Bei der IUGR (intrauterine growth restriction) werden grundlegend zwei Kategorien voneinander unterschieden. Die frühe, chronisch verlaufende Form ist mit einer Störung der plazentaren Entwicklung assoziiert. Der späten Form liegt mehr eine Diffusionsstörung bei nahezu normal entwickelter Plazenta zugrunde. Diese Zusammenfassung informiert über die neuesten Diagnosekriterien. Die Unterschiede im Management der early- und late-onset Form werden zusammen mit der Strategie zur Ermittlung des optimalen Entbindungszeitpunktes erklärt.
Definition: Was ist zu klein?
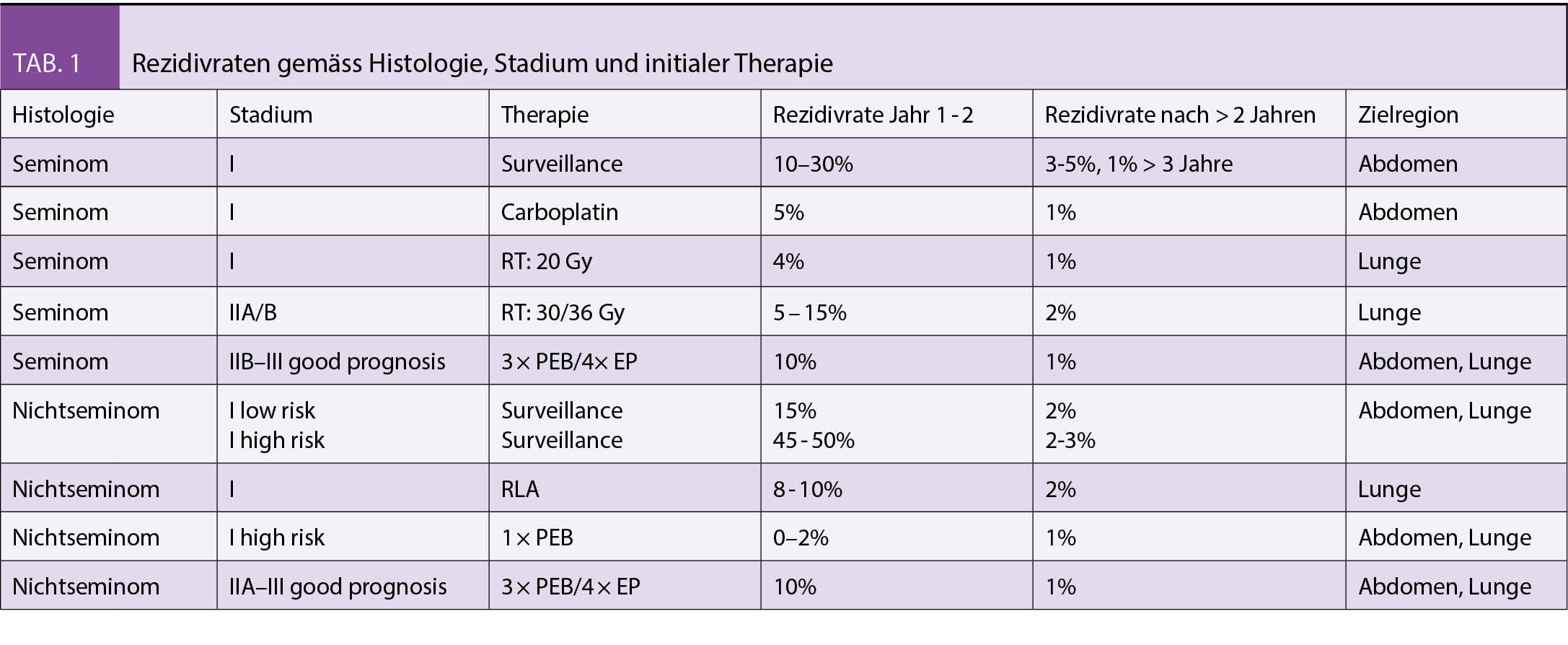
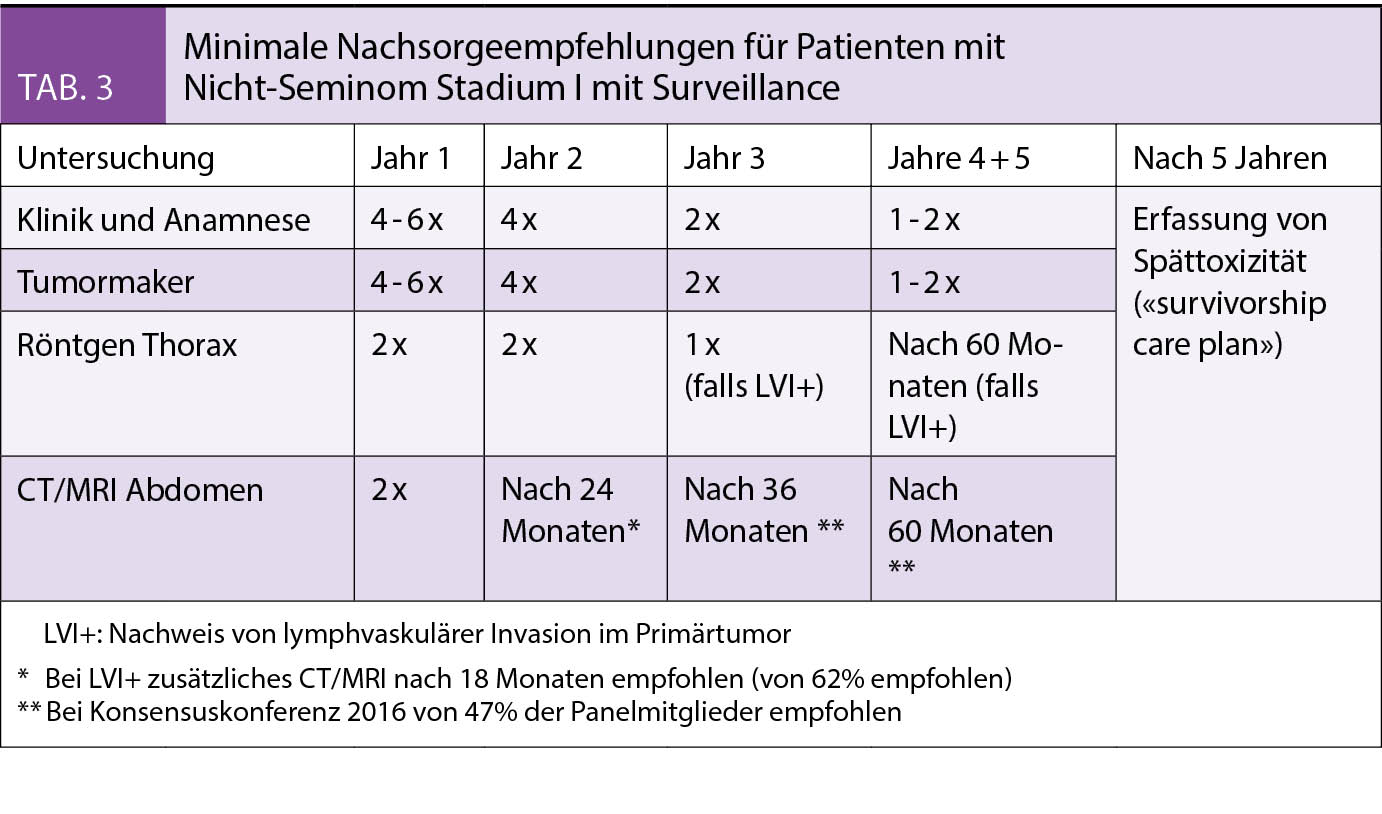
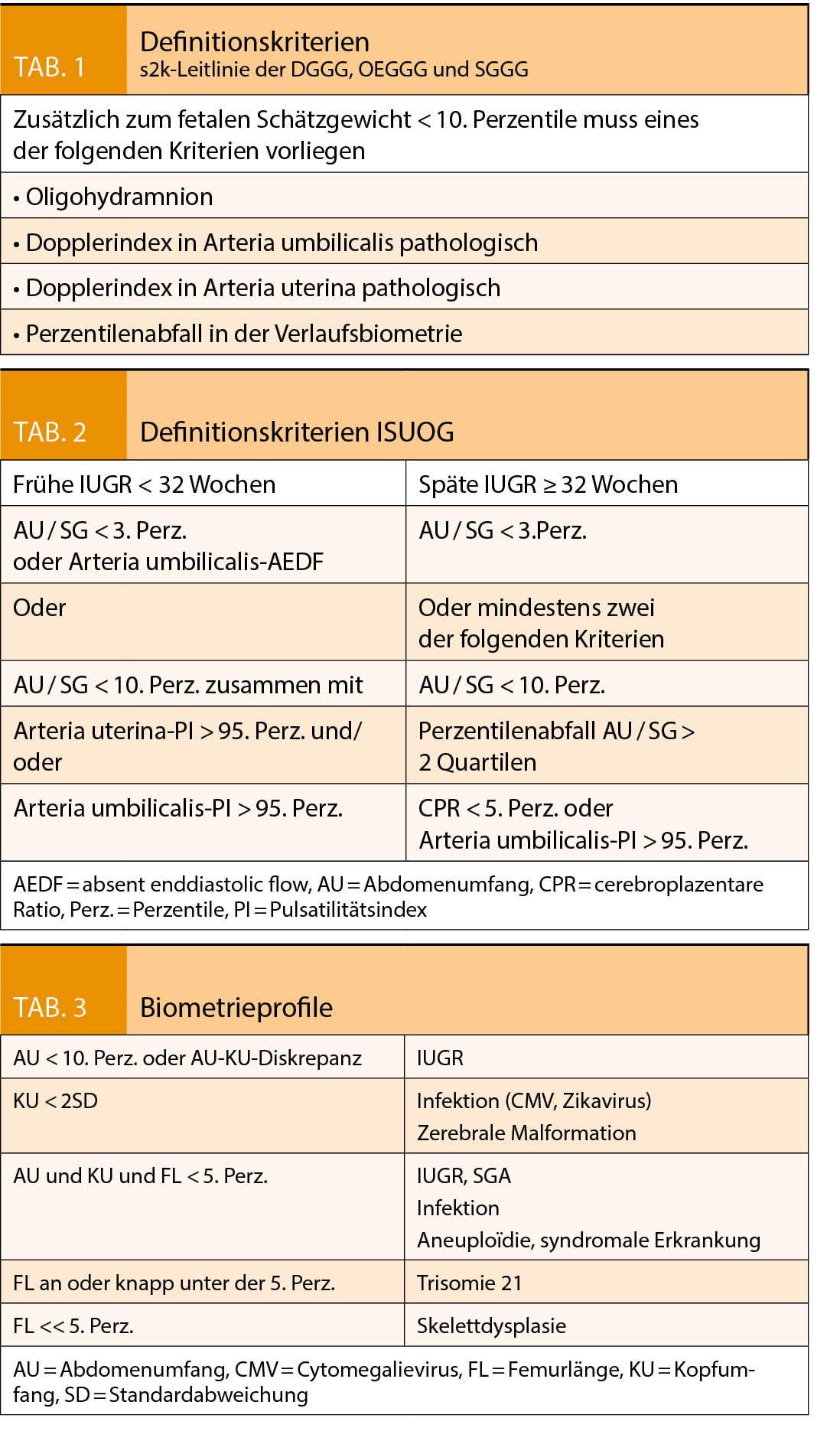
