu erfragen, bevor man spezifische Empfehlungen ausspricht. Es gibt z.B. Patientinnen, denen achtsamkeitsbasierte Verfahren zu «spürig» sind, während andere sich davon stark angesprochen fühlen.
Auch Vorerfahrungen spielen eine Rolle. Viele Frauen haben bereits vor ihrer Brustkrebserkrankung Komplementärmedizin genutzt. Daraus entstehen Erwartungen, die sich auf die Auswahl und möglicherweise auch die Wirksamkeit komplementärer Therapien während der Brustkrebserkrankung auswirken können. Bisherige Forschung zu Erwartungen an das Therapieergebnis zeigt, dass positive Vorerfahrungen sich auf die Erwartungen auswirken und dass hohe Erwartungen an den Therapieoutcome den Placeboeffekt steigern können. Umgekehrt kann die Erwartung von Nebenwirkungen auch zu mehr Nebenwirkungen führen. Eine Beobachtungsstudie mit 111 Patientinnen über 24 Monate (5) weist z.B. darauf hin, dass negative Erwartungen bzgl. einer endokrinen Behandlung bei Brustkrebs das Risiko von behandlungsspezifischen Nebenwirkungen, Nocebo-Nebenwirkungen und Non-Adhärenz erhöhen können. Placebo und Nocebo spielen in der Medizin eine wichtige Rolle (6) und die Aufklärung der Patientin darüber kann helfen, Nebenwirkungen zu reduzieren (7). Es ist kein primär komplementärmedizinisches Thema, lässt sich aber im Gespräch gut damit kombinieren.
Das Interesse an Komplementärmedizin und die Vorerfahrungen mit Komplementärmedizin lassen sich gut anhand von Fragebögen erfassen. Verwendet man Fragebögen in der Praxis, ist es aber zwingend, diese auch zusammen mit den Patientinnen in der Sprechstunde zu besprechen, um die Sinnhaftigkeit des Ausfüllens aufzuzeigen.
Die Patientin zur aktiven Teilnehmerin am Behandlungsprozess machen
Die aktive Einbindung der Patientinnen in die Behandlung ist ein wichtiges Element in der integrativen Onkologie und eine Stärke einiger komplementärmedizinischer Therapien. Eine Akupressur (z.B. bei Übelkeit oder Fatigue), welche die Patientin selbst durchführen kann, hat sehr viel mehr aktiven Anteil als eine vom Behandler oder von der Behandlerin durchgeführte Akupunktur. Bei der aktiven Teilnahme geht es auch um eine Verbesserung der Selbstwirksamkeit. Selbstwirksamkeit wird von Schwarzer beschrieben als die subjektive Überzeugung, kritische Anforderungssituationen aus eigener Kraft erfolgreich bewältigen zu können (8). Dabei wird an neue oder schwierige Situationen aus allen Lebensbereichen gedacht sowie an Barrieren, die es zu überwinden gilt. Dabei wird auch von der Annahme ausgegangen, dass Menschen ihre Erfolgs- und Misserfolgserfahrungen sich selbst zuschreiben und danach generalisieren. Dies zeigt auch, wie wichtig es ist, mit der Patientin realistische Ziele bei geplanten Lebensstiländerungen zu vereinbaren. Bei zu hoch gesteckten Zielen (z.B. ich mache ab morgen jeden Tag 30 Minuten Sport) ist die Gefahr einer Misserfolgserfahrung zu gross. Auch ist es wichtig, Lebensstiländerungen mit sog. «Behavioural Change Techniques» zu begleiten, um die langfristige Umsetzung zu sichern. Zu den bewährten Techniken gehören z.B. die Setzung von realistischen Zielen, die Identifikation von Barrieren, eine Umsetzungsplanung sowie die soziale Unterstützung bei der Durchführung.
Integration in die Praxis und Zugang zu Ressourcen
Komplementärmedizin kann man unterschiedlich in den gynäkologischen Praxisalltag integrieren. Man kann Patientinnen Informationen zu dem Thema geben, mit Patientinnen komplementäre Therapien auswählen oder auch Patientinnen mit komplementären Therapien behandeln (9).
Viele Patientinnen erwarten von ihrer Gynäkologin oder von ihrem Gynäkologen zumindest eine kompetente Beratung zum Thema Komplementärmedizin oder besser noch ein konkretes Angebot. Die Leitlinien zeigen, dass es dabei nicht um die Verordnung von Vitaminen und Nahrungsergänzungsmitteln geht, sondern zumeist um nicht-pharmakologische Verfahren. Es geht aber auch darum, der Patientin Zugang zu entsprechenden qualitätsgesicherten Angeboten vermitteln zu können, wenn diese in der eigenen Praxis oder Klinik nicht vorhanden sind. Unser in einem wissenschaftlichen Konsensusprozess (10) entwickelte Flyer für seriöse Anbieter kann hier heruntergeladen und mit einem Verweis auf die eigene Praxis oder Klinik versehen werden: http://www.iki.usz.ch/forschung/Seiten/kokon-kto.aspx.
Auch Online-Angebote können hilfreich sein. Laut S3-Leitlinie «Psychoonkologische Diagnostik, Beratung und Behandlung von erwachsenen Krebspatienten» sollen jeder Krebspatientin Entspannungsverfahren empfohlen werden (11). Jedoch haben nicht alle Patientinnen dazu Zugang. Auf unserer Webseite am Universitätsspital Zürich ermöglichen wir einen kostenlosen Zugang zu Audios mit Entspannungsübungen für Menschen mit Krebserkrankungen: https://www.mbm-usz.ch/krebs/.
Möchte man sich selbst fortbilden, so bietet die Kommission Integrative Medizin der Arbeitsgemeinschaft gynäkologische Onkologie ein strukturiertes Fortbildungscurriculum zur integrativen Medizin (12) für Ärztinnen und Ärzte an, welches auch unser evidenzbasiertes Training beinhaltet und international abgestimmte Kompetenzen für integrative Onkologie vermittelt (13, 14).
Bei diesem Artikel handelt es sich um einen Zweitabdruck des in der «info@gynäkologie» 04-2021 erschienenen Originalartikels.
Copyright bei Aerzteverlag medinfo AG
Prof. Dr. med. Claudia M. Witt, MBA
Institut für komplementäre und integrative Medizin
Universitätsspital Zürich
Sonneggstrasse 6
8091 Zürich
claudia.witt@uzh.ch
Renate Rugieri Stiftung; Schweizer Fachverband Mind Body Medicine; International Association for Mind-Body-Medicine and -Health e.V. (IAM); Arbeitsgruppe Supportive and Palliative Care der Swiss Group for Clinical Cancer Research (SAKK); Arbeitsgruppe «Interprofessionalität» der Schweizerischen Akademie der Medizinischen Wissenschaften (SAMW); Arbeitsgemeinschaft Gynäkologische Onkologie;
Society of Integrative Oncology (SIO); Society of Acupuncture Research.
