Introduction
Endoscopic procedures are increasingly performed on patients receiving antithrombotic therapy for high-risk thromboembolic conditions. This raises questions about the necessity of discontinuing antithrombotic drugs pre-procedure and whether coagulation assessment is essential to reduce the risk of post-interventional bleeding. Factors considered are anticoagulation type, indication, and type of endoscopic intervention. Commonly used procedures are gastroscopies with biopsies and colonoscopies with polypectomy.
This expert opinion statement provides updated guidance on antithrombotic drug use and coagulation testing before endoscopic procedures.
Pre-procedural assessment for bleeding diathesis – a questionnaire frequently exerts a greater impact compared to routine laboratory tests
Assessing the patient’s risk of bleeding before gastrointestinal endoscopies is crucial. Routine determination of the international normalised ratio (INR) and platelet count is common practice. If the INR is < 1.5 and platelet count > 50 G/l, the examination or intervention is performed.
However, evidence on the utility of routine laboratory tests before elective gastroenterological endoscopic examinations is limited, drawing from surgical patient experiences. Large prospective cohort studies (1, 2, 3) have shown that baseline coagulation tests (INR, aPTT, bleeding time, platelet count) are not predictive of intraoperative or postoperative bleeding in patients with an unremarkable bleeding history.
The American Society for Gastrointestinal Endoscopy (ASGE) published an expert opinion statement in 2014 emphasising the significance of medical history (4). Routine coagulation work-up is deemed unnecessary if the bleeding history is inconspicuous, even for interventions with high bleeding risk. Accordingly, in the recommendations of the Swiss Society of Gastroenterology (SSG) from 2016, bleeding history is prioritised in the coagulation work-up (5, 6). A negative response to all items in the questionnaire suggests no increased risk of bleeding, obviating the need for coagulation assessment (Table 1). However, in cases of a history or indication of increased bleeding risk (e.g. liver cirrhosis, severe renal insufficiency, malnutrition), determining INR and platelet count is recommended.
If two or more affirmative responses are elicited in the questionnaire, indicating the presence of major postoperative bleeding, a history of bleeding disorder, or a personal bleeding diathesis, there exists a heightened risk of bleeding. Therefore, prior to endoscopy, as well as biopsies or any interventions, a coagulation assessment (e.g. INR, aPTT, thrombin time, fibrinogen, platelet count, and possibly platelet function assay) should be conducted following consultation with a hematologist. Subsequently, the procedures and interventions should be deferred until the risk is mitigated.
However, the questionnaire is inapplicable to patients under antithrombotic therapy. Routine coagulation assessments are generally unnecessary for such patients, except when vitamin K antagonists (VKAs) are involved. In VKA-treated individuals, assessing the INR before endoscopies with planned biopsies or interventions (e.g. polypectomy) is prudent to avoid overlooking any cases of excessive anticoagulation.

