Monoklonale Gammopathien umfassen eine heterogene Gruppe von Neoplasien der terminalen B-Zelldifferenzierung, welche sich durch eine überschiessende Produktion monoklonaler (M) Immunglobuline und Immunglobulinleichtketten auszeichnen. Das Spektrum der Erkrankungen reicht dabei von «low-tumor-burden diseases» (Bsp.: AL-Amyloidose), über prämaligne Erkrankungen wie die monoklonale Gammopathie unklarer Signifikanz bis hin zu eindeutig malignen, hämatologischen Systemerkrankungen wie das asymptomatische («smouldering») Multiple Myelom, Multiple Myelom oder Morbus Waldenström.
Eine antikörpersezernierende Plasmazelle (PZ) eines gegebenen Klons bildet ausschliesslich einheitliche Antikörpermoleküle mit schweren Ketten eines einzigen Isotyps (α, γ oder μ) und Leichtketten einer einzigen Art (κ oder λ). Bei einer proliferativen Entartung nur jenes B-Zell-Klons, kommt es zur massenhaften Bildung von Immunglobulinen oder deren Fragmente mit identischer Primärstruktur, Funktion und physiko-chemischen Eigenschaften. Folglich zeigen M-Proteine desselben Klons ein identisches elektrophoretisches Verhalten, was sich dann typischerweise als enger Peak mit schmaler Basis, auch als M-Gradient bezeichnet, manifestiert. Entsprechend der Grenzen, in denen die polyklonalen Immunglobuline normalerweise migrieren, besitzen M-Proteine meist eine Mobilität innerhalb der γ-Globulinfraktion – daher der Begriff «monoklonale Gammopathie».