Bei einer IUGR schöpft der Fetus sein vorgegebenes Wachstums-potential nicht aus. Das Wachstum ist pathologisch restringiert und hat die Erhöhung der perinatalen Morbidität und Mortalität zur Folge. Davon unterschieden werden SGA Feten (small for gestational age). Sie sind konstitutionell klein, ihr Wachstum ist adäquat und liegt lediglich im untersten Normalbereich. Andererseits kann auch das Schätzgewicht eines IUGR-Feten im Normalbereich liegen. Guidelines ziehen teils unterschiedliche Definitionskriterien heran (Tab. 1 und 2). Ebenso wird auch ein unterschiedliches Zeitintervall zwischen Verlaufsuntersuchungen zur Definition der Wachstumsabflachung genannt, zwischen 2 und 3 Wochen je nach Guideline. (1, 2, 3)
Überprüfung der Fetometrie
In der 11. bis 14. SSW wird anhand der Biometrie bei einem Abweichen der Scheitelsteisslänge > 5 d das Gestationsalter korrigiert (4). Die S2k-Leitlinie empfiehlt eine Korrektur ab einer Abweichung von > 6 d (1). Ab der 12. SSW kann, besonders bei variabler Kopfhaltung, der BIP herangezogen werden. Die Korrektur erfolgt dann bei einer Differenz > 6 d (4). Liegt nach bereits erfolgter Terminkorrektur im 1. Trimenon erneut eine biometrische Differenz von > 6 d vor, sollte bei V.a. frühe IUGR und Chromosomenstörung zur spezialisierten pränatalen Diagnostik überwiesen werden. Bei sicherem Konzeptionstermin (z.B. ICSI) erfolgt keine Korrektur.