Antithrombotic management in the elective peri-procedural setting
The escalating prevalence of antithrombotic agents and its challenges
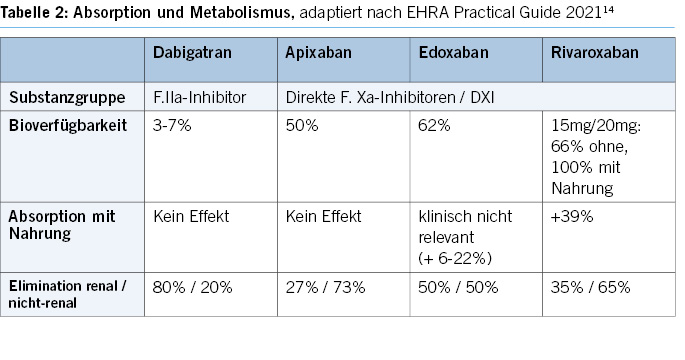
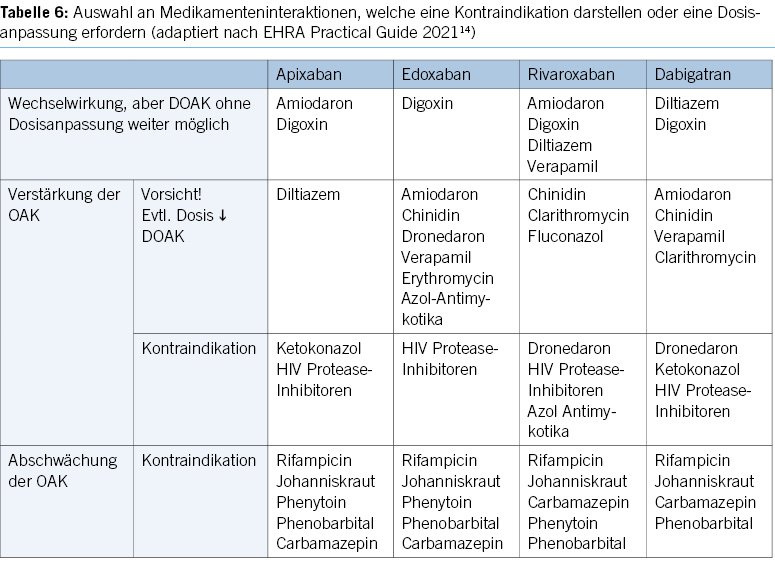
Oral anticoagulants and antiplatelet agents play a crucial role in modern cardiovascular medicine, and their combined use is becoming more common (Table 2).
Managing patients on antithrombotic therapy before and after endoscopic procedures presents challenges. Balancing the risk of intervention-related bleeding against the potential thromboembolic risk due to temporary discontinuation of antithrombotics requires careful consideration. Engaging in shared decision-making with patients is essential
to understand their preferences in such situations.
While preventing post-interventional bleeding is desirable, it is essential to note that the mortality associated with bleeding is very low. Most bleeding incidents