Die monoklonale Antikörperbildung ist unnatürlich und beruht häufig auf einer prämalignen oder malignen Transformation. Gelegentlich kommt es auch im Rahmen einer Reaktion gegen ein Immunogen, z.B. bei akutem Virusinfekt, zu einer limitierten, klonalen PZ-Proliferation, was vorübergehend zu einer schwach ausgeprägten monoklonalen Gammopathie führen kann. Damit ist auch ausgesagt, dass der Terminus «monoklonale Gammopathie» per se keine maligne Situation definiert. In mehr als der Hälfte der Fälle geht es nur um ein Laborphänom, welches auf Veränderungen hinweist, deren Dignität nicht eindeutig ist und die daher als Monoklonale Gammopathie unklarer Signifikanz (MGUS) bezeichnet werden.
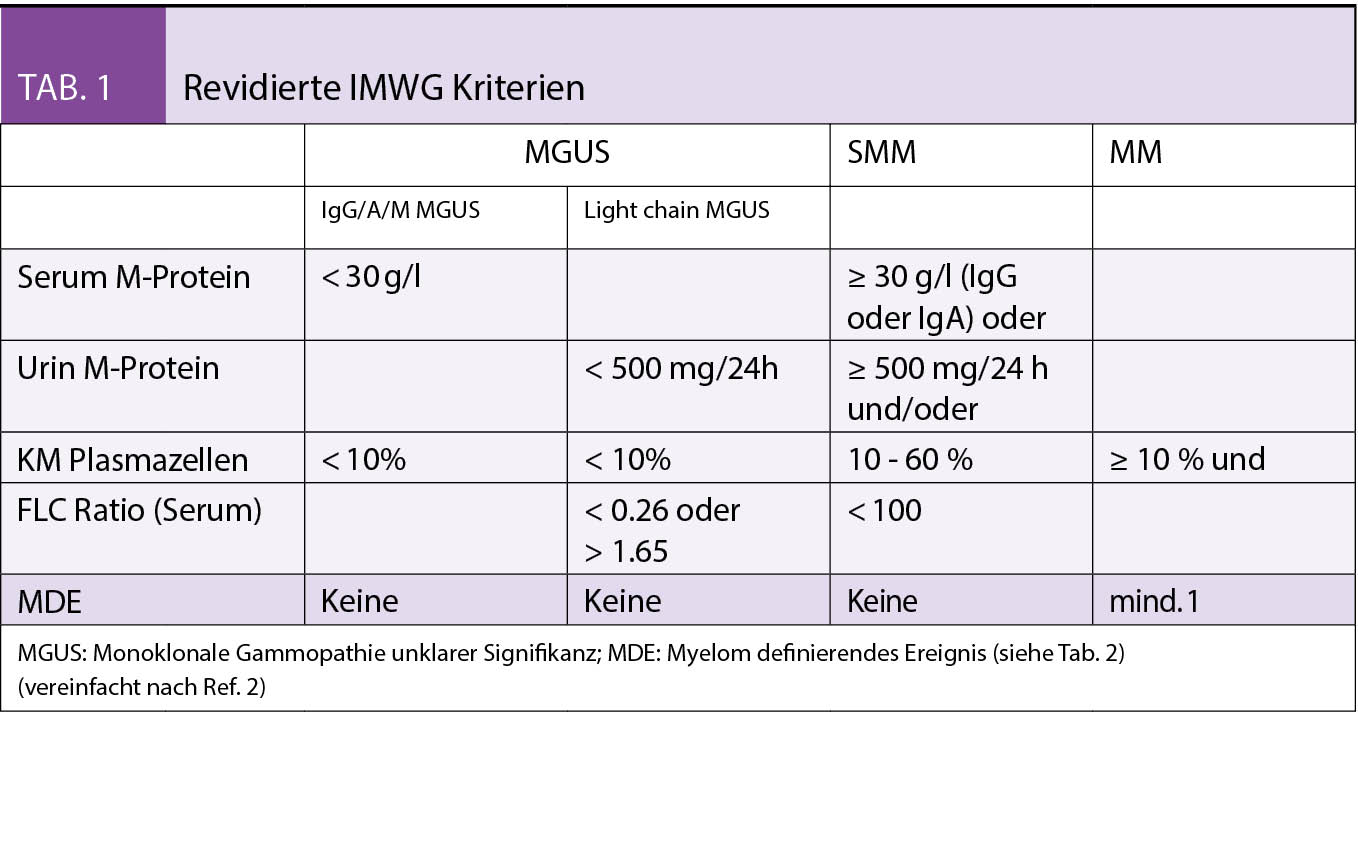
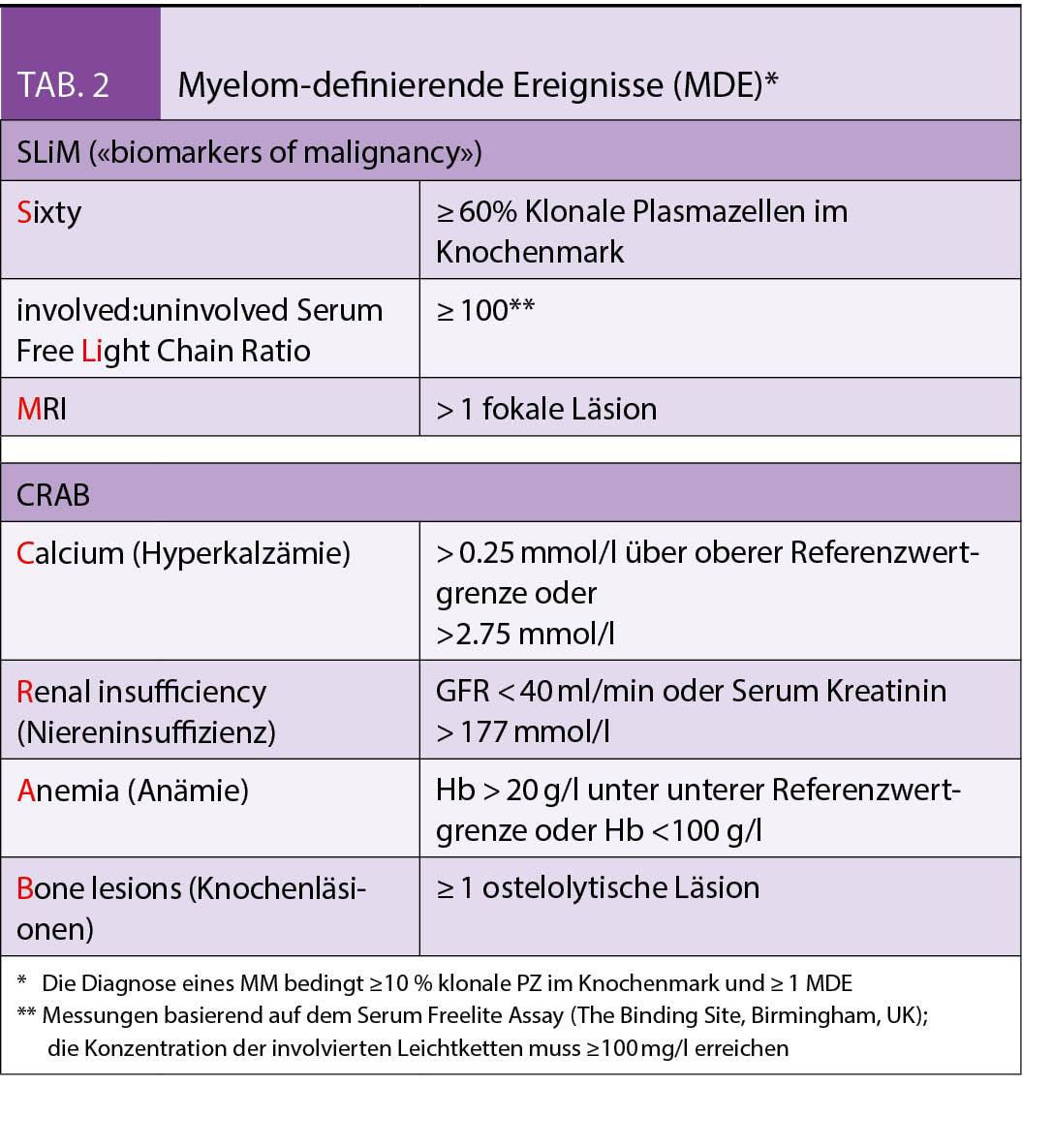
Jedem Multiplen Myelom (MM) geht eine MGUS voraus (1). Ob eine Transformation mit nachfolgender, permanenter Expansion klonaler Plasmazellen stattfindet, hängt von weiteren genetischen («second hit») und epigenetischen Läsionen ab. Bei einer durchschnittlichen Rate von 1% pro Jahr besteht ein relativ geringes Progressionsrisiko. Betroffen von einem MGUS sind bis zu 3% aller Menschen zwischen 50–70 Jahren und > 5% aller über 70-Jährigen. Formal liegt ein MM vor bei Nachweis von ≥ 10 % klonaler PZ im Knochenmark oder Myelom assoziierter Endorganschäden (CRAB-Kriterien, siehe Tab. 1 und 2).
Beim «smoldering» MM (SMM) handelt es sich klinisch um ein zwischen MGUS und MM anzusiedelndes intermediäres Stadium mit einem gegenüber dem MGUS deutlich erhöhten Progressionsrisiko. Mit den revidierten IMWG-Kriterien (2) wurden drei Früherkennungsmarker («biomarkers of malignancy») zur akkuraten Identifizierung von solchen SMM-Patienten eingeführt, die kurz vor der Ausbildung von Endorganschäden stehen (Tab. 2). Ist mindestens eines dieser neuen, auch unter dem Akronym «SLiM» bekannten, Kriterien erfüllt, besteht eine Therapieindikation.