◆ Brustkrebspatientinnen wünschen sich eine Integration von Komplementärmedizin in die Versorgung und das Thema sollte besprochen werden.
◆ Es ist wichtig, die Beweggründe der Patientin zu verstehen und die Erwartungen bezüglich des Gesprächs und an die komplementären Therapien vorher abzugleichen.
◆ Die Empfehlungen zur Komplementärmedizin sollten evidenzinformiert und patientenzentriert sein.
◆ Verfahren, die eine aktive Beteiligung der Patientinnen ermöglichen, sollten bevorzugt werden und Lebensstiländerungen sollten unterstützend begleitet werden.
Messages à retenir
◆ Les patientes atteintes d’un cancer du sein souhaitent l’intégration de la médecine complémentaire dans leur prise en charge. Ce sujet devrait être abordé avec elles.
◆ Il est très important de comprendre ce qui motive la patiente et ce qu’elle attend de l’entretien et des thérapies complémentaires afin de s’accorder avec elle.
◆ Les recommandations pour la médecine complémentaire devraient être éclairées par l’évidence scientifique et centrées sur l’individualité de la patiente.
◆ Des procédures permettant une participation active de la patiente devraient être envisagées en premier lieu, et un changement du style de vie devrait être accompagné et soutenu.
1. Keene MR, Heslop IM, Sabesan SS, Glass BD. Complementary and alternative medicine use in cancer: A systematic review. Complementary therapies in clinical practice 2019;35:33-47
2. Witt CM, Balneaves LG, Cardoso MJ, Cohen L, Greenlee H, Johnstone P, et al. A comprehensive definition for integrative oncology. J Natl Cancer Inst Monogr 2017;2017(52):lgx012
3. Lyman GH, Bohlke K, Cohen L. Integrative therapies during and after breast cancer treatment: ASCO endorsement of the SIO clinical practice guideline summary. Journal of oncology practice 2018;14(8):495-499
4. Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF. Leitlinienprogramm Onkologie: Leitlinie Komplementärmedizin in der Behandlung onkologischer PatientInnen. 2021 (Verfügbar: https://www.awmf.org/leitlinien/detail/ll/032-055OL.html)
5. Nestoriuc Y, von Blanckenburg P, Schuricht F, Barsky AJ, Hadji P, Albert US, et al. Is it best to expect the worst? Influence of patients‘ side-effect expectations on endocrine treatment outcome in a 2-year prospective clinical cohort study. Ann Oncol 2016;27(10):1909-1915
6. Schedlowski M, Enck P, Rief W, Bingel U. Neuro-Bio-Behavioral Mechanisms of Placebo and Nocebo Responses: Implications for Clinical Trials and Clinical Practice. Pharmacol Rev 2015;67(3):697-730
7. Evers AWM, Colloca L, Blease C, Gaab J, Jensen KB, Atlas LY, et al. What Should Clinicians Tell Patients about Placebo and Nocebo Effects? Practical Considerations Based on Expert Consensus. Psychotherapy and psychosomatics 2021;90(1):49-56
8. Schwarzer RJ, Jerusalem M. Allgemeine Selbstwirksamkeitserwartung (SWE). 2021 (Verfügbar: www.selbstwirksam.de)
9. Witt C, Müller T. Komplementärmedizin in der gynäkologisch-onkologischen Sprechstunde: Wie mache ich mich fit dafür? Gynäkologe 2021;54:32-37
10. Rogge AA, Baur I, Blettner G, Holtkamp U, Horneber M, Jahn P, et al. Defining criteria for guiding cancer patients to find a reputable complementary medicine provider: Results of a literature review and a consensus procedure. Patient Preference and Adherence 2020;14:747-755
11. Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF. Leitlinienprogramm Onkologie: Leitlinie Psychoonkologie. 2021 (Verfügbar: https://www.leitlinienprogramm-onkologie.de/leitlinien/psychoonkologie/)
12. Arbeitsgemeinschaft Gynäkologische Onkologie e.V. AGO. Kommission IMed (Integrative Onkologie in der Medizin). 2021 (Verfügbar: https://www.ago-online.de/ago-kommissionen/kommission-imed)
13. Witt CM, Balneaves LG, Carlson LE, Cohen M, Deng G, Fouladbakhsh JM, et al. Education Competencies for Integrative Oncology-Results of a Systematic Review and an International and Interprofessional Consensus Procedure. J Cancer Educ 2020
14. Witt CM, Helmer SM, Schofield P, Wastell M, Canella C, Thomae AV, et al. Training oncology physicians to advise their patients on complementary and integrative medicine: An implementation study for a manual-guided consultation. Cancer 2020;126(13):3031-3041
Das kutane Plattenepithelkarzinom (PEK) stellt nach dem Basalzellkarzinom den zweithäufigsten Hauttumor in der hellhäutigen Bevölkerung dar und ist histologisch charakterisiert durch die maligne Proliferation von epidermalen Keratinozyten (1). Das PEK tritt vornehmlich an UV-Licht exponierten Hautarealen auf (80%-90% in der Kopf-Hals-Region) und ist ein Tumor des höheren Alters (>70 Jahre) (2-4). Hauptrisikofaktor für die Entstehung des PEK stellt die kumulative UV-Belastung dar.
Après le carcinome basocellulaire, le carcinome épidermoïde cutané (CEC) est la deuxième tumeur cutanée la plus fréquente dans la population à peau claire et se caractérise histologiquement par la prolifération maligne des kératinocytes épidermiques (1). Le CEC apparaît principalement dans les zones cutanées exposées à la lumière ultraviolette (80 à 90 % dans la région de la tête et du cou) et est une tumeur d’âge avancé (> 70 ans) (2-4). Le principal facteur de risque pour le développement du CEC est l’exposition cumulative à la lumière ultraviolette.