can be effectively treated endoscopically, avoiding surgery or radiological intervention. This contrasts with the higher mortality risk associated with potential thromboembolic events after antithrombotic discontinuation, particularly coronary stent thrombosis.
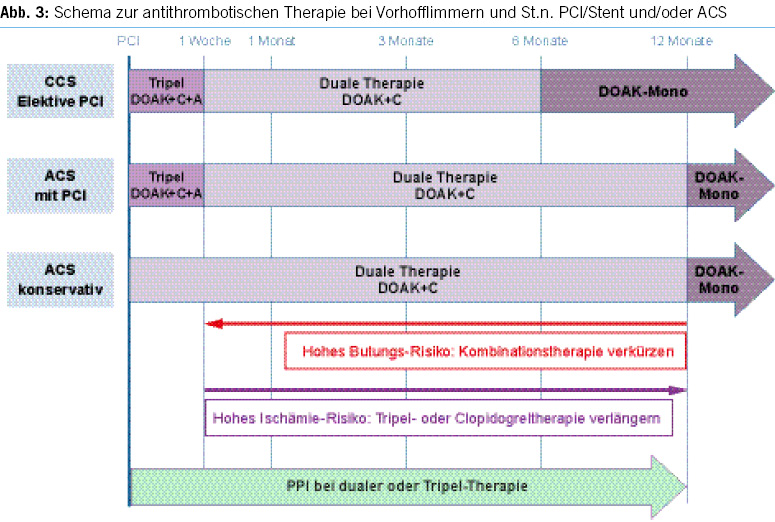
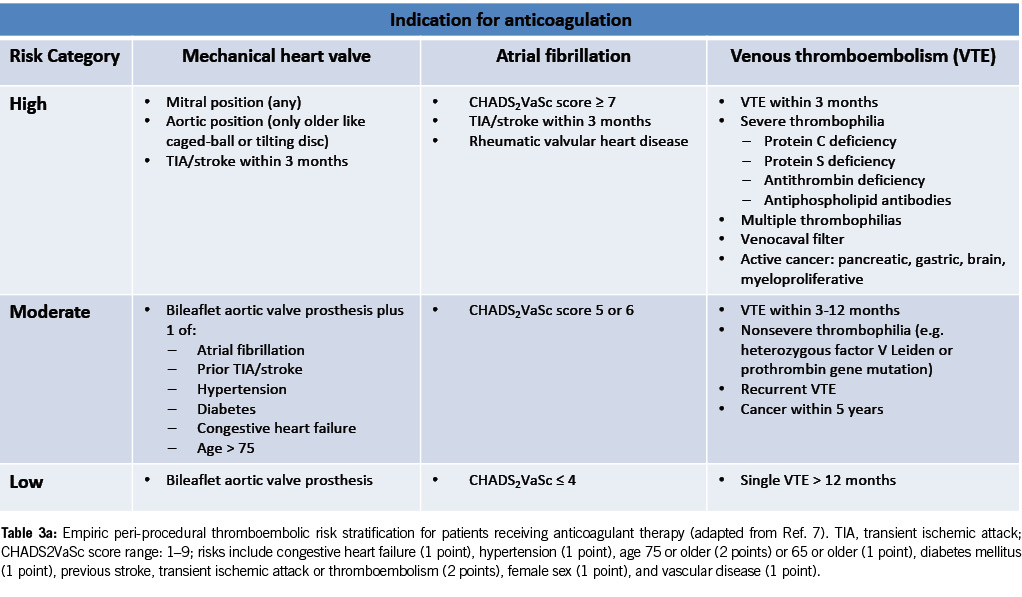
Risk stratification for patients on anticoagulant therapy
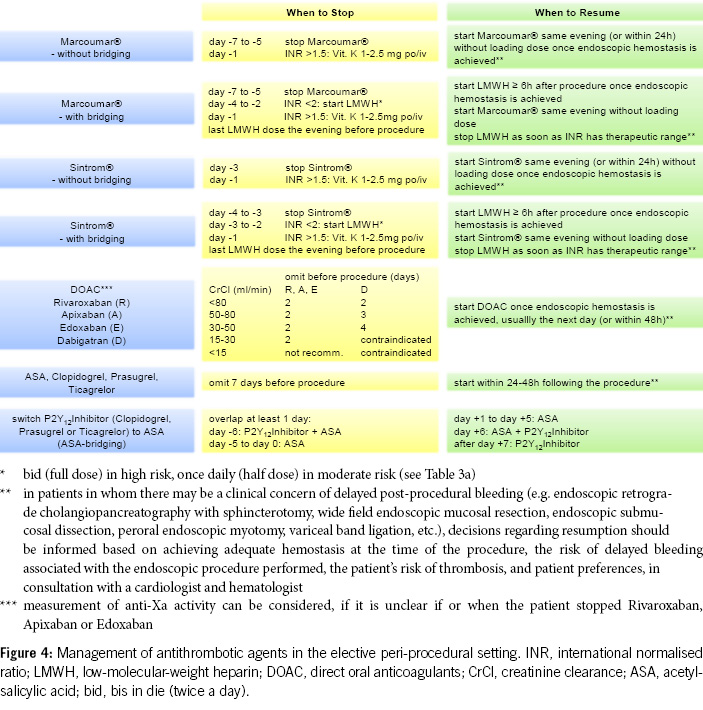
Table 3a provides risk stratification for patients on anticoagulant therapy based on thromboembolic risk. High-risk patients may require procedural deferral or bridge therapy during temporary VKA interruption for elective endoscopy (7).
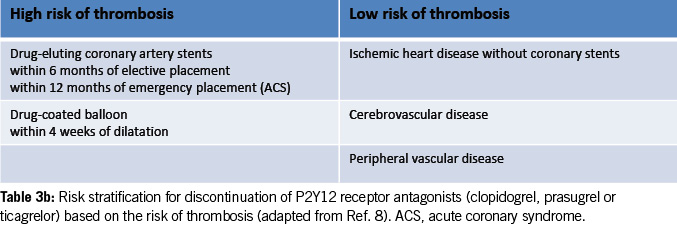
Risk stratification for discontinuing P2Y12 receptor antagonists
Table 3b provides risk stratification for discontinuing P2Y12 receptor antagonists (clopidogrel, prasugrel or ticagrelor) based on thrombosis risk (8).
