Obwohl die Krebsinzidenz mit dem Alter zunimmt und ihren Höhepunkt erst nach dem 50. Lebensjahr erreicht, wird jedes Jahr auch bei Tausenden von jungen Frauen und Männern Krebs diagnostiziert (1). So wird allein in den Vereinigten Staaten von Amerika (USA) bei mehr als 70.000 Jugendlichen und jungen Erwachsenen im Alter zwischen 15 und 39 Jahren ein Krebs diagnostiziert. Die Krebsinzidenz bei Kindern unter 15 Jahren beträgt ebenfalls etwa 10.000 Fälle pro Jahr (2). Moderne Krebsbehandlungen haben zu einer deutlichen Senkung der Sterblichkeit und dadurch indirekt auch zu einer Zunahme der unerwünschten Nebenwirkungen wie einer verminderten Fruchtbarkeit geführt.
Although cancer incidence increases with age, peaking after the age of 50, thousands of young women and men are also diagnosed with cancer each year (1). In the United States of America (USA) alone, more than 70,000 adolescents and young adults between the ages of 15 and 39 are diagnosed with cancer. The incidence of cancer in children under the age of 15 is also about 10,000 cases per year (2). Modern cancer treatments have led to a significant reduction in mortality, and thus indirectly to an increase in adverse side effects such as reduced fertility.
Key Words: cancer incidence, modern cancer treatments, reduced fertility
In Kombination mit einem höheren Alter bei der Geburt ihrer Kinder nehmen immer mehr Krebsüberlebende fruchtbarkeitserhaltende Massnahmen in Anspruch, um später ihre Familie vervollständigen zu können (3).
Die Bedrohung oder Erfahrung einer behandlungsbedingten Unfruchtbarkeit kann zu einer psychischen Belastung führen. Viele Patienten sind zum Zeitpunkt der Krebsdiagnose an der Aufrechterhaltung der Fruchtbarkeit und der zukünftigen Fortpflanzungsfähigkeit interessiert. Darüber hinaus können Bedenken hinsichtlich der gefährdeten Fruchtbarkeit auch ihre Behandlungsentscheidungen erheblich beeinflussen (4–6).
Nationale und internationale Leitlinien zeigen, dass Ärzte so früh wie möglich mit allen Patientinnen im gebärfähigen Alter ihr Risiko einer Unfruchtbarkeit aufgrund der Krankheit und/oder Behandlung und ihren Wunsch, nach einer Krebserkrankung Kinder zu bekommen, besprechen und die Patientin bei der fundierten Entscheidung zur Erhaltung der Fruchtbarkeit unterstützen sollten (7, 8). Wie von der American Society of Clinical Oncology (ASCO) und der European Society for Medical Oncology (ESMO) empfohlen, stellen heute die Kryokonservierung von Spermien/Eizellen und/oder Embryonen die beiden Standardstrategien zur Erhaltung der Fertilität bei männlichen bzw. weiblichen Patienten dar (9).
Behandlungskosten
Die Patientinnen und Patienten sollten darüber aufgeklärt werden, dass seit 01.07.2019 Behandlungen zum Erhalt der Fertilität gemäss dem Eidgenössischen Departement des Innern (EDI) von der obligatorischen Krankenpflegeversicherung (OKP) unter spezifischen Bedingungen vergütet werden (siehe Expertenbrief Nr. 59 der SGGG, Aktualisierung September 2020)). Der genaue Beitrag der Rückvergütung (CHF) aber ist noch in Diskussion.
Diese Übernahme der Kosten gilt unter folgenden Bedingungen:
- bei postpubertären Jugendlichen (weiblich und männlich) sowie weiblichen und männlichen Erwachsenen bis zum vollendeten 40. Lebensjahr (d.h. bis zum letzten Tag vor dem 40. Geburtstag). Die Fertilitätserhaltung bei präpubertären Kindern wird nicht rückerstattet.
- bei malignen Erkrankungen, die eine gonadotoxische Therapie erfordern, welche mit einem Risiko einer behandlungsinduzierten persistierenden Amenorrhoe/Azoospermie von über 20 % (d.h. mittleres bis hohes Risiko) einhergeht.
- bei nicht-malignen Erkrankungen, die eine gonadotoxische Therapie erfordern, welche mit einem Risiko einer behandlungsinduzierten persistierenden Amenorrhoe/Azoospermie von über 20 % (d.h. mittleres bis hohes Risiko) einhergeht.
- Bei Frauen und Männern, die eine Stammzelltransplantation erhalten.
Die Kosten folgender Behandlungen im Rahmen des Fertilitätserhaltes werden rückerstattet:
- Frauen: Ovarielle Stimulation mit Oozytenentnahme und Kryokonservierung von unbefruchteten Oozyten. Resektion und Kryokonservierung von Ovarialgewebe, sowie spätere Retransplantation bei persistierender II° Amenorrhoe. Ovarielle Downregulation mit GnRH-Analoga.
- Männer: Kryokonservierung von Spermien, testikuläre Spermienextraktion (TESE) und deren Kryokonservierung.
- Lagerung: Die Lagerung von Keimzellen (Oozyten/Spermien) und Gonadengewebe wird für bis zu 5 Jahre übernommen. Bei persistierender Ovarialinsuffizienz/Azoospermie werden die Kosten für die Lagerung für weitere 5 Jahre übernommen. Die Kostenübernahme einer weiteren Depotverlängerung erfolgt nur bei weiter bestehender Ovarialinsuffizienz bzw. Azoospermie auf vorgängige besondere Gutsprache des Versicherers, der die Empfehlung des Vertrauensarztes oder der Vertrauensärztin berücksichtigt.