In genomischen Analysen lässt sich für PEKs die typische UV-Mutationssignatur mit ihren charakteristischen C > T oder CC > TT Dinukleotid-Mutationen nachweisen, welche auch für das kutane Melanom charakteristisch sind. Im Vergleicht der verschiedenen Tumorentitäten weist das kutane Plattenepithelkarzinom bei weitem die höchsten Mutationsraten mit durchschnittlich 61,2/Megabase auf und liegt damit um das 4-5-fache höher als beim malignen Melanoms mit 13,2/Megabase (5). Die Heilungschancen sind für die meisten Patienten mit lokalisiertem Plattenepithelkarzinom nach adäquater Therapie (vollständige chirurgische Exzision +/- adjuvanter Radiotherapie im Falle einer R1-Situation oder ausgeprägter Perineuralscheiden-Infiltration) hoch, mit 10 Jahres krankheits-spezifischem Überleben von deutlich über 90% (6). Lediglich 2,8% des von Eigentler et al. prospektiv untersuchten Kollektivs von über 1400 Pateinten entwickelten nach Primärtherapie Metastasen und verstarben an den Folgen der Erkrankung (6). Eine Alternative zur Exzision stellt die primäre Radiotherapie dar mit lokalen Rezidivraten von 3-11% (7).
Systemische Behandlungsansätze des fortgeschrittenen PEK
Während für das lokalisierte PEK zwei Therapiemodalitäten mit ausgezeichneten Heilungschancen (> 90%) zur Verfügung stehen, ist die Behandlung des bereits fortgeschrittenen PEK deutlich problematischer. Dabei wird klinisch zwischen einem rein lokal fortgeschrittenen PEK und dem metastasierten (lymphogen oder peripher) PEK unterschieden. Für beide klinischen Szenarien sind die lokaltherapeutischen Massnahmen entweder wie im Falle des lokal fortgeschrittenen PEK ausgeschöpft (z. Bsp. lokales Rezidiv nach mehrmaliger Exzision oder vorangegangener Radiotherapie), oder aber nicht mehr sinnvoll wie im Falle beim Vorliegen von peripheren Metastasen.
Für diese Patientengruppen liegt eine palliative Gesamtsituation vor und eine systemische Behandlung ist primär indiziert. Bis zur Einführung der Immun-Checkpoint Inhibitoren (ICI) in den klinischen Alltag existierte kein Standard bei der Wahl der Systemtherapie für diese Patienten und die medianen Überlebenszeiten bei Einsatz von konventionellen Chemotherapeutika (z.Bsp. Cisplatin/5-FU, Carboplatin/Taxol) oder Medikamenten gegen den EGF-Rezeptor (Cetuximab, Panitumuab) und dessen Kinase-Domäne (Erlortinib, Gefitinib) betrugen nicht mehr als 16 Monate (8-10).
Immun-Checkpoint Inhibitoren als neue Therapiemöglichkeit des fortgeschrittenen PEK
Seit der Zulassung des ersten Immun-Checkpoint Inhibitors Ipilimumab (anti-CTLA4-Antikörper) im März 2011 durch die FDA für das metastasierte Melanom hat sich die Behandlungslandschaft in der Onkologie rasant verändert. Insbesondere Antikörper zur Blockierung der PD1/PD-L1 Achse, die ein besseres Wirkungs- und günstigeres Nebenwirkungsprofil als die anti-CTLA-4 Antikörper in der Monotherapie aufweisen finden heute Anwendung im klinischen Alltag.
Auf Grund der kausalen Beteiligung der kumulativen UV-Exposition an der Entstehung der PEKs weisen diese, wie bereits eingangs erwähnt, die höchste Mutationsrate unter den verschiedenen Tumorentitäten auf. Unter der Annahme, das für Tumoren mit hohen Mutationsraten, die Wahrscheinlichkeit für die Bildung von Neoantigenen steigt die in der Folge vom Körpereigenen Immunsystem als «fremd» erkannt werden können, sollten die PEKs für die Behandlung mit ICI geradezu prädestiniert sein. Erste klinische Hinweise für die Wirksamkeit von ICIs beim fortgeschrittenen PEK lieferten Fallbeschreibungen zum Einsatz der anti-PD1 Antikörper Pembrolizumab, Nivolumab oder Cemiplimab mit zum Teil langanhaltenden, kompletten Remissionen (> 24 Monate) (10-12). In Abbildung 1 ist der erfreuliche Verlauf eines 83-järigen Patienten mit multiplen PEK unter einer Behandlung mit Cemiplimab dargestellt. Basierend auf diesen vielversprechenden Fallbeschreibungen wurde eine erste prospektive, multizentrische Phase 1/2 Studie mit Cemiplimab lanciert (13): 26 Patienten (10 lokal fortgeschrittenes PEK + 16 metastasiert) aus der erweiterten Phase 1, sowie 59 Patienten mit metastasiertem (lymphogen + peripher) PEK erhielten 2-wöchentlich Cemiplimab in einer Dosierung von 3 mg/Kg Körpergewicht intravenös. Die Behandlung wurde in der Phase 1 bis maximal 48 Wochen in Phase 2 bis insgesamt 96 Wochen fortgeführt. Für 13 der 26 Patienten (50%) aus der Phase 1 und 28 der 59 Patienten (47%) aus der Phase 2 konnte unter der Behandlung ein objektives Ansprechen mit kompletten Remissionen für vier metastatische Patienten erzielt werden. Ein weiterer Zugewinn für einen Teil der Patienten war neben den hohen Ansprechraten auch die Dauer des Ansprechens von mindestens 6 Monaten in mehr als der Hälfte (57%) der «Responder». Als häufigste Nebenwirkungen traten die für eine ICI-Behandlung typischen Symptome wie Durchfall (27%, Müdigkeit (24%), Übelkeit (17%) und Hautauschlag (15%) auf. In den 12-Monats Verlaufsdaten der Kohorte für metastasierte PEK bestätigte sich die hohe Ansprechrate von rund 50% mit einer Dauer des Ansprechens von mehr als 12 Monaten für 75% der «Responder», das mediane Progressionsfreie Überleben betrug 18.4 Monate. (14).
Cemiplimab als neuer Standard in der Systemtherapie
Basierend auf diesen Daten wurde Cemiplimab am 28.9.2018 durch die FDA und von der EMA am 28.6.2019 als Monotherapie zur Behandlung von erwachsenen Patienten mit metastasiertem oder lokal fortgeschrittenem kutanen Plattenepithelkarzinom, die für eine kurative Operation oder kurative Strahlentherapie nicht in Betracht kommen zugelassen. Die Behandlung erfolgt in einer fixen Dosierung von 350mg in 3 wöchentlichen Intervallen gewichtsunabhängig, basierend auf pharmakokinetischen Studien.