High-risk patients may require procedural deferral and consultation with a cardiologist and hematologist before stopping the antiplatelet agents.
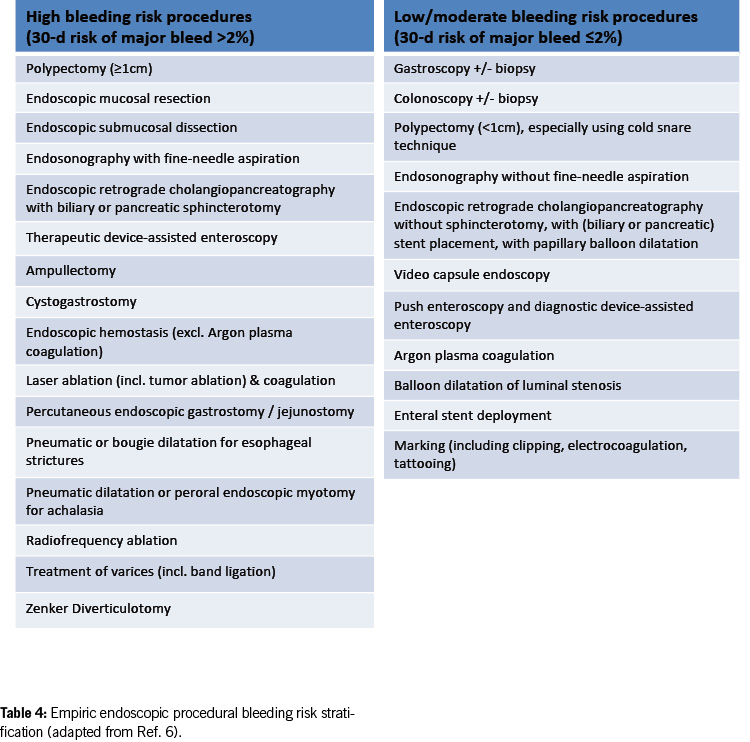
Empirical endoscopic procedural bleeding risk stratification
Table 4 presents an empirical framework for suggested intra- and post-procedural bleeding risk stratification (9). With advancements in endoscopic techniques, the current estimation of post-procedural bleeding risk is subject to change.
Existing data on endoscopic interventions under antithrombotic therapy are limited and controversial. Most studies focus on post-polypectomy bleeding in the colon and are retrospective. Prospective studies and randomised controlled trials are scarce; much of the evidence is based on expert opinion and extrapolation from other clinical situations. This is mainly due to the infrequency of relevant bleeding events after endoscopic procedures (0.07 – 1.7% after colonic polypectomy) (10), necessitating large case numbers to identify significant risk factors.
In 2016, new guidelines on antithrombotic therapy during endoscopic procedures were published by American and European / British authorities (11, 12). Switzerland also adapted its recommendations, particularly for colonic polypectomy, focusing on lower bleeding risk for endoscopic removal of very small (diminutive) polyps up to 5 mm, treating them similarly to biopsies (5).
The effect of thrombocyte inhibitors on colonic polypectomy and its implications
In 2016, we extended the acceptance of endoscopic removal of polyps up to 10 mm under clopidogrel therapy, basing this decision on a large prospective study involving 516 patients (13). This study demonstrated a higher rate of clinically relevant bleeding in the colon (after polypectomy) under thienopyridines (clopidogrel or prasugrel) compared to other antithrombotics (2.4% vs. 0%, p = 0.01). However, it is noteworthy that all bleeding cases in the thienopyridine group occurred in patients concurrently taking acetylsalicylic acid (ASA) and with larger polyps (mean size 12.8 mm, range 8-20 mm). None of the bleeding events were fatal or required surgical or radiological intervention.