Labordiagnostik monoklonaler Immunglobuline
Der Nachweis von Synthese und Sekretion monoklonaler Zellproteine gehört zu den charakteristischen Befunden einer monoklonalen Gammopathie und trägt richtungsweisend zur Diagnose, sowie zur Ermittlung von Stadium, Risiko und Therapieansprechen bei. In den meisten Fällen handelt es sich bei diesen hochspezifischen Tumorprodukten um intakte Immunglobuline und freie Leichtketten (Bence Jones Proteine). In nur ca. 5% aller Gammopathien stellen freie Leichtketten die einzigen monoklonalen Komponenten dar.
Screening
Im Gegensatz zu anderen serologischen Tumormarkern handelt es sich bei M-Proteinen um strukturell sehr variable Moleküle, deren proteinchemische Analyse den Einsatz unterschiedlicher Methoden verlangt. Die International Myeloma Working Group (3) empfiehlt denn auch für das erste Screening den kombinierten Einsatz von Proteinelektrophorese (ELP), Immunfixationselektrophorese (IFE) und freie Leichtketten (FLC) aus Serum. Die diagnostische Sensitivität dieser Testkombination für alle einer monoklonalen Gammopathie zugrunde liegenden Diagnosen liegt bei durchschnittlich 97.4 % (4) (Tab. 3). Reduziert man das First-line Screening auf die Anwendung von ELP und FLC, so sinkt die Sensitivität auf klinisch immer noch akzeptable 94.3%. Der Miteinbezug von Urinanalysen wird insbesondere bei Verdacht auf eine AL Amyloidose empfohlen.