Eine separate Phase 2 Studie untersuchte parallel die Wirksamkeit von Cimiplimab für rein lokal fortgeschrittene PEK (Patienten ohne lymphogene oder periphere Metastasen) für die eine kurativ intendierte chirurgische oder radiotherapeutische Behandlung nicht (mehr) in Frage kam (15). Insgesamt 78 Patienten erhielten Cemiplimab 3mg/kg in 2-wöchentlichen Intervallen für maximal 96 Wochen. Dabei konnte erneut eine hohe Ansprechrate von 44% (34 von 78 Patienten mit 10 kompletten Remissionen) erzielt werden. 23 der 34 «Responder» (68%) zeigten zudem eine Dauer des Ansprechens von mindestens 6 Monaten, das mediane progressionsfreie Überleben, sowie das mediane Gesamtüberleben ist für die Kohorte zum Zeitpunkt des Data-cutoffs noch nicht erreicht gewesen. Diese zweite Studie unterstützt nachdrücklich die Entscheidung der FDA und EMA für die Zulassung von Cemiplimab in entsprechender Indikation. Neben der Wirksamkeit wurde in dieser Studie auch der PD-L1 Status und die individuelle Mutationsrate des Tumors als mögliche prädiktive Marker untersucht. Weder der PD-L1 «tumor proportion score» (TPS), noch die Höhe der Mutationsrate (im Mittel 74/Megabase) erwiesen sich diesbezüglich als aussagekräftig.
Neben Cemiplimab werden auch Pembrolizumab (anti-PD1 Antikörper) und Avelumab (anit-PD-L1 Antikörper) in klinischen Studien für das fortgeschrittene PEK untersucht: Erste Daten zu Pembrolizumab als Erstlinien-Therapie zeigten ein Ansprechen für 15 der 39 behandelten Patienten (38.5%). Avelumab wird in einer randomisierten Phase 2 Studie als Monotherapie oder in Kombination mit Cetuximab untersucht und in einer weiteren Phase 2 Studie wird Cemiplimab in Kombination mit einer lokalen Therapie (intraläsionale Applikation eines oncolytischen Virus, RP1) getestet. Aktuell rekrutieren zudem auch zwei randomisierte Phase 3 Studien zur adjuvanten Behandlung mit entweder Cimiplimab oder Pembrolizumab (NCT03969004, NCT03833167).
Copyright bei Aerzteverlag medinfo AG
Dr. med. Michael Benzaquen
Klinik für Dermatologie
Inselspital Bern
3010 Bern
PD Dr. med. Julian Schardt, PhD
Klinik für Medizinische Onkologie
Inselspital Bern
3010 Bern
Julian.Schardt@insel.ch
Die Autoren haben im Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
Die Behandlung von Patienten mit fortgeschrittenem PEK hat sich mit der Einführung der Checkpoint-Inhibitoren deutlich verbessert. So lassen sich mit dem Einsatz von Cemiplimab Ansprechraten von bis zu 50% der behandelten Patienten erzielen, für ca. 2/3 der «Responder» ist zudem ein Langzeitansprechen von mindestens 6 Monaten möglich.
Weiterhin fehlen aber verlässliche prädiktive Marker: Weder der PD-L1 TPS-Score noch die Bestimmung der globalen Mutationsrate im Tumor sind in diesem Falle aussagekräftig. Ein weiterhin schwierig zu behandelndes Kollektiv bleibt das fortgeschrittene PEK von Organ-Empfängern unter Immunsuppression. Hier ist der Einsatz von ICI in der Regel mit einer Transplantatabstossung verbunden und es kommen konventionelle Chemotherapien oder anti-EGFR gerichtete Therapien zum Zuge.
Die Behandlung des fortgeschrittenen PEK mit Cemiplimab stellt bei fehlenden Kontraindikationen gegenwärtig den neuen Standard in der systemischen Therapie dar.
Messages à retenir
Le traitement des patients atteints du carcinome épidermoïde cutané (CEC) avancé s’est considérablement amélioré avec l’introduction des inhibiteurs de checkpoint. Par exemple, des taux de réponse allant jusqu’à 50 % des patients traités peuvent être obtenus par l’utilisation du Cemiplimab, et pour environ 2/3 des „répondeurs“, une réponse à long terme d’au moins 6 mois est également possible.
Cependant, il manque encore des marqueurs prédictifs fiables : ni le score TPS PD-L1 ni la détermination du taux de mutation global de la tumeur ne sont significatifs dans ce cas. Le CEC avancé des receveurs d’organes sous immunosuppression reste un collectif difficile à traiter. Ici, l’utilisation de l’ICI est généralement associée à un rejet de greffe et on utilise la chimiothérapie conventionnelle ou des thérapies dirigées contre l’EGFR.
Le traitement du CEC avancé par le cemiplimab représente actuellement la nouvelle norme en matière de thérapie systémique en l’absence de contre-indications.
Albert, M.R. and M.A. Weinstock, Keratinocyte carcinoma. CA Cancer J Clin, 2003. 53(5): p. 292-302.
Alam, M. and D. Ratner, Cutaneous squamous-cell carcinoma. N Engl J Med, 2001. 344(13): p. 975-83.
Nagarajan, P., et al., Keratinocyte Carcinomas: Current Concepts and Future Research Priorities. Clin Cancer Res, 2019. 25(8): p. 2379-2391.
Que, S.K.T., F.O. Zwald, and C.D. Schmults, Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J Am Acad Dermatol, 2018. 78(2): p. 237-247.
Pickering, C.R., et al., Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res, 2014. 20(24): p. 6582-92.
Eigentler, T.K., et al., Survival of Patients with Cutaneous Squamous Cell Carcinoma: Results of a Prospective Cohort Study. J Invest Dermatol, 2017. 137(11): p. 2309-2315.
Lansbury, L., et al., Interventions for non-metastatic squamous cell carcinoma of the skin: systematic review and pooled analysis of observational studies. BMJ, 2013. 347: p. f6153.
Cowey, L.e.a., Treatment patterns and outcomes among patients with advanced cutaneous squamous cell carcinoma (CSCC) in a US community oncology setting. J Clin Oncol, 2019. 37(no. 15_suppl).
Hillen, U., et al., Advanced cutaneous squamous cell carcinoma: A retrospective analysis of patient profiles and treatment patterns-Results of a non-interventional study of the DeCOG. Eur J Cancer, 2018. 96: p. 34-43.
Petersen, E.T., et al., Review of systemic agents in the treatment of advanced cutaneous squamous cell carcinoma. Future Oncol, 2019. 15(27): p. 3171-3184.
Borradori, L., et al., Rescue therapy with anti-programmed cell death protein 1 inhibitors of advanced cutaneous squamous cell carcinoma and basosquamous carcinoma: preliminary experience in five cases. Br J Dermatol, 2016. 175(6): p. 1382-1386.