In 2019, the first prospective, randomised, double-blind study on the impact of clopidogrel in colonic polypectomies was published (14). The study aimed to investigate whether continuous clopidogrel therapy significantly increased the risk of bleeding after colonic polypectomy. The study included 423 patients on clopidogrel treatment for seven days before a colonoscopy, who were randomly assigned to continue either clopidogrel (n = 208) or a placebo (n = 215) until the morning of the colonoscopy. One hundred and six patients on clopidogrel and 110 on the placebo underwent polypectomy. The bleeding rate within 30 days after the procedure was surprisingly low and similar (3.8% in the non-paused clopidogrel group, 3.6% in the 7-day paused group). All bleeding cases occurred in patients who were also taking ASA and were successfully treated endoscopically. No bleeding events were observed in patients who were solely on clopidogrel.
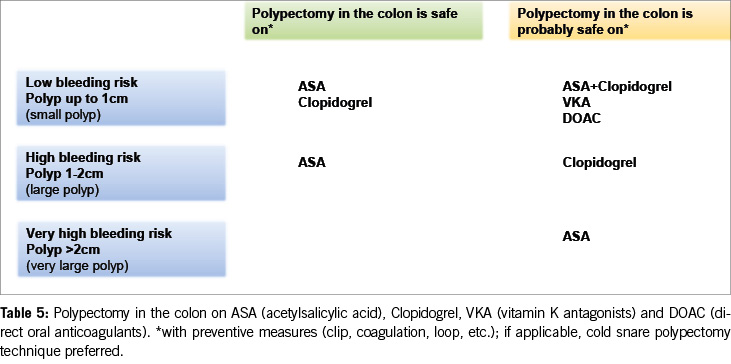
As a result, the 2016 guidelines‘ recommendation to pause clopidogrel before polypectomy is no longer supported. Current data suggest that clopidogrel does not significantly increase the risk of bleeding after colonic polypectomy compared to ASA. Therefore, medium-sized polyps (10–20 mm) can likely be resected under clopidogrel alone, particularly using the cold snare polypectomy technique (Table 5).
Limited data are available for the thrombocyte inhibitors prasugrel and ticagrelor in colonic polypectomy. These drugs are usually administered with ASA and do not need to be paused during diagnostic endoscopies with biopsies. A small randomised trial in patients undergoing cold snare polypectomy of polyps ≤ 10 mm reported similar delayed post-polypectomy bleeding rates between those continuing dual antiplatelet therapy (DAPT: ASA + clopidogrel, ticagrelor, or prasugrel) and those on ASA alone (2.4% vs 0%) (15).
For patients on dual thrombocyte inhibition with polyps larger than 10 mm, it might be advisable to wait until monotherapy is sufficient, if possible. Alternatively, the resection of polyps larger than 10 mm can be considered, accepting a higher risk of bleeding. Pausing clopidogrel before polypectomy in patients on concomitant ASA therapy does not appear to reduce the risk of post-polypectomy bleeding. An interesting alternative could be to pause P2Y12 receptor inhibitors for seven days after the day of polypectomy.
However, the randomised trials (13, 14, 15) had limited numbers of post-polypectomy bleeding events and wide confidence intervals, raising concerns about the sample size’s adequacy to detect a true difference. Further research may be needed to better understand the optimal approach in these cases.
The effect of oral anticoagulants on colonic polypectomy and its implications
A prospective, randomised, controlled study in Japan with 184 patients on oral anticoagulants (warfarin or direct oral anticoagulants, DOACs) examined endoscopic resection of non-sessile colon polyps < 10 mm (16). One group underwent cold snare polypectomy without interrupting anticoagulants, while the other group had anticoagulants paused and bridged with heparin during resection with diathermy. The primary endpoint was significant post-polypectomy bleeding, and no statistically significant difference was observed. Surprisingly, the oral anticoagulation group demonstrated reduced bleeding (4.7% vs. 12%), supporting a preference for resecting small colonic polyps under oral anticoagulation, particularly using the cold snare polypectomy technique.