Fertilitätserhalt
Fertilitätserhaltende Massnahmen bei Frauen vor Krebstherapie
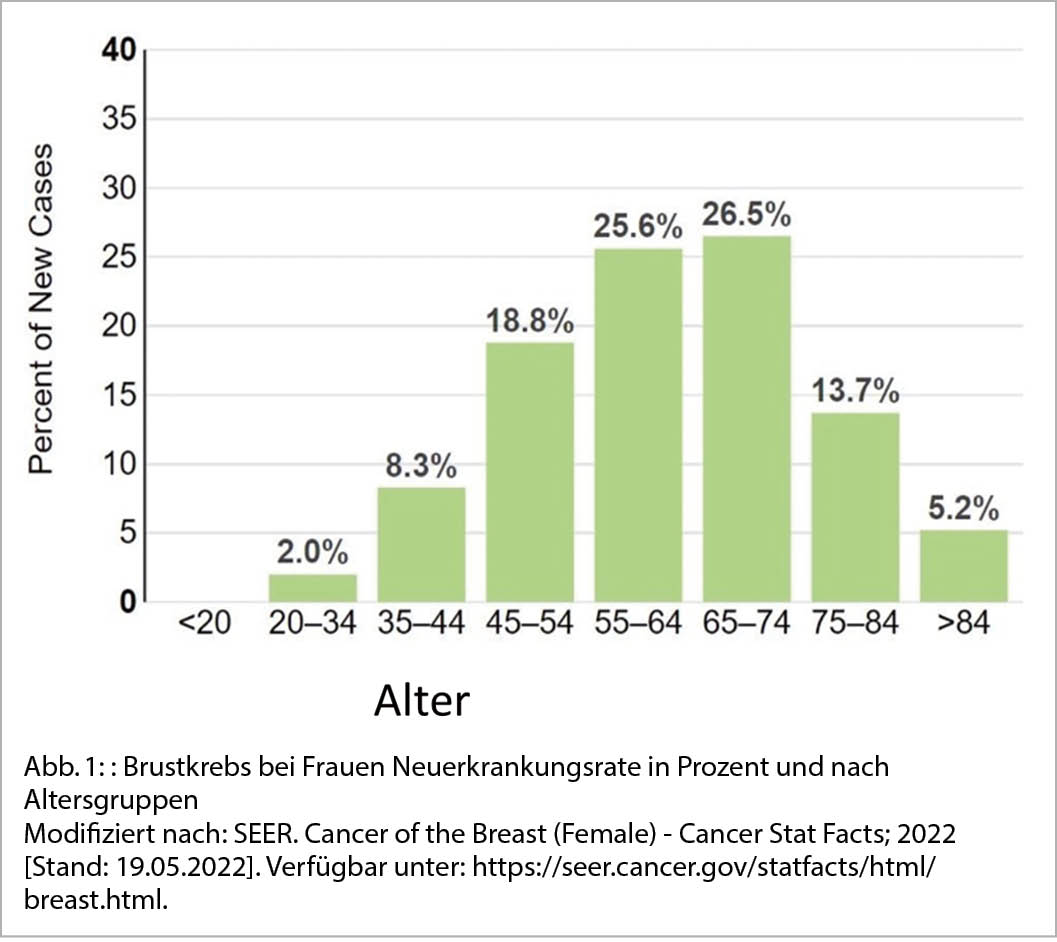
Die Inzidenz von Brustkrebs nimmt zwar vor allem mit dem Alter zu, doch ist er auch die am häufigsten diagnostizierte bösartige Erkrankung bei Frauen im gebärfähigen Alter. Frauen unter 45 Jahren erkranken in 10,5 % pro Jahr neu an Brustkrebs (12) (Abb. 1). Jüngere Frauen stellen sich häufiger mit einer aggressiveren Art des Brustkrebses vor, der eine gonadotoxische Chemotherapie und anschliessende endokrine Therapie beim Hormonrezeptor-positiven Brustkrebs erfordert, was die zukünftige Fertilität erheblich beeinträchtigen kann (13).
Während der Schwangerschaft ist die Diagnose Krebs eher ungewöhnlich. Brust-, Melanom- und Gebärmutterhalskrebs werden am häufigsten während der Schwangerschaft diagnostiziert, gefolgt von hämatologischen Malignomen (14, 15). Gewisse Studien deuten darauf hin, dass offensichtlich bei Frauen, die mittels ovariell stimulierenden Medikamenten gegen Unfruchtbarkeit behandelt werden, ein erhöhtes Risiko an Brustkrebs zu erkranken bestehen könnte (16–18). Demgegenüber aber steht eine Studie von Brinton et al. (19), die die Langzeitwirkung von den Medikamenten Clomifen oder Gonadotropinen untersuchte, welche zur ovariellen Stimulation verwendet werden. Nach einer mittleren Nachbeobachtungszeit von 30 Jahren entwickelten 749 von 9.892 Frauen (7.6%), die auf Unfruchtbarkeit untersucht wurden, Brustkrebs. Jegliche Anwendung von Clomifencitrat war nicht mit einem höheren Risiko, an Brustkrebs zu erkranken, verbunden. Ein signifikant erhöhtes Risiko wurde jedoch bei Frauen beobachtet, die sowohl eine hohe kumulative Dosis als auch mehrere Stimulationszyklen erhielten. Dies steht einer generellen Wahrscheinlichkeit von 12.5% gegenüber, mit welcher eine Frau im Laufe ihres Lebens einen Brustkrebs entwickelt (58). Trotz dieser Resultate aus Studien mit Sterilitätspatientinnen, die sich einem In-vitro-Fertilisations-Verfahren (IVF) unterziehen, ist Vorsicht geboten, da es schwierig ist, das schlussendliche Brustkrebsrisiko in diesem Umfeld einzuschätzen. Ein erheblicher Anteil der Patientinnen mit einer Krebsdiagnose im gebärfähigen Alter (ca. 50 %) wünscht sich zum Zeitpunkt der Krebsdiagnose eine Schwangerschaft. Krebsüberlebende haben jedoch aufgrund der zytotoxischen Therapie niedrigere Fruchtbarkeitsraten als die allgemeine Bevölkerung (20).