Falchook, G.S., et al., Responses of metastatic basal cell and cutaneous squamous cell carcinomas to anti-PD1 monoclonal antibody REGN2810. J Immunother Cancer, 2016. 4: p. 70.
Migden, M.R., et al., PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N Engl J Med, 2018. 379(4): p. 341-351.
Guminski, A., Phase 2 study of cemiplimab, a human monclonal anti-PD-1 antibody, in patients witih metastatic cutaneous squamous cell carcinomal (mCSCC; Group 1) 12-month follow-up. J Clin Oncol, 2019. 37(suppl):9526.
Migden, M.R., et al., Cemiplimab in locally advanced cutaneous squamous cell carcinoma: results from an open-label, phase 2, single-arm trial. Lancet Oncol, 2020. 21(2): p. 294-305.
Sarkome sind seltene Tumore: die altersstandardisierte Erkrankungsrate in der Schweiz lag 2011 – 2015 bei 4.43/100 000/Jahr für Weichgewebssarkome und bei 0.91/100 000/Jahr für Knochensarkome (1). Die aktuelle 5. Edition der WHO Klassifikation listet über 100 Entitäten intermediärer (lokal aggressiv, sehr selten metastasierend) und maligner Weichgewebs- und Knochentumore. Entsprechend ist die Inzidenz auch bei den häufigeren Entitäten wie dem Liposarkom und dem Leiomyosarkom < 1/100 000/Jahr.
Les sarcomes sont des tumeurs rares: le taux de maladie standardisé pour l’âge en Suisse en 2011 – 2015 était de 4,43/100 000 par an pour les sarcomes des tissus mous et de 0,91/100 000 par an pour les sarcomes osseux (1). La 5e édition actuelle de la classification de l’OMS énumère plus de 100 entités de tumeurs intermédiaires (localement agressives, très rarement métasta-tiques) et malignes des tissus mous et des os. En conséquence, l’incidence est < 1/100 000 par an pour les entités les plus fréquentes telles que les lipo-sarcomes et les léiomyosarcomes.
Um Fortschritte in der Behandlung von Sarkomen zu erzielen, ist – wie bei allen seltenen Erkrankungen – die internationale Zusammenarbeit in der Forschung und ein Wissenstransfer zentral, ebenso die Behandlung der Patienten durch erfahrene multidisziplinäre Teams. Dies bedingt den Aufbau von Referenzzentren respektive Referenznetzwerken sowie die entsprechende Zuweisung von Patienten. Bei den Sarkomerkrankungen relevant ist, dass die Zuweisung bereits zum Zeitpunkt der bildgebenden Verdachtsdiagnose und noch vor der Biopsie erfolgt, dies setzt das entsprechende Wissen um Abklärungsalgorithmen beim Primärbehandler voraus. Diagnostik und Behandlung in Referenzzentren korreliert positiv mit dem Gesamtüberleben der Patienten und hat entsprechend Eingang in die Guidelines gefunden (2, 3).
Fortschritte sind im letzten Jahr in verschiedenen Bereichen zu verzeichnen, worauf in der Folge eingegangen werden soll.
WHO Klassifikation 5. Edition
Seit diesem Jahr gilt die 5. Edition der WHO-Klassifikation der Weichgewebs- und Knochentumore. Die Fortschritte in der molekularen Charakterisierung verschiedener Sarkomentitäten finden darin Eingang. So wurde die Gruppe der ehemals Ewing-like Sarkome in die rundzelligen Sarkome mit EWSR1-non-ETS Fusionen, CIC-rearrangierte Sarkome und Sarkome mit BCOR Alteration aufgeteilt. Dies bei klinisch differentem, meist aggressiverem Verlauf verglichen mit dem klassischen Ewing Sarkom (4). Ebenso wurden neu die Gruppe der NTRK-rearranged spindel cell neoplasm und der EWSR1-SMAD3-positive fibroblastic tumors geschaffen. Nicht immer ist die molekulare Neuzuordnung bislang mit therapeutischen Konsequenzen verbunden. Die häufiger durchgeführten molekularen Analysen haben zudem die Anzahl bekannter Translokationen deutlich erhöht – dabei ist die Frage offen, wann diese Genfusionen auch Driver sind und damit in Zukunft therapeutisch angegangen werden können (5)
Ewing Sarkom
Am ASCO 2020 wurden die Resultate der Euro Ewing 2012 / R1-Randomisierung (6), der Ewing 2008 / R3-Arm (9) sowie die zweite Interimsanalyse der rEECur Studie (12) präsentiert.
Euro Ewing 2012: Annäherung in der Definition eines internationalen Standards für die Erstlinien Behandlung des Ewing Sarkoms Die R1 Randomisierung der Euro Ewing 2012 verglich den europäische Behandlungsstandard (Arm A: Induktion mit 6 Zyklen VIDE, Lokaltherapie, Konsolidierung mit 8 Zyklen VAC/VAI respektive bei lokalisierter Erkrankung mit hohem Risiko 1 Zyklus VAI/Hochdosischemotherapie mit Busulfan/Melphalan) mit der amerikanischen Standardbehandlung (Arm B: Induktion mit VDC/IE 2-wöchentlich, Lokaltherapie, Konsolidierung mit IE/VC 2-wöchentlich), wobei Patienten mit poor risk lokalisierter Erkrankung im Arm B analog dem europäischen Standard mit Hochdosischemotherapie behandelt wurden (Abb. 1). In der Schweiz haben die Zentren Bern, Zürich und St.Gallen an der Studie teilgenommen.
Der Arm B zeigte sich sowohl bezüglich Ereignis-freiem Überleben (event free survival, EFS) wie bezüglich Gesamtüberleben (overall survival, OS) klar überlegen, sodass die R1 Randomisierung nach Erreichen der statistisch geforderten Minimalanzahl Patienten im Mai 2019 geschlossen wurde. Die nun gezeigten Daten bestätigen mit einer Hazard Ratio (HR) von 0.70 (95% CI 0.51- 0.95) für das EFS und einer HR von 0.64 (95% CI 0.42 -0.96) für das OS den Vorteil des komprimierten VDC/IE Schemas. Die Toxizitäten waren vergleichbar (Arm A 68% serious adverse events (SAE) vs. Arm B 67% SAEs) (6). Somit wird der europäische Standard in der Erstlinientherapie des Ewing Sarkoms durch die amerikanische Standardbehandlung abgelöst, ergänzt durch eine Hochdosischemotherapie bei lokalisierter Erkrankung mit hohem Risiko.