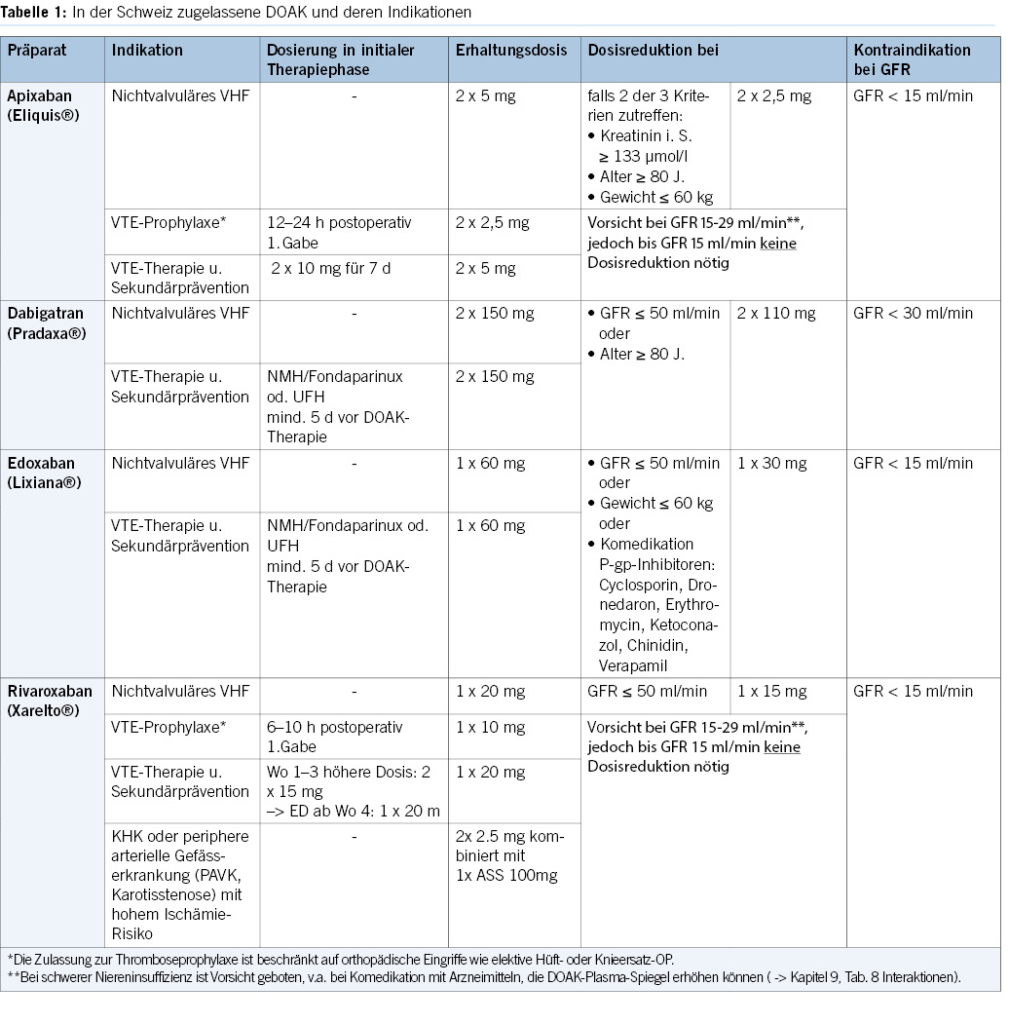
Furthermore, it is noteworthy that for elective colonoscopies, DOACs can be readily paused without significant risk in the overwhelming majority of instances.
The data discussed above indicate a decreasing trend in discontinuing antithrombotics before colonic polypectomy (Table 5).
The most recent international guidelines (8, 9) and a recent systematic review (17) now substantiate and endorse this emerging pattern. The 2021 European guidelines allow the removal of colonic polyps < 10 mm under clopidogrel, while the US-Canadian guideline in 2022 classifies polypectomy < 10 mm as a low/moderate bleeding risk procedure without the need to interrupt anticoagulant or antiplatelet therapy. Similar considerations apply to other endoscopic interventions with a high risk of bleeding, where ASA can be left in place. However, caution should be exercised in specific procedures (e.g. polypectomy/mucosectomy in the upper gastrointestinal tract, ampullectomy, wide-field endoscopic mucosal resection (EMR), endoscopic submucosal dissection (ESD), peroral endoscopic myotomy (POEM) and radiofrequency ablation (RFA)) (Table 4).