Dabei haben Brustkrebspatientinnen die niedrigste Schwangerschaftsrate unter den Krebsüberlebenden. Bei Ihnen ergibt sich eine um insgesamt 67% geringere Wahrscheinlichkeit, nach der Krebsbehandlung ein Kind zu bekommen im Vergleich zur Allgemeinbevölkerung (21). Es zeigte sich jedoch auch, dass Brustkrebspatientinnen, die nach Diagnose und Behandlung schwanger wurden, ein um 41 % geringeres Sterberisiko hatten als Frauen, die nicht schwanger wurden (22). Zu den Hauptsorgen jüngerer Patientinnen im fortpflanzungsfähigen Alter nach einer Krebstherapie zählen auch mögliche Komplikationen für ihren Nachwuchs. Es hat sich jedoch herausgestellt, dass sich die Ergebnisse von Neugeborenen sowohl bei männlichen wie auch bei weiblichen Überlebenden nach einer Krebsdiagnose nicht von den Ergebnissen der gesunden Bevölkerung unterscheiden (23, 24). Allerdings ist bei Patientinnen, die nach der Brustkrebsdiagnose schwanger wurden, eine hohe Rate von Schwangerschaftsabbrüchen zu beobachten (25). Ebenso konnte eine höhere Inzidenz von Geburtskomplikationen (z.B. Kaiserschnitt, Frühgeburt, Kinder mit niedrigem Geburtsgewicht) bei Patientinnen nach einer Krebstherapie im Vergleich zu Kontrollen beobachtet werden. Bei Frauen, die sich einer Beckenbestrahlung unterziehen mussten, kann es zu Gebärmutterschäden mit einem erhöhten Risiko für Fehlgeburten, Frühgeburten und niedrigem Geburtsgewicht kommen (26, 27). Krebsbehandlungen (Operation, Strahlentherapie, zytotoxische Chemotherapie und endokrine Therapie) können die männliche und weibliche Fertilität vorübergehend oder dauerhaft beeinträchtigen (8). Eine Operation kann einen direkten Einfluss auf die Fertilität haben und anatomische oder vaskuläre Probleme verursachen (z.B. retrograde Ejakulation oder beeinträchtigte Ejakulation, Veränderungen der Anatomie von Gebärmutter, Gebärmutterhals oder Vagina). Chemotherapie und Strahlentherapie haben eine direkte gonadotoxische Wirkung, indem sie bei der Frau die Ovarialfollikel und damit den ovariellen Reservepool zerstören und beim Mann die Anzahl, Beweglichkeit, Morphologie und DNA-Integrität der Spermien beeinträchtigen (28, 29). So ist die onkologische Beratung bezüglich Fruchtbarkeit stets von Frau zu Frau zu individualisieren und diese persönlich auf ihren Fall spezifisch zu beraten. Dabei ist auch zwischen dem absoluten Nutzen der vorgeschlagenen Krebstherapie und dem Risiko einer dauerhaften Unfruchtbarkeit abzuwägen (30). Die Wahrnehmung des Risikos für eine Kinderlosigkeit ist immer individuell und auch der eigene Wunsch der Frau ist zu berücksichtigen. Auch ist zu beachten, dass bei Behandlungen mit einer eigentlich nur geringen Gonadotoxizität (z.B. Doxorubicin, Bleomycin, Vinblastin, Dacarbazin bei Hodgkin-Lymphompatientinnen) die ovarielle Reserve je nach individueller Ausgangslage (weibliches Alter, Ovarielle Reserve) trotzdem reduziert und die Fertilität beeinträchtigt werden kann (31).
Möglichkeiten fertilitätserhaltender Massnahmen bei Frauen
In der 20. Woche der Embryogenese erreichen Frauen mit etwa sechs bis sieben Millionen potenziellen Eizellen, dem sogenannten Primordialfollikelpool, die maximale Anzahl von Keimzellen in ihren Genitalien. Im weiteren Verlauf kommt es zu einem stetigen atretischen Untergang von Eizellen, sodass bei Geburt noch gut eine Million und bei Eintritt der Pubertät noch ca. 400.000 Eizellen vorhanden sind. Diese Zahl an Eizellen, die sogenannte ovarielle Reserve, nimmt in der fruchtbaren Lebensphase der Frau weiter stetig ab und erreicht zum Zeitpunkt der Menopause, d.h. nach insgesamt ungefähr 450 monatlichen Ovulationszyklen und dem atretischen Zugrundegehen der allermeisten Eizellen, einen Wert von 0 bis 1.000 Eizellen (32). Die Erhaltung der ovariellen Reserve mit ihrer Hormonproduktion bei einer prämenopausalen Frau ist aber auch notwendig, um die allgemeine Gesundheit der Frau zu erhalten. Sie spielt nicht nur bei der Entwicklung der Eizelle im Rahmen der Fruchtbarkeit eine wichtige Rolle, sondern auch durch die Hormonproduktion in anderen Hormonsystemen wie dem Herz-Kreislauf-System und dem Knochenstoffwechsel. Der Grad der Erschöpfung der ovariellen Reserve und damit der Verlust der hormonellen Funktion, unterscheidet sich zwischen Chemotherapie und Strahlentherapie. Die Auswirkung einer Chemotherapie variiert je nach weiblichem Alter (je jünger die Frau, desto geringer das Risiko einer Ovarialinsuffizienz), dem verwendeten Chemotherapeutikum, der Dauer sowie Anzahl der Behandlungszyklen. Eizellen sind sehr empfindlich gegenüber Strahlen. Eine Strahlenbelastung von 20–30Gy im Bereich des Beckens oder eine Ganzkörperbestrahlung von 15Gy führen bereits zum kompletten Verlust der Ovarialfunktion (33). Mittels einer fertilitäterhaltenden Massnahme soll versucht werden, Eizellen/Ovarialgewebe vor einer Chemotherapie/Radiotherapie extrakorporell zu erhalten und sie nicht der zytotoxischen Therapie auszusetzen.