Diese Resultate bestätigen den Benefit einer dosisintensiveren Behandlung beim Ewing Sarkom, wie dies bereits vom Benefit der Hochdosischemotherapie bekannt ist. In welchem Ausmass damit vermehrt Langzeittoxizitäten in Kauf genommen werden müssen wird sich weisen, sicherlich sind diesbezüglich gewisse Limiten erreicht. Dringend sollten auch deswegen neue Substanzen in die Behandlung des Ewing Sarkoms integriert werden können. Die Voraussetzung einer einheitlichen Erstlinienbehandlung für zukünftige Studien in möglichst interkontinentaler Zusammenarbeit ist hiermit nun gegeben (7, 8).
Hochdosischemotherapie beim Ewing Sarkom: indiziert ausschliesslich bei lokalisierter Erkrankung mit hohem Risiko
Die Ewing 2008 Studie untersuchte in ihrem dritten Studienarm bei Patienten mit primär disseminierter Erkrankung den Nutzen einer zusätzlichen Hochdosischemotherapie mit Treosulfan und Melphan (Abb. 2). Das 3-Jahres EFS war mit und ohne Hochdosistherapie in der Gesamtkohorte vergleichbar bei 20.9% (95% CI 11.5 – 37.9%) vs. 19.2% (95% CI 10.8 – 34.4.%). Nur für Patienten < 14 Jahre zeigte sich ein möglicher Vorteil der Hochdosischemotherapie (3 Jahre EFS 39.9% vs. 9%) (9).
Der Stellenwert der Hochdosischemotherapie in der Erstlinienbehandlung des Ewing Sarkoms nach Induktionstherapie mit VIDE hat sich somit geklärt und ist einzig bei Patienten mit lokalisierter Hochrisiko Erkrankung gegeben. Eine lokalisierte Erkrankung wird der Hochrisikogruppe zugeordnet, wenn sich nach Induktionschemotherapie ein schlechtes pathologisches Ansprechen zeigt (≥ 10% residuelle Tumorzellen) respektive initial ein grosser Tumor vorliegt (≥ 200 ml), welcher wegen primärer oder fehlender Resektion respektive präoperativer Radiotherapie bezüglich des pathologischen Ansprechens nicht evaluiert werden kann. Zudem bei Vorliegen eines unresezierten Tumors < 200 ml mit schlechtem radiologischen Ansprechen auf die Induktionschemotherapie. In dieser Patientengruppe konnte durch die Busulfan und Melphalan Hochdosischemotherapie ein klar verbessertes Gesamtüberleben erreicht werden: OS nach 8 Jahren 64.5% (95% CI, 54.4% – 73.5%) versus 55.6% (95% CI, 45.8% – 65.1%) (10).
rEECur, Therapie des rezidivierten und primär refraktären Ewing Sarkoms
In der rEECur Studie werden erstmals verschiedene Chemotherapieschemata in der rezidivierten und primär refraktären Situation beim Ewing Sarkom untersucht. Dabei erfolgte initial eine Randomisierung in folgende 4 Arme: Topotecan / Cyclophosphamid (TC), Irinotecan/Temozolomid (IT), Gemcitabin/Docetaxel (GD) und hochdosiertes Ifosfamid (IFOS) (Abb. 3). In der ersten geplanten Interimsanalyse zeigte sich eine geringere Effizienz der Therapie mit Gemcitabine/Docetaxel verglichen mit den übrigen 3 Armen (RR 11.5%, median PFS 3.0 Monate, medianes OS 13.7 Monate), sodass dieser Arm geschlossen wurde (11). In der zweiten Interimsanalyse war nun der IT-Arm unterlegen mit einer Ansprechrate von 20%, einem medianen progressionsfreien Überleben (progression free survial, PFS) von 4.7 Monaten und einem medianen Gesamtüberleben von 13.9 Monaten (12). In die beiden verbleibenden Arme (TC und IFOS) können weiterhin Patienten eingeschlossen werden.
In der Schweiz wird die Studie durch die Schweizerische Pädiatrische Onkologie Gruppe (SPOG) durchgeführt, ein Einschluss von Patienten > 18 Jahren in SPOG-verantworteten Studien ist bislang nicht möglich. Für seltene Erkrankung wie das Ewing Sarkom ist zwingend, möglichst allen Patienten den Zugang zu Studien zu ermöglichen. Für altersübergreifende Erkrankungen sollte hierfür rasch eine möglichst administrationsarme Lösung zwischen Schweizerische Arbeitsgemeinschaft für Klinische Krebsforschung (SAKK) und der SPOG gefunden werden.
Dies erhält zusätzlich Gewicht, gerät die Altersgruppe der Adoleszenten und jungen Erwachsenen (Adolescents and Young Adults, AYA, Alter 15 – 39 Jahre) zunehmend in den Fokus. Die dieser Altersgruppe eigenen oft komplexen medizinischen, psychologischen und sozialen Aspekte bedürfen sowohl in Studien wie im klinischen Alltag einer gesonderten Betrachtung. So ist es auch eine Tatsache, dass die Studienteilnahme von Patienten dieses Alters oft geringer ist als bei pädiatrischen Patienten resp. älteren Erwachsenen. Dies nicht zuletzt auf Grund der herkömmlich limitierten Zusammenarbeit zwischen Studiengruppen der pädiatrischen und der Erwachsenen Onkologie (13, 14).
Perioperative Systemtherapie
Die Resultate einer japanischen Phase II/III Studie zur perioperativen Systemtherapie beim Weichteilsarkom stützen erneut die Wirksamkeit einer entsprechenden Behandlung (15). In dieser randomisierten Non-Inferioritätstudie wurde bei operablen G2/G3 Weichteilsarkomen eine perioperative Therapie mit Doxorubicin/Ifosfamid 10g/m2 (AI) mit Gemcitabine/Docetaxel (GD) verglichen. Es erfolgte die Verabreichung von 3 Zyklen präoperativ und 2 Zyklen postoperativ. In der zweiten geplanten Interimsanalyse zeigte sich ein 2-Jahresüberleben für AI von 94.3%, für GD von 91.6%. Das PFS für AI lag bei 81.9%, für GD bei 64% bei einer HR von 2.32 (95% CI 1.22 – 4.39). Die Studie wurde auf Grund der fehlenden Non-Inferiorität von GD geschlossen. Somit bleibt AI Standard für eine perioperative Systemtherapie, falls eine solche durchgeführt wird (16). Die Resultate sind erneut kein formaler Beweis für die Wirksamkeit von AI (neo)adjuvant, legen eine mindestens PFS-relevante Wirkung jedoch sehr nahe – es sei denn, eine Therapie mit GD stellt einen PFS-relevanten Nachteil dar.