Das biometrische Profil (Tab. 3) hilft bei der ersten differentialdiagnostischen Einschätzung. Referenzkurven von Einlingsschwangerschaften können vor allem im letzten Trimenon nicht ohne weiteres auf Zwillingsschwangerschaften übertragen werden (5).
Infektionsscreening
Bei schwerer SGA oder IUGR sollte ein serologisches Screening auf CMV und Toxoplasmose veranlasst werden, bei Risikopatientinnen auch auf Malaria und Syphilis.
Ultraschallfeindiagnostik und genetische Abklärung
Die Fetometrie < 10. Perzentile ist eine Indikation zur sonographischen Diagnostik durch einen spezialisierten Untersucher (1). Malformationen oder Hinweiszeichen auf eine syndromale Erkrankung sollten verlässlich ausgeschlossen werden. Die Plazenta sollte morphologisch sowie hinsichtlich einer velamentösen Nabelschnurinsertion beurteilt werden. Feten mit genetischen Anomalien fallen oft durch eine vermehrte Fruchtwassermenge zusammen mit normaler Dopplersonographie auf. Eine Amniozentese sollte dann erwogen und diskutiert werden. Bei normalem Karyotyp sollte eine hochauflösende molekulare Karyotypisierung (Array CGH) angefordert werden. Mikrodeletionssyndrome wie das Wolf-Hirschhorn (4q) oder das Cri-du-Chat Syndrom (5 q) sind Beispiele für genetische Ursachen einer Wachstumsstörung beim Feten. Darüber hinaus kann ein auf die Plazenta beschränktes chromosomales Mosaik (z.B. 16 oder 9 q) Ursache für Plazentainsuffizienz mit konsekutiver fetaler Wachstumsstörung eines genetisch unauffälligen Feten sein (6). Solche plazentaren Mosaiktrisomien können mit NIPD (Nicht invasive Pränataldiagnostik) entdeckt werden, die nachfolgende Amniozentese zeigt einen normalen fetalen Karyotyp. Die NIPD kann in diesen Fällen im Sinne einer Plazentauntersuchung die erhöhte Wahrscheinlichkeit für deren Dysfunktion vorhersagen.
Doppleruntersuchung
Mit dem Nachweis erhöhter Gefässwiderstände in der Art. uterina und Art. umbilicalis lassen sich IUGR und SGA voneinander unterscheiden.
Die frühe IUGR ist durch eine Minderentwicklung und Obliteration von Plazentazotten charakterisiert. Die Erhöhung der Pulsatilität der Art. umbilicalis tritt erst nach Verschluss von 30% der Zottenarterien ein. Bei fehlendem enddiastolischem Fluss (AEDF , absent enddiastolic flow) oder reversem enddiastolischen Fluss (REDF , reversed enddiastolic flow) sind mehr als 60% der Zottenarterien verschlossen (7).
Als Zeichen der fetalen Kompensation bei Hypoxie sinkt der Widerstandsindex in der Art. cerebri media, typischerweise bei early-onset-IUGR. Die Pulsatilität der Art. cerebri media ist erniedrigt. Die Messung erfolgt nahe am Circulus arteriosus Willisii. Die cerebroplazentare Ratio sinkt unter die 5. Perzentile.
Die späte IUGR ist weniger durch eine Erhöhung des Gefässwiderstands in der Art. umbilicalis gekennzeichnet. Die Doppleruntersuchung der Art. umbilicalis kann normal ausfallen. Führendes und oft einziges Zeichen einer fetalen Verschlechterung ist die Erniedrigung der Pulsatilität in der Art. cerebri media.
Zudem kompensiert der Fetus die suboptimale plazentare Perfusion durch eine verstärkte Neubildung von Erythrozytenvorstufen. Die konsekutive Polyglobulie kann beim Neugeborenen sehr ausgeprägt sein. Bei fortgeschrittenen Fällen kommt es jedoch durch Downregulation der Erythropoese zur fetalen Anämie. Diese wird bei chronischer IUGR über die erhöhte Maximalgeschwindigkeit in der Art. cerebri media nachgewiesen (8).