Kryokonservierung von befruchteten Eizellen. d.h. Embryonen
Die Kryokonservierung von Embryonen ist eine weit verbreitete und zuverlässige Methode. Frauen werden einer kontrollierten ovariellen Stimulation mit Gonadotropin-Injektionen unterzogen, um ein multifollikuläres Wachstum zu erreichen. Nach ca. 10 – 14 Tagen Stimulation erfolgt die Eizellentnahme mittels transvaginaler ultraschallgesteuerter Nadelaspiration und normalerweise unter Analgosedierung (10). Heutzutage werden die Eizellen im IVF-Labor grösstenteils unbefruchtet und denudiert vitrifiziert (Schock-Gefriemethode auf -196° in flüssigem Stickstoff). Eher selten und bei Wunsch des Paares kann bei stabiler Partnerschaft auf deren Wunsch auch eine Befruchtung (Fertilisierung wird nicht von der OKP übernommen) erfolgen, um die dann entstehenden Embryonen im Blastozystenstadium zu vitrifizieren (7, 32, 34). Eine hormonelle ovarielle Stimulation kann dabei zu jedem Zeitpunkt des weiblichen Zyklus erfolgen, sowohl in der frühen Follikelphase (32) als auch in der präovulatorischen oder lutealen Zyklusphase. Es gibt drei Hauptverfahren zur Kryokonservierung: langsames Einfrieren (slow freezing), ultraschnelles Einfrieren und Vitrifikation (7). Langsames Gefrieren beinhaltet eine schrittweise programmierte Temperaturabnahme, wobei ein Gefriergleichgewicht aufgrund des Austauschs der extra- und intrazellulären Flüssigkeiten erreicht wird, ohne bedeutende osmotische und zelluläre Deformationseffekte zu verursachen. Innerhalb der Eizelle können sich jedoch Eiskristalle bilden, die beim Auftau zur Lyse der Zelle führen können. Die Vitrifikation wandelt die Eizelle mit grossem Wassergehalt in feste glasähnliche Zellen um und vermeidet so die Bildung von Eiskristallen, sowohl intrazellulär als auch extrazellulär (7). Die Vitrifikation wird heutzutage als Standardmethode bevorzugt.
Kryokonservierung von unbefruchteten Eizellen
Als Alternative zur Kryokonservierung von Embryonen, ist diese Technik die heute bevorzugte Option für postpubertäre und jugendliche Frauen. Dank der Vitrifikation von unbefruchteten Eizellen können diese später ohne Lyse aufgetaut und mit dem zukünftigen Partner befruchtet werden. So kann sich die Frau ihre Fortpflanzungsautonomie für später bewahren. Eine solche ovarielle Stimulation ist jedoch nicht geeignet für Patienten, die eine sofortige Krebsbehandlung benötigen und somit die Zeit für die Stimulation fehlt, oder bei Frauen mit hormonsensitivem Krebsleiden, da hier ein fragliches Risiko durch die hormonelle Stimulation auf das Verhalten des Primärtumors bestehen könnte. Eizellen können als reife Eizellen oder auch als unreife Eizellen vitrifiziert werden. Unreife Eizellen sind eine mögliche Option für Frauen mit hormonsensitiven Krebserkrankungen, wo keine hormonelle Stimulation möglich ist. Dies ermöglicht eine sofortige Krebsbehandlung (7, 32) ohne Zeitverzögerung. Die Daten bezüglich der Schwangerschaftsraten mittels dieser unreifen Eizellen sind aber aufgrund fehlender Expertise und mangelhafter Technik sehr ernüchternd und die entsprechenden Schwangerschaftschancen sind sehr gering.
Kryokonservierung von Ovarialgewebe
Die Methode der Kryokonservierung von Eierstockgewebe zur späteren Retransplantation ist mittlerweile von einem experimentellen Stadium zu einem Standardprozedere geworden. Nach der S2-Richtlinie der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF) ist sie mittlerweile sogar eine etablierte Methode zur temporären Wiederherstellung der Fertilität nach einer Krebstherapie bei Frauen (7). Diese Methode kann unabhängig vom Zyklus sowie auch schon vor der Menarche bei präpubertären Mädchen durchgeführt werden. Eine relevante Verzögerung der Krebstherapie ist somit nicht nötig. Es ist jedoch zu bedenken, dass zur Entnahme des Ovarialgewebes eine Intubationsnarkose erforderlich ist und der allgemeinzustand der Frau diese erlauben muss. Eine grosse Sorge bei diesem Verfahren ist die Möglichkeit einer potentiellen Retransplantation karzinogener Zellen oder eine mögliche nachfolgende bösartige Transformation des Eierstockgewebes (7, 35). Das Risiko ovarieller Metastasen ist für keine Tumorentität auszuschliessen. Ein diesbezüglich bekanntes erhöhtes Risiko besteht bei Leukämien, Neuroblastomen, Burkitt-Lymphomen und malignen Ovarialtumoren (7).
Transposition der Ovarien
Die Zahl der Fälle von Gebärmutterhalskrebs bei jungen Frauen nimmt zu. Dies zeigen diverse Studien aus verschiedenen Nationen. So stieg die Inzidenz von Gebärmutterhalskrebs z.B. bei Frauen im Alter von 20–24 Jahren in Neuseeland in den Jahren von 1985 bis 2013 signifikant an (36). In Korea stiegen die Inzidenz- und Mortalitätsraten von Gebärmutterhalskrebs bei jungen Frauen unter 30 Jahren von 1993 bis 2012 signifikant an (37). Laut Statistiken aus dem Jahr 2010 traten in China fast 15,7 % der Fälle von Gebärmutterhalskrebs bei Frauen unter 40 Jahren auf (38). Bei Frauen, bei denen ein lokal fortgeschrittener Gebärmutterhalskrebs diagnostiziert wurde, beinhaltet die Standardbehandlung die gleichzeitige Strahlentherapie und Chemotherapie des Beckens. Die Bestrahlung des weiblichen Beckens kann in einem hohen Prozentsatz zu einem kompletten Versagen der Eierstockfunktion führen. Eine erloschene Ovarialfunktion kann nicht nur zu menopausalen Symptomen führen, sondern auch Osteoporose, Herz-Kreislauf-Erkrankungen und Urogenitalatrophie verursachen (39). Die ovarielle Transposition hat sich als gute Empfehlung für den Erhalt der Ovarialfunktion bei Patientinnen mit malignen Erkrankungen des Beckens vor Radiotherapie erwiesen. Zu den chirurgischen Ansätzen gehören die intra- und retroperitoneale Transposition auf oder lateral des Psoas-Muskels, die perkutane Nadeltransposition der Eierstöcke und die Exteriorisation in das subkutane Fettgewebe. Hier ist zu erwähnen, dass Ovarien und Tuben am Uterusansatz abgesetzt werden müssen. Nach einer späteren Ovar-Reposition müssen dann entweder die Tuben refertilisiert werden, um eine spontane Konzeption wieder zu ermöglichen, oder aber es muss eine IVF/ICSI-Therapie durchgeführt werden. Mittlerweile hat sich die Methode der ovariellen Transposition als eine zuverlässige Methode herausgestellt (40, 41). Die Methodik der ovariellen Transposition ist beschränkt auf junge Patientinnen mit operablen zervikalen Tumoren im Frühstadium, die eine primäre oder adjuvante Strahlentherapie benötigen. Sie kann zusätzlich bei ovariellen Dysgerminomen, Vaginalkarzinomen, aber auch nicht- gynäkologischen Malignomen wie Ependymome, Morbus Hodgkin, Sarkomen und Rektumkarzinomen durchgeführt werden (42).