Sehr spannend in diesem Kontext der perioperativen Behandlung sind auch die präliminären Daten einer Phase II Studie zur konkomitierenden neoadjuvanten Checkpoint Blockade (Nivolumab oder Ipilimumab / Nivolumab) mit Radiotherapie bei dedifferenziertem retroperitonealen Liposarkom (DDLPS) und undifferenziertem pleomorphen Sarkom (UPS) der Extremiät oder des Rumpfs (17). Es zeigte sich beim UPS ein sehr hohes pathologisches Ansprechen von 95% (95% CI 85% – 99%), beim DDLPS von 22.5%. Dies unterstreicht die Wertigkeit der Checkpointinhibitortherapie bei gewissen Sarkomentitäten. Weitere zu beachten ist, dass das pathologische und radiologische Ansprechen gemäss RECIST Kriterien nicht korrelieren – diese erweisen sich somit zur Evaluation des Therapieansprechens bei Sarkomen erneut als ungenügend.
Ebenso erfreulich sind die Resultate der TRASTS Studie, in welcher Patienten mit myxoidem Liposarkom eine präoperative Radiochemotherapie erhielten: 3 Zyklen Trabectedin sowie Bestrahlung mit 45 Gy (18). Diese Kombination liegt bei bekannter Sensitivität des myxoiden Liposarkoms sowohl auf Radiotherapie als auch auf Trabectedin sowie radiosensitivierender Wirkung von Trabectedin nahe. Im Resektat zeigte sich bei 23/43 Patienten residueller Tumor >10%, in den restlichen 47% ≤ 10% respektive bei 14% der Patienten (6/43) ein komplettes pathologisches Ansprechen.
Fortgeschrittenes Sarkom
Für die Behandlung fortgeschrittener Sarkome wurden am ASCO 2020 hauptsächlich Daten zu Kombinationstherapien bereits bekannter Substanzen präsentiert. Weiterhin eher im Hintergrund steht die Immuntherapie, wenn diese auch in einzelnen Entitäten gute Ansprechraten aufweist, wie bereits erwähnt.
So bestätigt die finale Analyse der LMS-02 Studie mit den OS-Daten die Wirksamkeit einer Kombinationstherapie mit Doxorubicin und Trabectedin beim Leiomyosarkom (LMS). Diese Phase II Studie prüfte beim uterinen LMS (u-LMS) wie beim Weichgewebs LMS (st-LMS) eine Therapie mit Doxorubicin + Trabectedin als Erstlinientherapie. Es zeigte sich ein Ansprechen (complete remission, CR und partial remisson, PR) für das u-LMS von 59.6% (47 Patienten), für das st-LMS von 39.4% (61 Patienten) (19). Nach einem medianen Follow-up von 7.2 Jahren betrug das mediane PFS 10.1 Monate und das mediane OS 34.4 Monate (20). Im indirekten Vergleich fand sich in der Phase III Announce Studie im Therapiearm Doxorubicin mono beim metastasierten LMS ein PFS von 6.9 Monaten und ein OS von 21.9 Monaten. Eine entsprechende randomisierte Phase III Studie läuft (LMS04).
Eine Phase Ib/II Studie zur Kombinationstherapie mit Lenvatinib und Eribulin beim fortgeschrittenen Lipo- und Leiomyosarkom nach maximal zwei Vortherapielinien weckt weitere Hoffnung, erreichte die Ansprechrate gemäss CHOI Kriterien doch sehr gute 67% (Eribulin mono overall response rate 4%) mit einem medianen progressionsfreien Überleben von 56 Monaten (Eribulin mono 2.6 Monate) (21).
Von der FDA wurden im letzten Jahr zudem verschiedene neue Substanzen für die Behandlung von Sarkomen zugelassen:
2019 Entrectinib, nach Larotrectinib der zweite TRK-Inhibitor für NTRK-fusionsassoziierte Tumore, basierend auf den drei zulassungsrelevanten Studien ALKA, STARTRK-1 und STARTRK-2 sowie Pexidartinib (CSF-1 Rezeptor Inhibitor) für die Behandlung tenosynovialer Riesenzelltumore (22).
2020 erfolgte bislang die Zulassung von Tazemetostat (EZH Inhibitor) für epithelioide Sarkome (23) sowie von Pomalidomid für das HIV-positive wie -negative Kaposi Sarkom (accelerated approval, basierend auf der NIH Studie 12-C-0047). Weiter zugelassen wurde Selumetinib, ein MEK-Inhibitor, für die Behandlung plexifomer Neurofibrome der Neurofibromatose Typ 1 (24), sowie in der Therapie des gastrointestinalen Stromatumors (GIST) Avapritinib (25), mit Wirksamkeit bei PDGFRA D842V Mutation, sowie Ripretinib (26) in der späteren Therapielinie.
Die Behandlung der Sarkome als Gesamtentität gehört der Vergangenheit an. Digitalisierung mit verstärkter elektronischer Vernetzung sowie die Behandlung der Patienten in national und international kollaborierenden Netzwerken bilden die Voraussetzung, den therapeutischen Fortschritt dieser teils ultrararen Erkrankungen möglichst rasch weiter voranzutreiben.
Copyright bei Aerzteverlag medinfo AG
Dr. med. Christina Appenzeller
Klinik für Medizinische Onkologie und Hämatologie
Kantonsspital St.Gallen
Rorschacher Strasse 95
9007 St.Gallen
PD Dr. med. Christian Rothermundt
Leitender Arzt / Leitung Ambulatorium St. Gallen & Rorschach
Kantonsspital St. Gallen
Klinik für Med. Onkologie/Hämatologie
Rorschacher Strasse 95
9007 St. Gallen
Die Autoren haben im Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
Ab 2020 gilt die neue WHO Klassifikation 5. Edition der Weichgewebs- und Knochentumore.
Die optimale Erstlinientherapie des Ewing Sarkoms ist heute die Induktion mit VDC/IE, Lokaltherapie, Konsolidation mit IE/VC. Patienten mit lokalisierter Hochrisikoerkrankung erhalten zusätzlich eine Hochdosistherapie.
Checkpoint Inhibitoren insbesondere in Kombination mit Radiotherapie zeigen bei einigen Weichteilsarkomen ein sehr gutes pathologisches Ansprechen.
Neue Substanzen für die personalisierte Therapie spezieller Weichteilsarkome wie Larotrectinib und Entrectinib (NTRK+ Sarkome), Pexidartinib (tenosynoviale Riesenzelltumore), Tazemetostat (epithelioide Sarkome), Pomalidomid (Kaposi Sarkome), Selumetinib (Neurofibromatose Typ1), Avapritinib und Ripretinib (GIST) sind teilweise bereits zugelassen.