However, pausing is still recommended for other antithrombotic drugs in high-risk bleeding procedures (8, 9, 18, 19, 20).
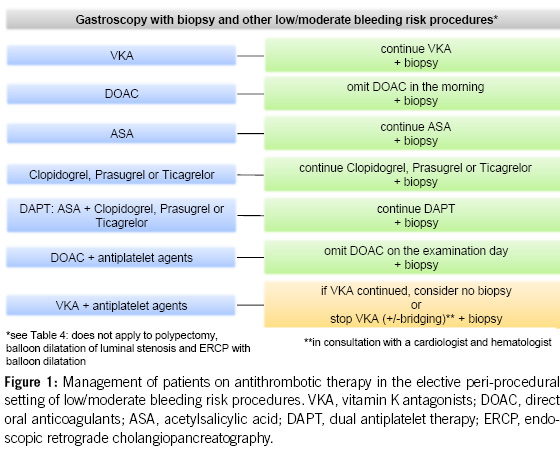
For diagnostic endoscopies with biopsies, as well as for other endoscopic examinations with a low bleeding risk (e.g. endoscopic retrograde cholangiopancreatography (ERCP) without papillotomy/with stenting, endosonography without fine needle puncture (FNP), diagnostic device-assisted enteroscopy, gastrointestinal stents, argon plasma coagulation (APC), marking, capsule endoscopy; see Table 4), antithrombotic drugs can generally be left in place, including DAPT.
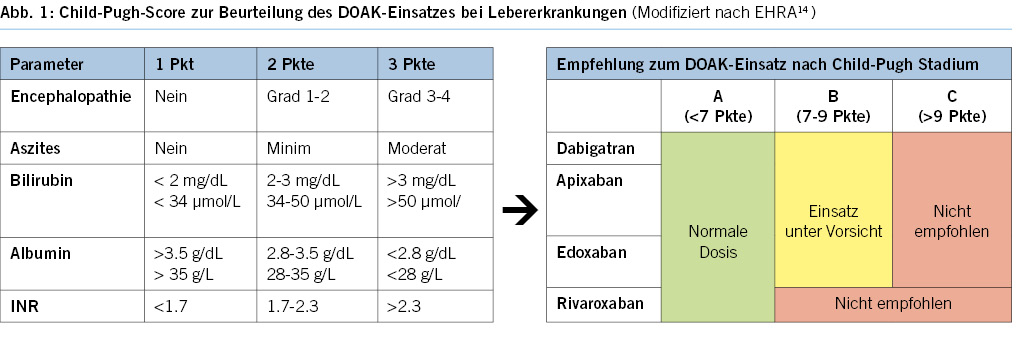
DOACs can be omitted in combination with antiplatelet agents on the morning of the examination, without issues. Caution is advised when VKAs are combined with antiplatelet agents, especially during biopsies (8, 9, 21).
ERCP with papillary balloon dilatation and balloon dilatation of luminal stenosis are considered low/moderate bleeding risk procedures in the latest US-Canadian guideline (9). However, data on these procedures under antiplatelet agents (especially P2Y12 receptor inhibitors) and anticoagulants are still limited, and a cautious approach is recommended, categorising them as high-risk procedures in such cases.
Polypectomy < 10 mm in the colon using the cold snare technique and preventive measures (e.g. coagulation, clips) is considered a procedure with a low risk of bleeding. However, in the upper gastrointestinal tract, a more cautious approach is advised; such interventions are still categorised as high-bleeding-risk procedures.
The recommendations for clinical practice regarding antithrombotic medication and endoscopic procedures can be summarised as follows:
1. Routine checks for INR and platelets before endoscopies are not necessary. However, the bleeding history should be thoroughly assessed (using a questionnaire, Table 1) to identify relevant coagulation disorders.