Weitere Fertilitätsprotektive Massnahmen
Ein weiterer Ansatz zur Erhaltung der Fertilität besteht darin, die ovarielle Aktivität während einer onkologischen Behandlung durch die Gabe von möglicherweise protektiven Medikamenten zu schützen. Dies kann durch die Anwendung von Gonadotropin-Releasing-Hormone Agonisten (GnRHa), die optimal mind. 7 Tage vor Beginn der Chemotherapie verabreicht werden, versucht werden. GnRH- Analoga führen nach initialer Hyperstimulation zu einer Downregulation der Hypothalamus-Hypophysen-Gonaden-Achse und hemmen die ovarielle Aktivität, indem sie die Gonadotropin-Spiegel unterdrücken. Der Nutzen der GnRH-Analoga wird seit Jahren kontrovers diskutiert. In einer Cochrane-Analyse basierend auf der Auswertung von 4 randomisierten Studien aus den Jahren 1987 bis 2007 zeigten die Autoren, dass die Patientinnen von der Verwendung von GnRH-Analoga profitieren. Es konnten erhöhte Menstruations- (RR 1,90, 95%-KI 1,30 – 2,79) und Ovulationsraten (RR 2,70, 95%-KI 1,52 – 4,79) nach abgeschlossener Chemotherapie gezeigt werden. Eine Metaanalyse aus dem Jahr 2011 konnte ebenfalls einen protektiven Effekt durch GnRH-Analoga auf die ovarielle Funktion nachweisen. Eine Metaanalyse aus dem Jahr 2015 untersuchte insgesamt 1231 prämenopausalen Mammakarzinompatientinnen. Die temporäre Suppression der Ovarialfunktion durch GnRH-Analoga war mit einem geringeren Risiko eines preterm ovarian failure (POF) assoziiert (OR 0,36, 95%-KI 0,23 – 0,57; p < 0,001). Bei der PROMISE-GIM6-Studie kommt es bei Mammakarzinompatientinnen zur Schlussfolgerung, dass von einem Schutz der Ovarialfunktion durch GnRH- Analoga bei gleichzeitig unveränderter Prognose ausgegangen werden kann. Zu einem anderen Resultat kamen die Autoren Elgindy und Mitarbeiter im Jahr 2015. Die Daten von 907 Patientinnen gingen in die Metaanalyse ein. Es zeigten sich keine signifikanten Unterschiede zwischen den zusätzlich mit GnRH-Analoga und den allein mittels Chemotherapie behandelten Patientinnen (68,4 vs. 59,9%). Die Patientinnen profitierten weder hinsichtlich des FSH-Spiegels (p = 0,27), des Anti- Müller-Hormons (p = 0,40), noch bez. der Antralfollikelzahl (p = 0,17) von der GnRH-Analoga-Gabe. Vor diesem Hintergrund der widersprüchlichen Aussagen bez. der Wirksamkeit von GnRH-Analoga sollte der Einsatz mit der Patientin kritisch und ausführlich diskutiert werden (43,44,45,46,47,48).
Allerdings kommt die AWMF (7) zu dem Ergebnis, dass GnRHa alleine zur Fertilitätsprotektion derzeit nicht ausreichend zu sein scheinen.
Mögliche zukünftige Optionen (aktuell experimentell)
Kryokonserviertes Ovarialgewebe von präpubertären Mädchen enthält unreife Primordialfollikel, die später aktiviert werden müssen, um eine hormonelle Produktion und eine Eizellentwicklung bei der pubertären jungen Frau zu erreichen. Dies wird in vivo oder auch in vitro vor der Autotransplantation durch Aktivierung der Phosphatidylinositol-3-Kinase (PI3K)/Phosphatase und Tensin-Homolog (PTEN)/Proteinkinase B (AKT) versucht (49). Die Möglichkeit eines artifiziellen Ovars für eine Transplantation wird ebenfalls zur Wiederherstellung der Fertilität in Erwägung gezogen. Beide Techniken befinden sich aber derzeit noch im tierexperimentellen Stadium (50). Ebenfalls in der Entwicklung sind Nanopartikel und fertilitätsprotektive Wirkstoffe. Das Ziel wäre, die Ovarialzellen während gonadotoxischer onkologischer Behandlungen zu schützen (28, 33, 51)
Fertilitätserhaltende Massnahmen beim Mann
Bei Männern, die sich einer gonadotoxischen Behandlung unterziehen, sind derzeit sowohl die Spermien- als auch Hodengewebe-Kryokonservierung verfügbar. Vor einer zytotoxischen Therapie ist die Kryokonservierung von ejakulierten Spermien die empfohlene Technik für erwachsene Männer und pubertierende Jungen, die Spermien im Ejakulat produzieren und zur Ejakulation befähigt sind. Für Männer, die eine Strahlentherapie erhalten, kann eine Gonadenabschirmung eine Option sein, wenn eine Spermiengewinnung nicht möglich ist. Bei der Spermienabgabe sind individuell ca. 1-3 Samenproben nach einer Abstinenzzeit von optimal 48 Stunden zwischen den Proben sinnvoll (7). Im Falle eines Ejakulationsversagens oder wenn keine Spermien im Ejakulat gefunden werden, können Spermien durch epididymale oder/und testikuläre Spermienextraktion, selten auch durch Elektrostimulation, gewonnen werden (52,53). Präpubertäre Knaben durchlaufen noch keine Spermatogenese und haben daher keine reifen Spermien in ihren Hoden. Daher ist hier die Kryokonservierung von Spermien nicht möglich. Die einzige Möglichkeit ist die Kryokonservierung von noch unreifem Hodengewebe, das spermatogone Stammzellen enthält. Analog zur Kryokonservierung von Ovarialgewebe bei präpubertären Mädchen kann das Hodengewebe durch eine Hodenbiopsie gewonnen und in Form von Spermatogonien oder in Form von Hodengewebe, beides momentan noch rein experimentell, unter Verwendung von Slow-Freeze- oder Ultra-Rapid-Techniken, kryokonserviert werden (54).