Messages à retenir
À partir de 2020, la nouvelle classification de l’OMS (5e édition) des tumeurs des tissus mous et des os s’appliquera.
La thérapie optimale de première ligne pour le sarcome d’Ewing aujourd’hui est l’induction avec VDC/IE, la thérapie locale, la consolidation avec IE/VC. Les patients présentant une maladie localisée reçoivent en outre une thérapie à forte dose.
Les inhibiteurs de checkpoint, en particulier en combinaison avec la radiothérapie, montrent une très bonne réponse pathologique dans
certains sarcomes des tissus mous.
De nouvelles substances pour la thérapie personnalisée des sarcomes spéciaux des tissus mous, telles que le Larotrectinib et l’Entrectinib (sarcomes NTRK+), le Pexidartinib (tumeurs ténosynoviales à cellules géantes)
Le Tazemetostat (sarcomes épithélioïdes), le Pomalidomide (sarcome de Kaposi), le Selumetinib (neurofibromatose de type 1), l’Avapritinib et le Ripretinib (GIST) sont déjà app-rouvés dans certains cas.
1. Kollar A et al. Incidence, mortality, and survival trends of soft tissue and bone sarcoma in Switzerland between 1996 and 2015. Cancer Epidemiol. 2019; 63: 101596.
2. Loong et al. International collaborations and regional challenges in providing specialist multidisziplinary sarcoma care. ASCO Educational Book 2019
3. Blay J-Y et al. Surgery in Reference Centers Improves Survival of Sarcoma Patients: A Nationwide Study. Ann Oncol 2019; 30: 1143.
4. Palmerini E et al. Graceful Project: a global collaboration on CIC-DUX4, BCOR-CCNB3, high grade undif-ferentiated round cell sarcoma (URCS), CTOS 2019
5. Johansson B et al. Most gene fusions in cancer are stochastic events. Genes Chromosomes Cancer 2019; 58: 607.
6. Brennan B et al. Comparison of chemotherapy regimens in Ewing sarcoma (ES): Overall and subgroup results of the Euro Ewing 2012 randomized trial (EE2012). J Clin Oncol 2020; 38 (suppl; abstr 11500).
7. Anderton J et al. International randomised controlled trial for the treatment of newly diagnosed EWING sarcoma family of tumours. Trials 2020; 21: 96.
8. Gorlick R et al. Dose Intensification Improves the Outcome of Ewing Sarcoma.
J Clin Oncol 2018; 36: 3072.
9. Dirksen U et al. Efficacy of add-on treosulfan and melphalan high-dose therapy in patients with high-risk metastatic Ewing sarcoma: Report from the International Ewing 2008R3 trial. J Clin Oncol 2020; 38 (suppl; abstr 11501).
10. Wehlan J et al. High-Dose Chemotherapy and Blood Autologous Stem-Cell Rescue Compared With Standard Chemotherapy in Localized High-Risk Ewing Sarcoma: Results of Euro-E.W.I.N.G.99 and Ewing-2008. J Clin Oncol 2018; 36: 3110.
11. McGabe M et al. Results of the first interim assessment of rEECur, an international randomized controlled trial of chemotherapy for the treatment of recurrent and primary refractory Ewing sarcoma.J Clin Oncol 2019; 37 (suppl; abstr 11007).
12. McGabe M et al. Results of the second interim assessment of rEECur, an international randomized con-trolled trial of chemotherapy for the treatment of recurrent and primary refractory Ewing sarcoma (RR-ES). J Clin Oncol 2020; 38 (suppl; abstr 11502).
13. Osborn M et al. Models of care for adolescent and young adult cancer programs. Pediatr Blood Cancer 2019; 66: e27991.
14. Reed DR et al. Sarcoma as a Model for Adolescent and Young Adult Care. J Oncol Pract 2019; 15: 239.
15. Tanaka K et al. Results of a randomized phase II/III study comparing perioperative adriamycin plus ifosfamide and gemcitabine plus docetaxel for high-grade soft tissue sarcomas: Japan Clinical Oncology Group study JCOG1306. J Clin Oncol 2020; 38 (suppl; abstr 11504).
16. Rothermundt C. Perioperative treatment of soft-tissue sarcoma. Memo 2020;13:174.
17. Roland CL et al. Preliminary results of a phase II study of neoadjuvant checkpoint blockade for surgically resectable undifferentiated pleomorphic sarcoma (UPS) and dedifferentiated liposarcoma (DDLPS). J Clin Oncol 2020; 38 (suppl; abstr 11505.
18. Gronchi A et al. Trabectedin and radiotherapy in soft-tissue sarcoma (TRASTS) study: An international, prospective, phase II trial in localized myxoid liposarcoma—A collaborative Spanish (GEIS), Italian (ISG) and French (FSG) group study. J Clin Oncol 2020; 38 (suppl; abstr 11514).
19. Pautier P et al. Trabectedin in combination with doxorubicin for first-line treatment of advanced uterine or soft-tissue leiomyosarcoma (LMS-02): a non-randomised, multicentre, phase 2 trial. Lancet Oncol 2015; 16: 457.
20. Patricia Pautier A single-arm multicenter phase II trial of doxorubicin (Doxo) in combination with trabecte-din (Trab) given as first-line treatment to patients with metastatic/advanced uterine (U-LMS) and soft tissue leiomyosarcoma (ST-LMS): Final results of the LMS-02 study J Clin Oncol 2020; 38 (suppl; abstr 11506).
21. Tom Wei-Wu Chen et al. A Ib/II study of the combination of lenvatinib (L) and eribulin (E) in advanced liposarcoma (LPS) and leiomyosarcoma (LMS) (LEADER).
J Clin Oncol 2020; 38 (suppl; abstr 11507)
22. Tap WD et al. Pexidartinib versus placebo for advanced tenosynovial giant cell
tumour (ENLIVEN): a randomised phase 3 trial. Lancet. 2019; 394: 478.
23. Stacchiotti S et al. Safety and efficacy of tazemetostat, a first-in-class EZH2 inhibitor, in patients with epi-thelioid sarcoma. J Clin Oncol 2019; 37 (suppl; abstr 11003).
24. Gross AM et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N Engl J Med 2020; 382:1430
25. Heinrich M et al. Clinical activity of avapritinib in ≥ fourth-line (4L+) and PDGFRA Exon 18 gastrointestinal stromal tumors (GIST). J Clin Oncol 2020; 38 (suppl 4; abstr 826)
26. Blay J-Y et al. Ripretinib in Patients With Advanced Gastrointestinal Stromal Tumours (INVICTUS): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol 2020; 21: 923