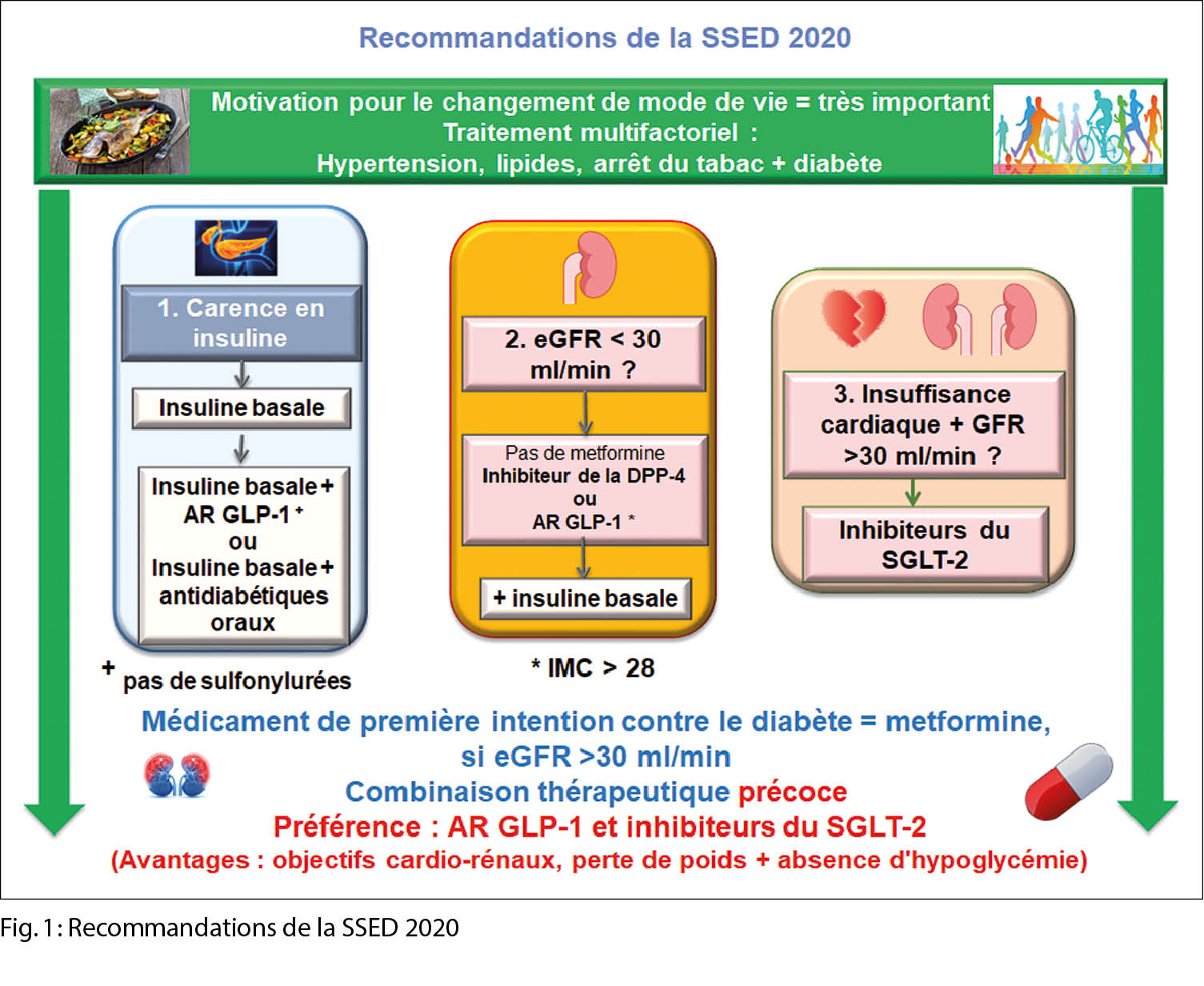
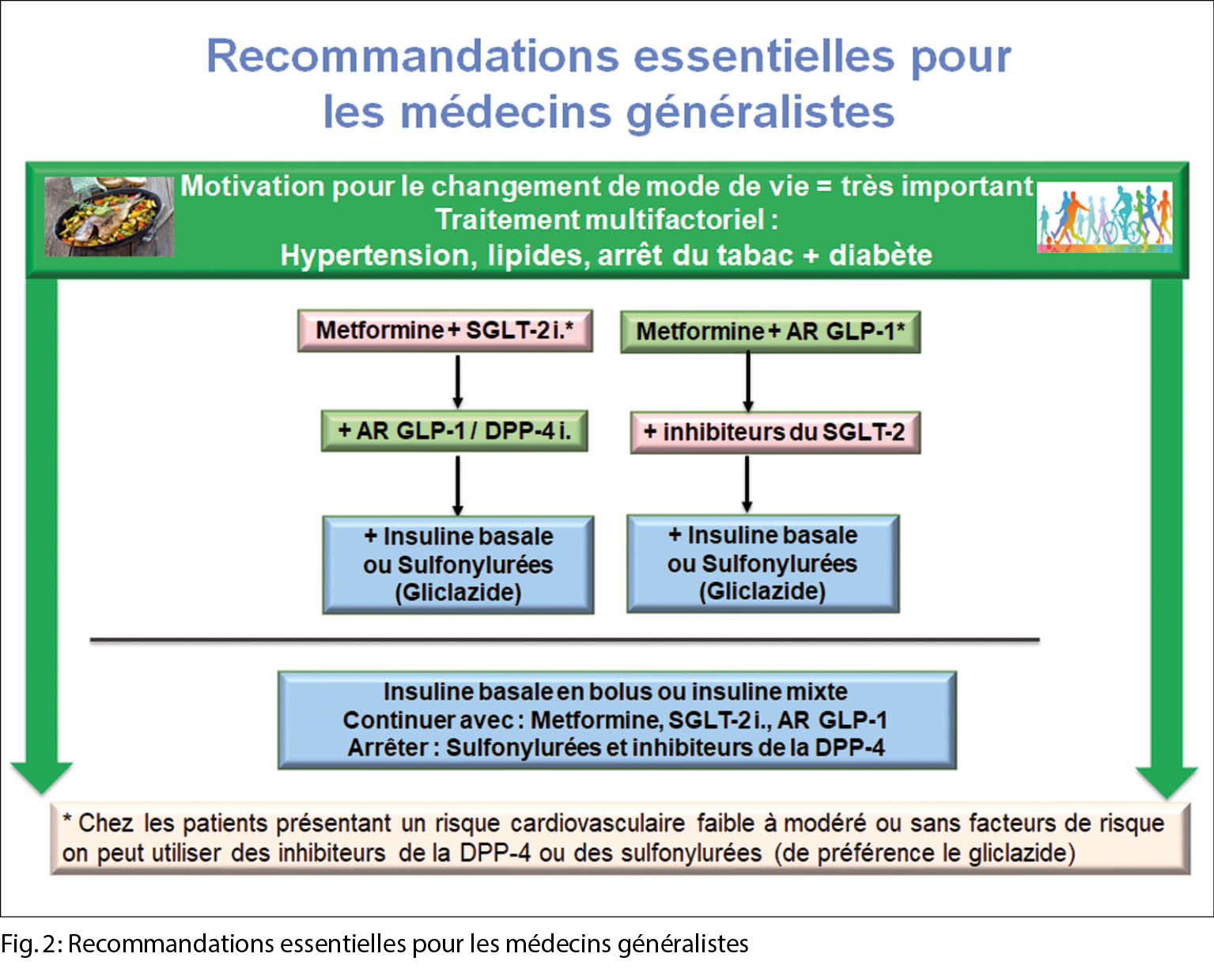
La Société Suisse d’ Endocrinologie et de Diabétologie (SSED/SGED) a été l’ une des premières sociétés professionnelles à publier des recommandations modernes qui se focalisent sur l’ état actuel des connaissances concernant le risque cardiovasculaire.