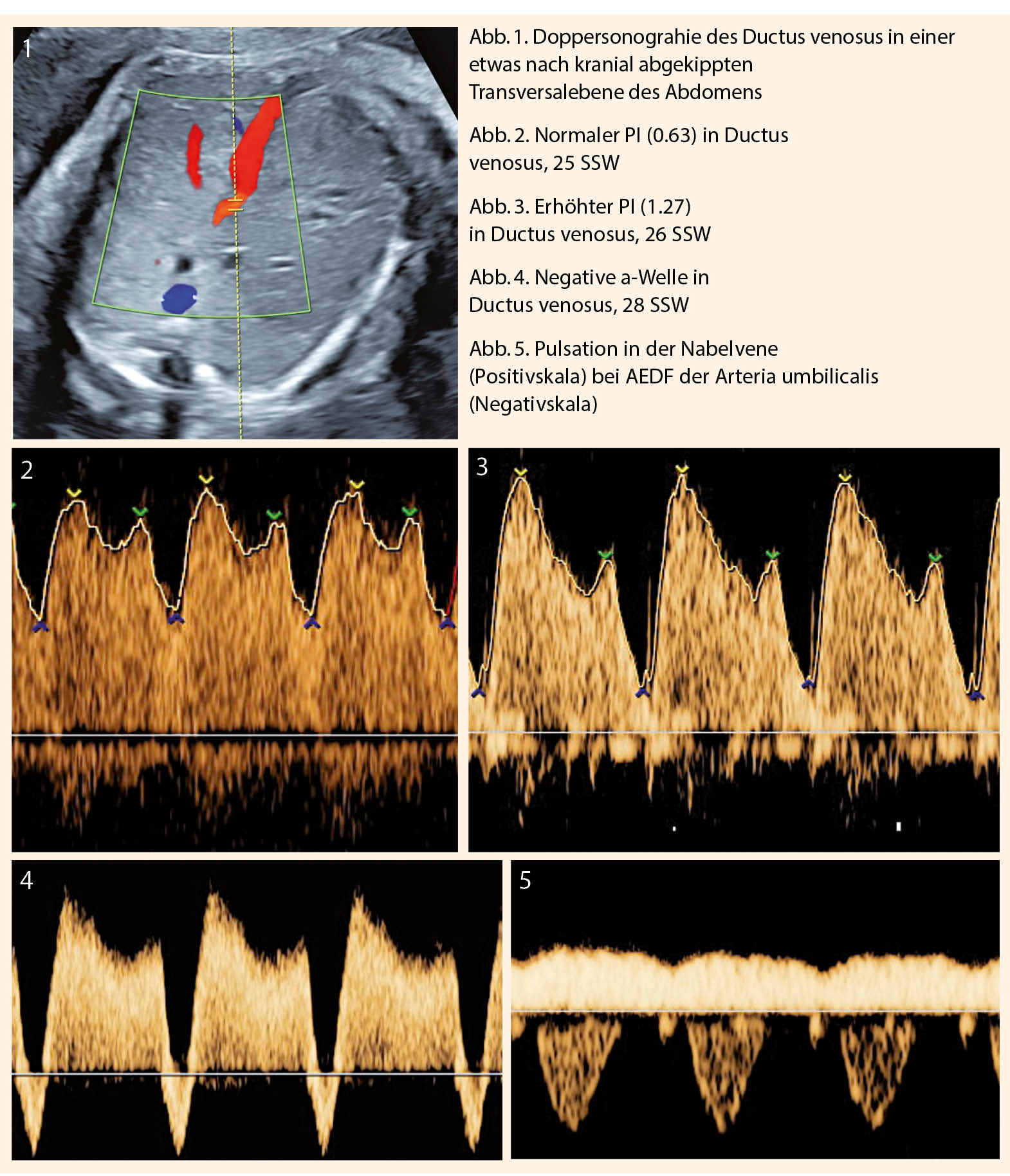
Bei pathologischem Widerstand in der Art. umbilicalis ist die Dopplersonographie des Ductus venosus Teil der Überwachung (Abb.1). Die Erhöhung der Pulsatilität bis hin zum Verlust der a-Welle reflektiert die Flusseinschränkung während der Vorhofkontraktion. Eine reverse a-Welle oder Erhöhung des PI ≥ 95. Perz. im Ductus venosus ist ein Hinweis auf eine drohende Azidose. Die kardiale Funktion des Feten ist eingeschränkt und das Risiko für intrauterinen Fruchttod (IUFT) verdoppelt sich täglich (9).
Pulsationen in der Nabelvene sind Ausdruck fortgeschrittener Hypoxie und können präterminal gesehen werden.