Schlussfolgerungen
Die onkologische Gesundheitsversorgung ist heute weit davon entfernt, nur den Krebs zu heilen. Die Hoffnung auf zukünftige Fruchtbarkeit nach einer onkologischen Behandlung erhöht die Lebensqualität der Patienten erheblich und hilft ihnen, die Krebserkrankung emotional besser zu bewältigen. Der Erhalt der Fertilität ist heute sowohl bei weiblichen als auch bei männlichen onkologischen Patienten möglich und sollte in die onkologische Betreuung integriert werden. Es gibt verschiedene Techniken zum Fertilitätserhalt, wobei die am besten geeignete für den jeweiligen Patienten gewählt werden soll, je nachdem ob dieser weiblich oder männlich und präpubertär oder postpubertär ist. Die Spermienasservierung gilt heute als erste Option zum Erhalt der Fertilität beim Mann. Die Vitrifikation der Eizellen gilt derzeit als Erstlinienoption für postpubertäre Frauen, wenn die entsprechende Zeit zur ovariellen Stimulation trotz Grunderkrankungen vorhanden ist. Sollte aus zeitlichen Gründen diese Stimulation nicht möglich sein, gilt die Kryokonservierung von Ovargewebe als heute etablierte Therapie der Wahl. Die Kryokonservierung von Embryonen gerät beim Erhalt der Fertilität in einen ethischen Konflikt, da das Ziel der autonome Fertilitätserhalt der Frau sein soll. So werden heute nur noch selten Embryonen des Paares als Fertilitätserhalt für die Frau vitrizifiert. Zudem bestätigt der wachsende Erfolgt der Vitrifikationstechnik bez. Sicherheit und Effizienz, dass die Vitrifikation von unbefruchteten Eizellen bevorzugt werden sollte. Der Vorteil der Kryokonservierung von Ovarialgewebe wiederum ist, dass so eine spätere spontane intrakorporelle Fertilisierung möglich gemacht wird, welche gemäss aktueller Datenlage eine selbige kumulative Schwangerschaftsrate von 30 – 35% pro Gewebetransfer (55) erbringt wie diejenige mit Verwendung vitrifizierter Eizellen. Vitrifizierte Eizellen wiederum können später nur mittels einer künstlichen Befruchtung verwendet werden.
Abkürzungen: ASCO American Society of Clinical Oncology, AWMF Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften, ESMO European Society for Medical Oncology, IVF In-vitro-Fertilisation, GnRH Gonadotropin-Releasing-Hormon
Copyright bei Aerzteverlag medinfo AG
Frauenklinik, Kantonsspital St. Gallen
Rorschacher Str. 95/Haus 06
9007 St. Gallen
Fiore Praxis AG
Brauerstrasse 95
9016 St. Gallen
Frauenklinik
Kantonsspital St. Gallen
Rorschacher Str. 95/Haus 06
9007 St. Gallen
Die Autorinnen haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. American Cancer Society. Cancer facts and figures; 2008.
2. Del-Pozo-Lérida S, Salvador C, Martínez-Soler F, Tortosa A, Perucho M, Giménez-Bonafé P. Preservation of fertility in patients with cancer (Review). Oncol Rep 2019; 41(5):2607–14. doi: 10.3892/or.2019.7063.
3. Johnson J-A, Tough S, Wilson RD, Audibert F, Blight C, BrockS J-A et al. Delayed Child-Bearing. J Obstet Gynaecol Can 2012; 34(1):80–93. doi: 10.1016/S1701-2163(16)35138-6.
4. Canada AL, Schover LR. The psychosocial impact of interrupted childbearing in long-term female cancer survivors. Psychooncology 2012; 21(2):134–43. doi: 10.1002/pon.1875.
5. Rosen A, Rodriguez-Wallberg KA, Rosenzweig L. Psychosocial distress in young cancer survivors. Semin Oncol Nurs 2009; 25(4):268–77. doi: 10.1016/j.soncn.2009.08.004.
6. Senkus E, Gomez H, Dirix L, Jerusalem G, Murray E, van Tienhoven G et al. Attitudes of young patients with breast cancer toward fertility loss related to adjuvant systemic therapies. EORTC study 10002 BIG 3-98. Psychooncology 2014; 23(2):173–82. doi: 10.1002/pon.3384.
7. AWMF. Fertility preserv ation for patients with malig nant disease. Guideline of the DGGG, DGU and DGRM: S2k-Level, AWMF Registry No.015/082. Berlin; 2017. Verfügbar unter: http://www.awmf.org/leitlinien/detail/ll/015-082.html.
8. Lee SJ, Schover LR, Partridge AH, Patrizio P, Wallace WH, Hagerty K et al. American Society of Clinical Oncology recommendations on fertility preservation in cancer patients. J Clin Oncol 2006; 24(18):2917–31. doi: 10.1200/JCO.2006.06.5888.
9. Loren AW, Mangu PB, Beck LN, Brennan L, Magdalinski AJ, Partridge AH et al. Fertility preservation for patients with cancer: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol 2013; 31(19):2500– 10. doi: 10.1200/JCO.2013.49.2678.
10. Matthews TJ, Hamilton BE. Delayed childbearing: more women are having their first child later in life. NCHS Data Brief 2009; (21):1–8.
11. Pentheroudakis G, Pavlidis N. Cancer and pregnancy: poena magna, not anymore. Eur J Cancer 2006; 42(2):126–40. doi: 10.1016/j.ejca.2005.10.014.