Die ELP ist ein kostengünstiger, automatisierbarer Erstlinienentest, welcher rasch einen Überblick über die quantitativ wichtigsten Proteinfraktionen in Form eines Densitogramms liefert. Typische, schmalbasige M-Gradienten wie eingangs beschrieben, sind mühelos erkennbar. Schwieriger ist die Interpretation, wenn die M-Komponente, niedrig konzentriert ist (< 10 g/l), bei Überlagerung durch eine starke polyklonale Reaktion oder bei Migration ausserhalb der γ-Fraktion. Mit einer diagnostischen Sensitivität von 79.0 % ist der alleinige Einsatz der ELP zur Erstdiagnose deshalb nicht zu empfehlen.
Mittels der IFE wird das Vorhandensein eins M-Proteins bestätigt und deren Schwer- und Leichtkettentyp charakterisiert. Die Methode ist zwar deutlich sensitiver als die ELP, jedoch stellt sich auch hier das Problem der Interpretation subtiler Abnormalitäten. Ein ausgeprägtes M-Protein kann an den scharf fokussierten und intensiv gefärbten Banden leicht ausgemacht werden. Nicht selten jedoch präsentieren sich in der IFE diskrete oder wenig fokussierte, inhomogene Zonen, deren isolierte Beurteilung die Unterscheidung zwischen einer potentiell malignen von einer immunreaktiven Situation nicht zulässt. Solche Zonierungen dürfen also einerseits nicht übergangen, anderseits auch nicht überbewertet werden.
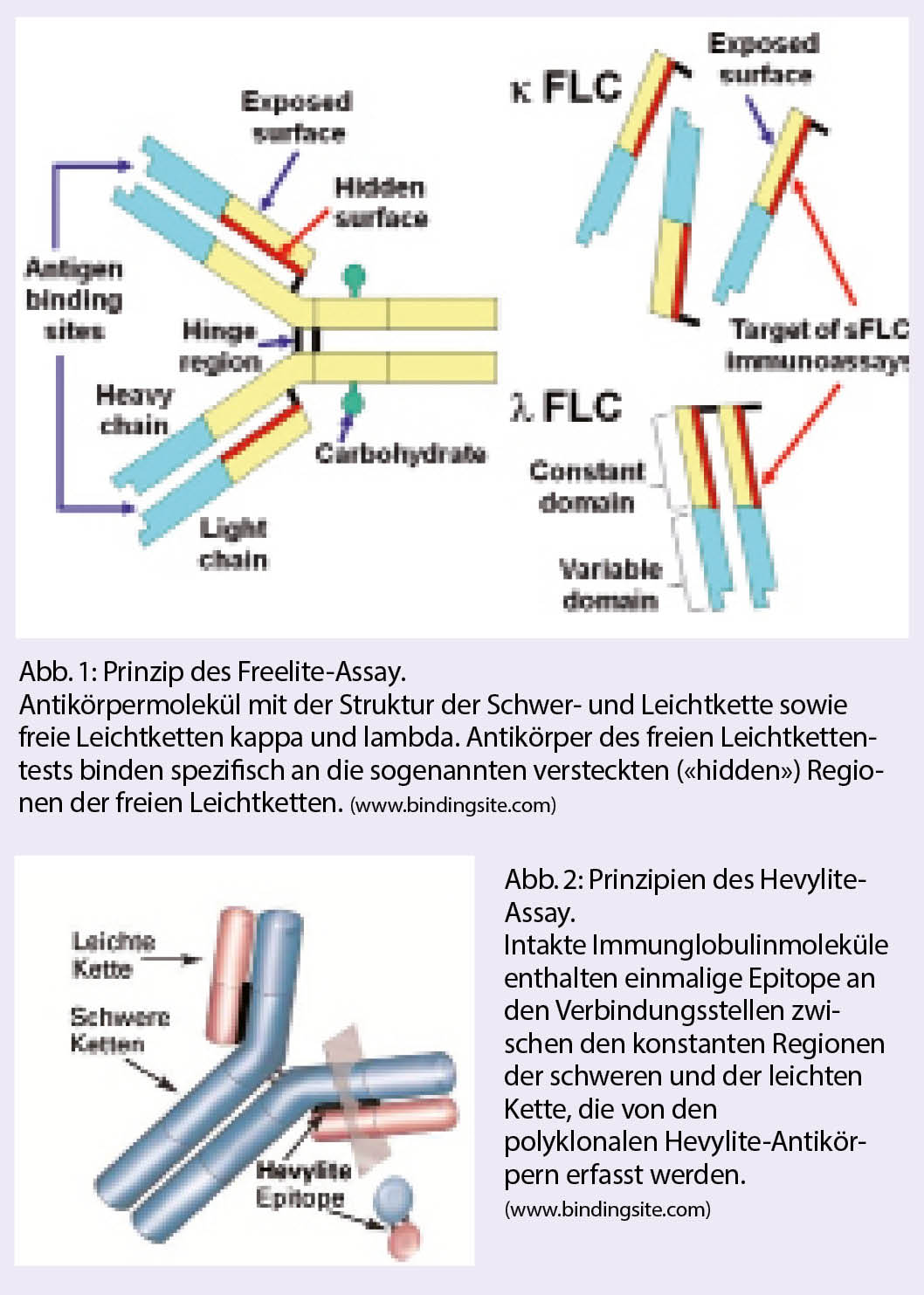
Mittels stark aviden Antisera gegen die einzigartigen, «kryptischen» Epitope (Abb. 1) freier Immunglobulin-Leichtketten ist es möglich, diese getrennt nach κ- und λ-Isotyp und ohne Kreuzreaktivität mit gebundenen Leichtketten nephelometrisch oder turbidimetrisch zu erfassen. Klinische Kernindikationen dieses mittlerweile gut etabilierten, quantitativen Tests bestehen insbesondere in den Fällen, in denen die Leichtketten, die einzigen sezernierten monoklonalen Proteine darstellen (Bsp.: Leichtkettenmyelom). Die κ- und λ-FLC Konzentrationen sind einerseits abhängig von deren Produktion durch die Plasmazellen bzw. den Tumor und andererseits deren renalen Elimination. Das