Computerisiertes CTG
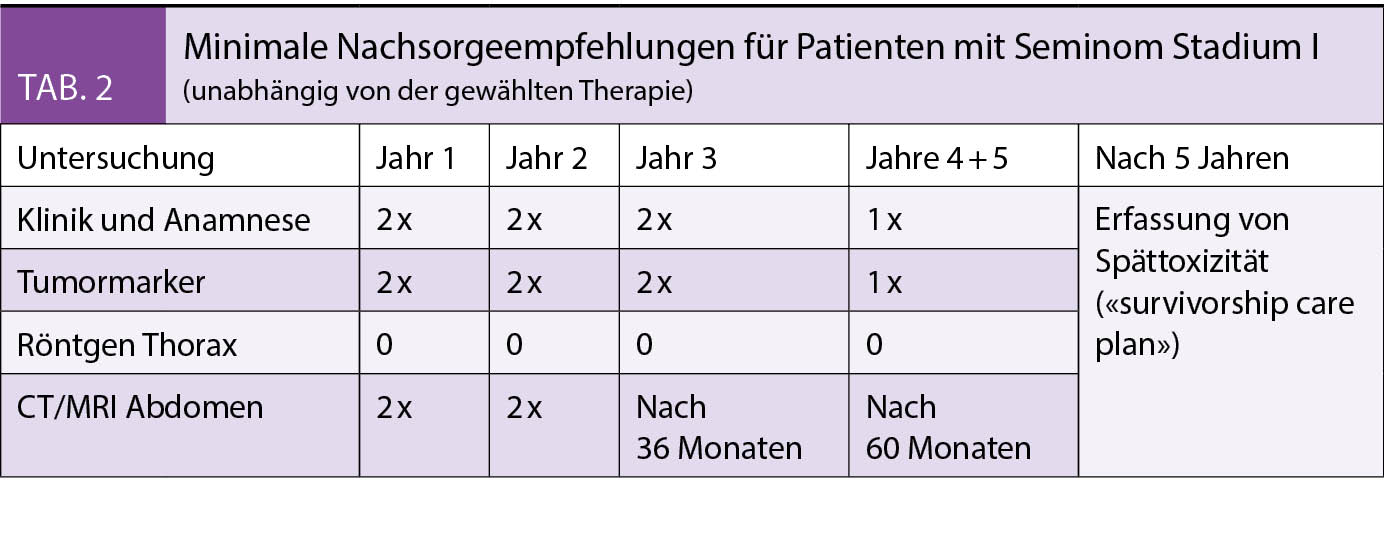
Das konventionelle CTG vermag zwar eine akute fetale Hypoxie anzuzeigen, ist aber wenig hilfreich, um den optimalen Entbindungszeitpunkt zu ermitteln. Es sollte nicht als alleinige Überwachungsmethode herangezogen werden (3, 10). Im computerisierten CTG (cCTG, auch: Oxford-CTG) wird mittels einer computerbasierten Analyse die Kurzzeitvariabilität (KZV) ermittelt. Sie ist Ausdruck fetaler Hirnfunktion, reflektiert den fetalen Säure-Basen-Status und steigt mit zunehmendem Gestationsalter an. Die Beurteilung ist objektiv. Die KZV trägt somit zur besseren Terminierung des Entbindungszeitpunktes bei. Das Erreichen der sogenannten Dawes-Redman-Kriterien oder eine KZV > 4,5 ms haben hohen prädiktiven Wert für einen stabilen metabolischen Status des Feten.
Management
Wie kontrollieren und wann entbinden?
- Bei unauffälligem Doppler in der Art. umbilicalis: alle 2 Wochen bei früher IUGR (1)
- Bei PI ≥ 95. Perz. in Art. umbilicalis: wöchentlich ambulant
- Bei AEDV bis REDV in Art. umbilicalis: < 32 SSW: Hospitalisation in Perinatalzentrum, Lungenreifung. Empfohlen ist die tägliche Ductus-venosusDoppleruntersuchung, jedoch wurde die Untersuchungsfrequenz in der TRUFFLE-Studie nicht getestet. (11)
Unabhängig von der fetalen Überwachung ist bei schwerer Präeklampsie oder HELLP die mütterliche Indikation zur Entbindung gegeben. Die fetale Indikation zur unmittelbaren Entbindung liegt bei spontanen rezidivierenden Dezelerationen oder persistierendem Variabilitätsverlust im konventionellen CTG vor.