12. SEER. Cancer of the Breast (Female) – Cancer Stat Facts; 2022 [Stand: 19.05.2022]. Verfügbar unter: https://seer.cancer.gov/statfacts/html/breast.html.
13. Kasum M, Beketić-Orešković L, Orešković S. Subsequent pregnancy and prognosis in breast cancer survivors. Acta Clin Croat 2014; 53(3):334–41. doi: Review.
14. Mieog JSD, van der Hage JA, van de Velde CJH. Neoadjuvant chemotherapy for operable breast cancer. Br J Surg 2007; 94(10):1189–200. doi: 10.1002/bjs.5894.
15. Stensheim H, Møller B, van Dijk T, Fosså SD. Cause-specific survival for women diagnosed with cancer during pregnancy or lactation: a registry-based cohort study. J Clin Oncol 2009; 27(1):45–51. doi: 10.1200/JCO.2008.17.4110.
16. Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Collaborative Group on Hormonal Factors in Breast Cancer. Lancet 1997; 350(9084):1047–59.
17. Fortner RT, Eliassen AH, Spiegelman D, Willett WC, Barbieri RL, Hankinson SE. Premenopausal endogenous steroid hormones and breast cancer risk: results from the Nurses‘ Health Study II. Breast Cancer Res 2013; 15(2):R19. doi: 10.1186/bcr3394.
18. Calderon-Margalit R, Friedlander Y, Yanetz R, Kleinhaus K, Perrin MC, Manor O et al. Cancer risk after exposure to treatments for ovulation induction. Am J Epidemiol 2009; 169(3):365–75. doi: 10.1093/aje/kwn318.
19. Brinton LA, Scoccia B, Moghissi KS, Westhoff CL, Niwa S, Ruggieri D et al. Long-term relationship of ovulation-stimulating drugs to breast cancer risk. Cancer Epidemiol Biomarkers Prev 2014; 23(4):584–93. doi: 10.1158/1055- 9965.EPI-13-0996.
20. Letourneau JM, Ebbel EE, Katz PP, Katz A, Ai WZ, Chien AJ et al. Pretreatment fertility counseling and fertility preservation improve quality of life in reproductive age women with cancer. Cancer 2012; 118(6):1710–7. doi: 10.1002/cncr.26459.
21. Stensheim H, Cvancarova M, Møller B, Fosså SD. Pregnancy after adolescent and adult cancer: a population-based matched cohort study. Int J Cancer 2011; 129(5):1225–36. doi: 10.1002/ijc.26045.
22. Azim HA, Santoro L, Pavlidis N, Gelber S, Kroman N, Azim H et al. Safety of pregnancy following breast cancer diagnosis: a meta-analysis of 14 studies. Eur J Cancer 2011; 47(1):74–83. doi: 10.1016/j.ejca.2010.09.007.
23. Azim HA, Kroman N, Paesmans M, Gelber S, Rotmensz N, Ameye L et al. Prognostic impact of pregnancy after breast cancer according to estrogen receptor status: a multicenter retrospective study. J Clin Oncol 2013; 31(1):73– 9. doi: 10.1200/JCO.2012.44.2285.
24. Winther JF, Olsen JH, Wu H, Shyr Y, Mulvihill JJ, Stovall M et al. Genetic disease in the children of Danish survivors of childhood and adolescent cancer. J Clin Oncol 2012; 30(1):27–33. doi: 10.1200/JCO.2011.35.0504.
25. Lawrenz B, Henes M, Neunhoeffer E, Fehm T, Huebner S, Kanz L et al. Pregnancy after successful cancer treatment: what needs to be considered? Onkologie 2012; 35(3):128–32. doi: 10.1159/000336830.
26. Critchley HOD, Wallace WHB. Impact of cancer treatment on uterine function. J Natl Cancer Inst Monogr 2005; (34):64–8. doi: 10.1093/jncimonographs/lgi022.
27. Dalberg K, Eriksson J, Holmberg L. Birth outcome in women with previously treated breast cancer–a population-based cohort study from Sweden. PLoS Med 2006; 3(9):e336. doi: 10.1371/journal.pmed.0030336.
28. Kalich-Philosoph L, Roness H, Carmely A, Fishel-Bartal M, Ligumsky H, Paglin S et al. Cyclophosphamide triggers follicle activation and „burnout“; AS101 prevents follicle loss and preserves fertility. Sci Transl Med 2013; 5(185):185ra62. doi: 10.1126/scitranslmed.3005402.
29. Wallace WHB, Thomson AB, Saran F, Kelsey TW. Predicting age of ovarian failure after radiation to a field that includes the ovaries. Int J Radiat Oncol Biol Phys 2005; 62(3):738–44. doi: 10.1016/j.ijrobp.2004.11.038.
30. Abusief ME, Missmer SA, Ginsburg ES, Weeks JC, Partridge AH. Relationship between reproductive history, anthropometrics, lifestyle factors, and the likelihood of persistent chemotherapy-related amenorrhea in women with premenopausal breast cancer. Fertil Steril 2012; 97(1):154–9. doi: 10.1016/j.fertnstert.2011.10.005.
31. van der Kaaij MAE, Heutte N, Meijnders P, Abeilard-Lemoisson E, Spina M, Moser EC et al. Premature ovarian failure and fertility in long-term survivors of Hodgkin’s lymphoma: a European Organisation for Research and Treatment of Cancer Lymphoma Group and Groupe d’Etude des Lymphomes de l’Adulte Cohort Study. J Clin Oncol 2012; 30(3):291–9. doi: 10.1200/JCO.2011.37.1989.
32. Donnez J, Dolmans M-M. Fertility preservation in women. Nat Rev Endocrinol 2013; 9(12):735–49. doi: 10.1038/nrendo.2013.205.
33. Kim S-Y, Kim SK, Lee JR, Woodruff TK. Toward precision medicine for preserving fertility in cancer patients: existing and emerging fertility preservation options for women. J Gynecol Oncol 2016; 27(2):e22. doi: 10.3802/jgo.2016.27.e22.