κ- /λ-Verhältnis bewegt sich normalerweise innerhalb enger Grenzen (0.26–1.65). Zu beachten ist, dass sich der Quotient bei Patienten mit geringer monoklonaler Produktion einerseits und Leichtkettenanstieg bei Niereninsuffizienz sowie bei entzündlichen Reaktionen andererseits überlappen können. Aus diesem Grund erscheint die Bestätigung der Monoklonalität bei der Erstdiagnose allein aufgrund eines wenig verschobenen κ/λ-Verhältnisses als problematisch. In der malignen Situation kann dagegen eine deutliche Konzentrationserhöhung der involvierten Leichtketten im Serum (> 100 mg/l) mit eindeutiger Verschiebung der Leichtkettenratio als Zeichen der Monoklonalität (> 100 oder < 0,01) erwartet werden (2).
Monitoring
Die Konzentration des M-Proteins wird klassischerweise mittels Integration des M-Proteinpeaks im Densitogramm der ELP ermittelt. Als Indikator für das Therapieansprechen sollte dies alle 3–6 Monate erfolgen (5). Bei niedriger Konzentration (< 10 g/L) oder unzuverlässiger Abgrenzbarkeit der M-Komponente ist die densitometrische Quantifizierung ungenau, wenn nicht gar irreführend. In solchen Fällen ist die wiederholte Messung der Serum-FLC zu empfehlen, vorausgesetzt die initiale Konzentration der involvierten FLC ist > 100 mg/L und der FLC-Quotient ist abnorm. Vorteil gegenüber der densitometrischen M-Proteinquantifizierung ist zudem die kurze Halbwertszeit freier Leichtketten von 2–6 h, welche eine engmaschigere Verlaufskontrolle während einer Therapie ermöglicht. Durch die modernen Therapiemöglichkeiten und das längere Patientenüberleben steigt auch die Möglichkeit von sog. «tumor escape mutants» im Rezidiv, was sich im Falle eines «light-chain escape» nur am isolierten Anstieg monoklonaler FLC erkennen lässt. Bei hoher renaler FLC-Ausscheidungsrate (> 200 mg/24h) wird offiziell die densitometrische Quantifizierung mittels der ELP im Urin empfohlen. Die FLC-Messung im Serum hat nach neueren Studien demgegenüber den Vorteil, deutlich sensitiver und zudem prognostisch aussagekräftiger zu sein (6).