Early onset IUGR
Bei AEDF in der Art. umbilicalis: sollte die Entbindung per Sectio spätestens mit 34 + 0 SSW erfolgen. Bei REDV in der Art. umbilicalis: spätestens mit 32 + 0 SSW oder entsprechend dem lokalen Protokoll ggf. auch schon früher (1,11). Pulsationen in der Nabelschnurvene gelten jedoch immer als Indikation für die zügige Entbindung (12). Das optimale Timing der Entbindung hat das normale entwicklungspädiatrische und neurologische Langzeit-outcome zum Ziel. Die Resultate der TRUFFLE Studie bieten Hilfe bei der Entscheidungsfindung. Der optimale Entbindungszeitpunkt wird mit seriellen Doppleruntersuchungen des Ductus venosus und Monitoring mittels cCTG ermittelt. Bei negativer a-Welle im Ductus venosus sollte die Entbindung indiziert werden. Zusätzlich und unabhängig von Ductus-venosus-Doppler gelten folgende cCTG-Veränderungen als Entbindungskriterium: Zum einen kontinuierliche Abnahme der KZV in seriellen Messungen (1) oder, zum anderen, eine KZV < 2,6 ms (26 + 0 bis 28 + 6 SSW) oder < 3 ms (29+0 bis 32 + 0 SSW). Die Abnahme der KZV ist Hinweis auf eine fetale metabolische Azidose und reflektiert den fetalen Zustand über einen anderen Mechanismus als die venöse Dopplersonographie. (10, 11)
Late onset IUGR
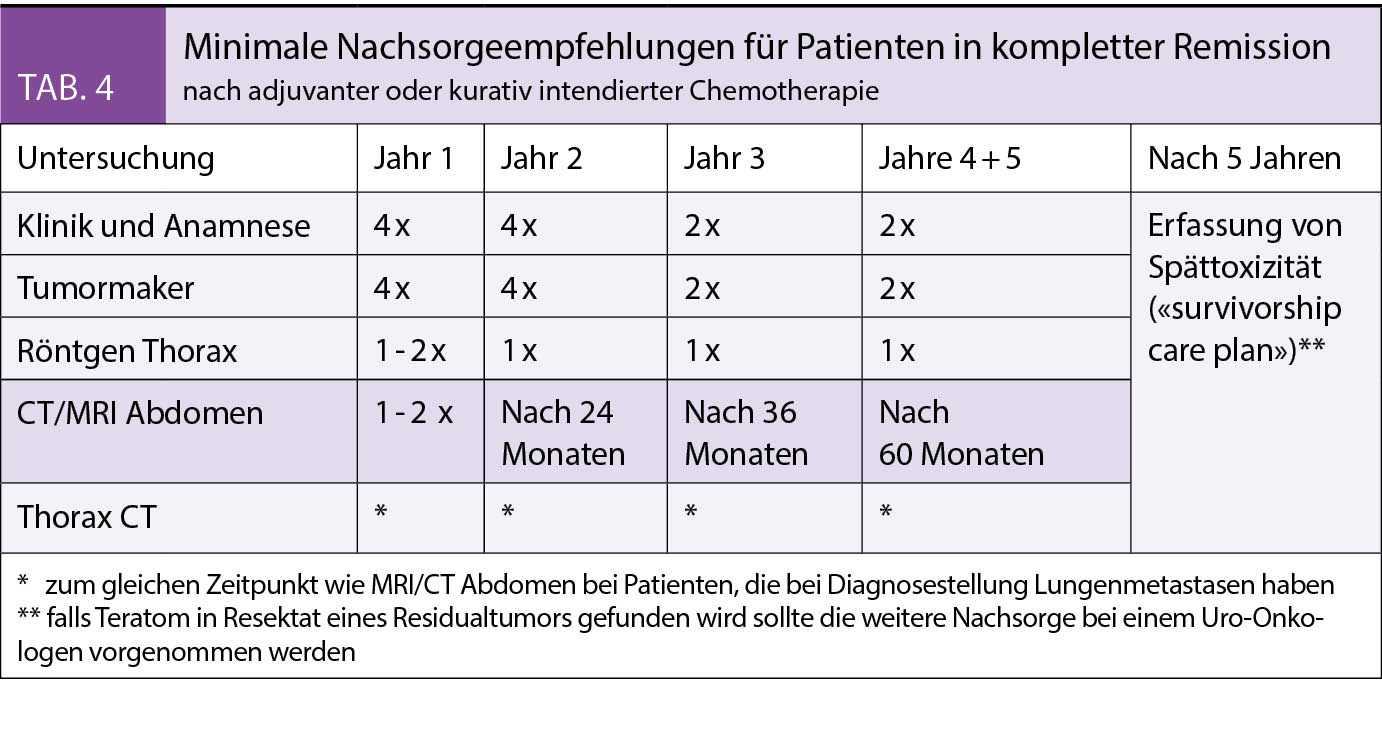
Die späte IUGR ist durch die Wachstumsabflachung gekennzeichnet. Die Fetometrie kann dabei noch in der Norm sein, die Wachstumsstörung ist weniger ausgeprägt (13). Die Diagnose ist deswegen erschwert. Wegen des hohen Stoffwechselumsatzes des Feten am Termin ist die
Intoleranz von Hypoxie deutlich höher. Die Latenz zum IUFT ist bei fortgeschrittenem Gestationsalter kürzer als bei der frühen IUGR < 32 SSW. Es liegt weniger ein Problem der Obliteration von Zottenarterien vor, sondern eher eine Diffusionsrestriktion. Es gibt keine klaren Cut-Offs für die
Definition der frühen versus späten IUGR (13). Für die Ermittlung des optimalen Entbindungszeitpunktes gibt es für das Gestationsalter 32 – 37 SSW wenig Evidenz. Ab 37 SSW ist die cerebroplazentare Ratio bzw. der PI in der Art. cerebri media wichtig für das Timing der Geburt, auch bei normalem Umbilicalarteriendoppler (14, 15, 16). Bei
schwerer SGA < 3.Perzentilie mit normalem Doppler sollte auch mit 37 SSW entbunden werden.
Bei SGA mit normalem Doppler sollte die 40. SSW nicht überschritten werden.