2. For diagnostic gastroscopies with potential biopsies, antiplatelet agents (monotherapy and dual therapy) and oral anticoagulants (VKAs, DOACs) in monotherapy can be continued without the need for routine discontinuation. We recommend to omit DOACs in the morning of the examination. In the concurrent administration of antiplatelet agents, DOACs should be avoided on the day of the examination (Figures 1 & 4).
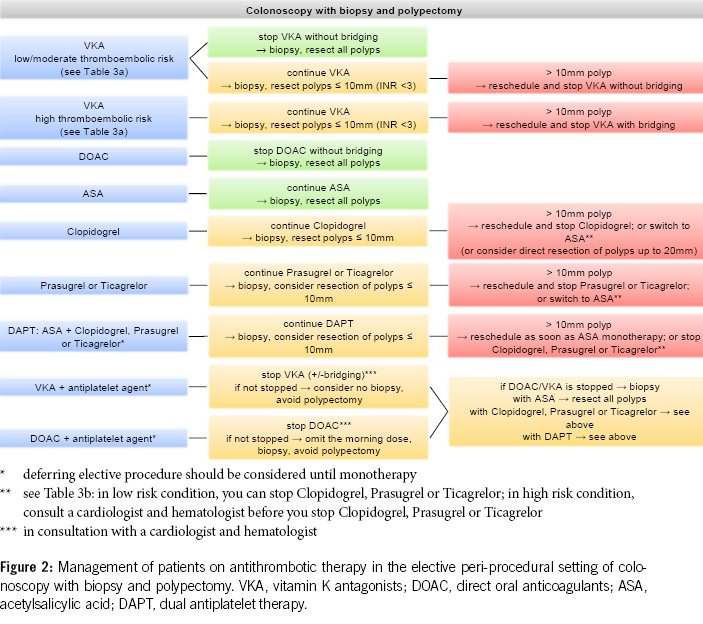
3. Before diagnostic colonoscopies, antithrombotic drugs can generally be continued, as the majority of polyps found are small (< 10 mm) and can be directly resected, especially using the cold snare polypectomy technique. It is recommended to pause DOACs routinely, as this can be done with minimal risk (Figures 2 & 4).
4. In patients with a high thromboembolic risk, VKAs should not routinely be stopped before diagnostic colonoscopies. In low-risk patients, VKAs can be stopped 5–7 days before the colonoscopy without bridging, but stopping may not be necessary, and polyps up to 10 mm can be directly resected using the cold snare technique (Figures 2 & 4).
5. Antiplatelet agents do not need to be stopped before colonoscopy. With ASA, most polyps, including larger ones (> 20 mm), can be removed. Under P2Y12 receptor inhibitors, polyps up to 10 mm can be resected, possibly in combination with ASA. If switching from dual thrombocyte inhibition to monotherapy is anticipated, the colonoscopy should be postponed until then (Figure 2, Table 5).
6. For other endoscopic procedures with an increased risk of bleeding (e.g. EMR, ESD, percutaneous endoscopic gastrostomy, percutaneous endoscopic jejunostomy), antithrombotic drugs should be paused, except for ASA (Figures 3–4, Table 4).
7. There is no one-size-fits-all approach; decisions should be individualised, considering the risk of intervention-related bleeding versus thromboembolism caused by temporary discontinuation of antithrombotics (Table 3a & 3b). Shared
decision-making with patients is essential in such situations.
Overall, these recommendations represent a suitable approach to continue with antithrombotic medication in spite of planned endoscopic procedures and aim to balance between the risks of bleeding and thromboembolism. In complex cases it may be helpful to have interdisciplinary discussions involving cardiologists and hematologists.

Disclaimer
These recommendations have been developed in accordance with the current guidelines and recommendations from America, Europe and the United Kingdom (ASGE, ESGE, BSG), in collaboration with a hematologist experienced in hemostasis (W. Wuillemin).
These recommendations may need to be adjusted and revised in future, depending on new data, new technologies, and practical experience.
The recommendations are intended to provide guidance for clinical practice and should not be applied as universally
valid rules. The clinical situation may require deviation from the currently proposed recommendations.