Seit wenigen Jahren ist auch die Quantifizierung von M-Gradienten niedriger Konzentration in einer festen Schwer- und Leichtkettenkombination («Hevylite®») möglich geworden (Abb. 2). Das sensitive Monitoring bei monoklonalen IgA-Proteinen, welche häufig eine β-Mobilität aufweisen und densitometrisch schwer zu erfassen sind, ist bereits in offiziellen Empfehlungen aufgenommen worden (7). Analog zur FLC-Ratio liefert das Verhältnis des involierten zum nicht-involierten Immunglobulin-Isotyp auch prognostische Informationen (8).
Patientenmanagement
Wie sollen Patienten mit einem MGUS im weiteren Verlauf kontrolliert werden?
Risikofaktoren für eine Progression eines MGUS
Die Voraussage, wie sich ein MGUS verhalten wird, ist schwierig. Als Risikofaktoren gelten Höhe und Typ des M-Proteins, Anteil Plasmazellen im KM und FLC Ratio. Aus diesen Faktoren entwickelte Rajkumar ein MGUS- Risikostratifizierungsmodell (9) (Tab. 4).
Kontrollintervalle Niedrig-Risiko MGUS, 0 Punkte im Risikostratifizierungsmodell, ca 40% der Patienten: Sofern klinisch keine Hinweise auf einen Organschaden vorliegen, bedürfen diese Patienten keiner weiteren Abklärungen, auch keine Knochenmarkpunktion. Jedoch muss eine Nachkontrolle nach 6 Monaten erfolgen mit Bestimmung des Blutbildes, Serum-ELP, sFLC, Calcium und Kreatinin. Bei stabilem Verlauf sind danach alle 2-3 Jahre oder bei Symptomatik Verlaufskontrollen empfohlen.
Intermediäres und Hochrisiko-MGUS, 1-3 Punkte im Risikostratifizierungsmodell: Diese Patienten bedürfen einer Knochenmarkpunktion inklusive Flowzytometrie und Zytogenetik / FISH sowie Bildgebung mittels Osteoscan oder Ganzkörper-MRI. Gemäss aktuellen Daten ist keine der beiden Bildgebungsmodalitäten klar überlegen. Auch bei Verdacht auf eine AL-Amyloidose sollte eine Knochenmarkpunktion erfolgen. Liegt nach diesen Untersuchungen weiterhin ein MGUS vor, empfiehlt sich eine Verlaufskontrolle alle 3-6 Monate im ersten Jahr und bei stabilem Verlauf danach alle 6-12 Monate (Blutbild, Serum-ELP, sFLC, Calcium und Kreatinin).
Abteilung für Hämatologie und Onkologie / Pathologie
Kantonsspital St. Gallen
Rorschacherstrasse 95
9007 St. Gallen
thomas.lehmann2@kssg.ch
Zentrum für Labormedizin
Frohbergstrasse 3
9001 St. Gallen
siegfried.stranders@zlmsg.ch
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Beitrag deklariert.
1. Landgren O. et al. Monoclonal gammopathy of undetermined significance consistently preceds multiple myeloma. Blood 2009;113(22):5412-7
2. Rajkumar SV et al. International Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-48
3. Dispenzierie A et al. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia 2009;23(2):215-24
4. Katzmann JA et al. Screening Panels for Detection of Monoclonal Gammopathies. Clin Chem. 2009;55(8):1517-22
5. Durie BG et al. International uniform response criteria for multiple myeloma. Leukemia 2006;20(9):1467-73
6. Dejoie T et al. Serum free light chains, not urine specimens, should be used to evaluate response in light-chain multple myeloma. Blood 2016;128(25):2941-48
7. Kumar S et al. IMWG consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol.2016;17(8):e328-e346
8. Bradwell A et al. Prognostic utility of intact immunglobulin Ig’kappa/Ig’lambda ratios in mutiple myeloma patients. Leukemia 2013;27(1):202-7
9. Rajkumar SV et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood 2005;106(3):812–7