34. Cakmak H, Rosen MP. Ovarian stimulation in cancer patients. Fertil Steril 2013; 99(6):1476–84. doi: 10.1016/j.fertnstert.2013.03.029.
35. Ernst EH, Offersen BV, Andersen CY, Ernst E. Legal termination of a pregnancy resulting from transplanted cryopreserved ovarian tissue due to cancer recurrence. J Assist Reprod Genet 2013; 30(7):975–8. doi: 10.1007/s10815-013-0026-x.
36. Smith MA, Edwards S, Canfell K. Impact of the National Cervical Screening Programme in New Zealand by age: analysis of cervical cancer trends 1985-2013 in all women and in Māori women. Cancer Causes Control 2017; 28(12):1393–404. doi: 10.1007/s10552-017-0967-y.
37. Moon E-K, Oh C-M, Won Y-J, Lee J-K, Jung K-W, Cho H et al. Trends and Age-Period-Cohort Effects on the Incidence and Mortality Rate of Cervical Cancer in Korea. Cancer Res Treat 2017; 49(2):526–33. doi: 10.4143/crt.2016.316.
38. Cai H-B, Liu X-M, Huang Y, Li X-N, Lie D-M, Zhou Q et al. Trends in cervical cancer in young women in Hubei, China. Int J Gynecol Cancer 2010; 20(7):1240–3. doi: 10.1111/igc.0b013e3181ecec79.
39. Mascarenhas-Melo F, Sereno J, Teixeira-Lemos E, Ribeiro S, Rocha-Pereira P, Cotterill E et al. Markers of increased cardiovascular risk in postmenopausal women: focus on oxidized-LDL and HDL subpopulations. Dis Markers 2013; 35(2):85–96. doi: 10.1155/2013/724706.
40. Clough KB, Goffinet F, Labib A, Renolleau C, Campana F, La Rochefordiere Ad et al. Laparoscopic unilateral ovarian transposition prior to irradiation: Prospective study of 20 cases. Cancer 1996; 77(12):2638–45. doi: 10.1002/(SICI)1097-0142(19960615)77:12<2638::AID-CNCR30>3.0.CO;2-R.
41. Fujiwara K, Mohri H, Yoshida T, Yamauchi H, Kohno I. Subcutaneous transposition of the ovary following hysterectomy. Int J Gynaecol Obstet 1997; 58(2):223–8. doi: 10.1016/s0020-7292(97)00087-8.
42. Cowles RA, Gewanter RM, Kandel JJ. Ovarian repositioning in pediatric cancer patients: Flexible techniques accommodate pelvic radiation fields. Pediatr Blood Cancer 2007; 49(3):339–41. doi: 10.1002/pbc.20652.
43. Chen H, Li J, Cui T. et al. Adjuvant gonadotropin-releasing hormone analogues for the prevention of chemotherapy induced premature ovarian failure in premenopausal women. Cochrane Database Syst Rev 2011; (11) CD008018
44. Bedaiwy MA, Abou-Setta AM, Desai N. et al. Gonadotropin-releasing hormone analog cotreatment for preservation of ovarian function during gonadotoxic chemotherapy: a systematic review and meta-analysis. Fertil Steril 2011; 95: 906 – 14.e1-906 – 14.e4
45. Moore HC, Unger JM, Phillips KA. et al. POEMS/S0230 Investigators. Goserelin for ovarian protection during breast-cancer adjuvant chemotherapy. N Engl J Med 2015; 372: 923-932
46. Gerber B, Ortmann O. Muss der Ovarschutz mit GnRHa nach dem ASCO 2014 neu bewertet werden?. Frauenarzt 2015; 56: 142-145
47. Lambertini M, Ceppi M, Poggio F. et al. Ovarian suppression using luteinizing hormone-releasing hormone agonists during chemotherapy to preserve ovarian function and fertility of breast cancer patients: a meta-analysis of randomized studies. Ann Oncol 2015; 26: 2408-2419
48. Lambertini M, Boni L, Michelotti A. et al. Ovarian suppression with triptorelin during adjuvant breast cancer chemotherapy and long-term ovarian function, pregnancies, and disease-free survival: a randomized clinical trial. JAMA 2015; 314: 2632-2640
49. Hsueh AJW, Kawamura K, Cheng Y, Fauser BCJM. Intraovarian control of early folliculogenesis. Endocr Rev 2015; 36(1):1–24. doi: 10.1210/er.2014- 1020.
50. Laronda MM, Jakus AE, Whelan KA, Wertheim JA, Shah RN, Woodruff TK. Initiation of puberty in mice following decellularized ovary transplant. Biomaterials 2015; 50:20–9. doi: 10.1016/j.biomaterials.2015.01.051.
51. Roness H, Kashi O, Meirow D. Prevention of chemotherapy-induced ovarian damage. Fertil Steril 2016; 105(1):20–9. doi: 10.1016/j.fertnstert.2015.11.043.
52. Köhn FM, Schroeder-Printzen I, Weidner W, Montag M, van der Ven H, Schill WB. Testicular sperm extraction in a patient with metachronous bilateral testicular cancer. Hum Reprod 2001; 16(11):2343–6. doi: 10.1093/humrep/16.11.2343.
53. Chan PTK, Palermo GD, Veeck LL, Rosenwaks Z, Schlegel PN. Testicular sperm extraction combined with intracytoplasmic sperm injection in the treatment of men with persistent azoospermia postchemotherapy. Cancer 2001; 92(6):1632–7. doi: 10.1002/1097-0142(20010915)92:6<1632::AID- CNCR1489>3.0.CO;2-I.
54. Ning L, Meng J, Goossens E, Lahoutte T, Marichal M, Tournaye H. In search of an efficient injection technique for future clinical application of spermatogonial stem cell transplantation: infusion of contrast dyes in isolated cadaveric human testes. Fertil Steril 2012; 98(6):1443-8.e1. doi: 10.1016/j.fertnstert.2012.08.023.
55. Beckmann Matthias W. et al: Concept Paper on the Technique of Cryopreservation, Removal and Transplantation of Ovarian Tissue for Fertility Preservation. Geburtshilfe Frauenheilkunde, Jan 2019; 79(1): 53-62