FMH für Fetomaternale Medizin
Ultraschalltutorin SGUMGG | DEGUM II
Luzerner Kantonsspital
Frauenklinik, Spitalstrasse
6000 Luzern 16
alice.winkler@luks.ch
Die Autorin hat keine Interessenskonflikte im Zusammenhang mit diesem Beitrag deklariert.
1. Intrauterine Wachstumsrestriktion, s2k-Leitlinie der DGGG, OEGGG und SGGG, AWMF Register Nr. 015/080. 2016
2. Gordijn SJ et al. Consensus definition of fetal growth restriction. Ultrasound Obstet Gynecol 2016
3. Vayssière C et al. Fetal growth restriction and intra-uterine growth restriction: guideline for clinical practice from the French College of Gynaecologists and Obstetricians. Eur J Obstet Gynecol Reprod Biol 2015
4. Empfehlungen der Ultraschalluntersuchung in der Schwangerschaft, 3. Auflage, SGUMGG 2011
5. Stirrup OT et al. Fetal growth reference ranges in twin pregnancy: analysis of the Southwest Thames Obstetric Research Collaborative multiple pregnancy cohort. Ultrasound Obstet Gynecol 2014
6. Sparks TN et al. Mosaic trisomy 16: what are the obstetric and long-term childhood outcomes? Genet Med 2017
7. Morrow RJ et al. Effect of placental embolization on the umbilical arterial velocity waveform in fetal sheep. Am J Obstet Gynaecol 1989
8. Baschat AA. Fetal responses to placental insufficiency: an update. Br J Obstet Gynaecol 2004
9. Turan OM et al. Duration of persistent abnormal ductus venosus flow and its impact on perinatal outcome in fetal growth restriction. Ultrasound Obstet Gynecol 2011
10. Lees CC et al. TRUFFLE study group. 2 year neurodevelopmental and intermediate perinatal outcomes in infants with very preterm fetal growth restriction: a randomized trial. Lancet 2015
11. Bilardo MC et al. Severe fetal growth restriction at 26-32 weeks: key messages from the TRUFFLE study. Ultrasound Obstet Gynecol 2017
12. RCOG greentop guideline. The investigation and management of the Small-for-gestational-Age Fetus. 2014
13. Figueras F, Gratacos E. Update on the Diagnosis and Classification of Fetal Growth Restriction and Proposal of a Stage-Based Management Protocol. Fet Diagn Ther 2014
14. Flood K et al. Results of the multicenter PORTO study. Am J Obstet Gynaecol 2014
15. O’Dwyer V et al. Defining the risks of adverse perinatal outcome in growth restricted fetuses with normal umbilical artery blood flow. Am J Obstet Gynaecol 2014