Depuis 2008, la Federal Drug Administration (FDA) exige des études cliniques sur la sécurité cardiovasculaire qui recourent à des critères de jugement (primaires et secondaires) significatifs. Cette évolution a conduit à des développements complètement nouveaux dans les recommandations de thérapie pour le diabète sucré de type 2. À ce jour, environ deux tiers des patients atteints de diabète sucré de type 2 meurent d’ un événement cardiovasculaire. En conséquence l’ accent sera mis à l’ avenir sur les thérapies qui réduisent le risque cardiovasculaire et préviennent les décès.
Actuellement, ces données existent pour les inhibiteurs du SGLT2 et pour les analogues du GLP-1, dont il a été démontré qu,ils réduisent le risque d’ événements cardiovasculaires ainsi que la mortalité et qu’ ils protègent les reins. Il a également été démontré que les inhibiteurs du SGLT2 ont un effet positif sur l’ insuffisance cardiaque et la progression de l’ insuffisance rénale. Les analogues du GLP1 ont en outre montré une réduction du risque d’ accident ischémique cérébral. Pour l’ empagliflozine, un inhibiteur du SGLT2, et le liraglutide, un analogue du GLP-1, une réduction de la mortalité totale a été démontrée. Une étude de comparaison directe des deux classes ou une étude des paramètres cardiovasculaires avec une combinaison des deux classes de substances n’ est pas disponible à ce jour. Néanmoins, il existe des analyses post hoc qui indiquent un effet additif sur l’ abaissement de l’ HbA1c, la réduction du poids et la réduction du risque cardiovasculaire (événements et mortalité).
La procédure dans la pratique quotidienne
Nous allons maintenant présenter systématiquement les aspects les plus importants du traitement du diabète sucré de type 2 sur la base des maladies et des facteurs de risque antérieurs. En outre, une modification notable du mode de vie (arrêt de la nicotine, exercice physique, etc.) est importante en tant que fondement de la thérapie. Les questions suivantes devraient servir d’ aide-mémoire dans la pratique quotidienne. En résumé, les figures 1 et 2 contiennent toutes les informations importantes.
1. Le patient a-t-il besoin d’ insuline ?
S’ il existe des indications d’ un état métabolique catabolique comme expression d’ une carence en insuline, telles que la perte de poids, la polyurie, la polydipsie ou si le taux d’ HbA1c est supérieur à 10 %, l’ insuline comme thérapie primaire n’ est jamais mauvaise. L’ insuline doit être administrée en cas de pancréatite chronique et/ou de cétonurie, vu qu’ il s’ agit d’ une situation de Redflag. Lors du choix de l’ insuline de base, une insuline à action ultra-lente (l’ insuline dégludec (Tresiba) ou l’ insuline glargine U300 (Toujeo)) est appropriée, étant donné que ce régime produit manifestement moins d’ hypoglycémies.
2. Dans quel état se trouve la fonction rénale ? Une question importante, puisque la plupart des produits thérapeutiques ne peuvent plus être admi-nistrés si le débit de filtration glomérulaire estimé (DFGe/eGFR) est au-dessous de 30ml/min :
eGFR 30-45ml/min
Selon le Compendium, les inhibiteurs du SGLT2 peuvent être administrés jusqu’ à un eGFR de 45 ml/min (sauf l’ ertugliflozine, qui ne peut être administrée que jusqu’ à un eGFR de 60 ml/min). Les données récentes des études sur les résultats cardiovasculaires avec l’ empagliflozine (Jardiance) et la canagliflozine (Invokana) montrent que les deux médicaments peuvent être prescrites sans danger jusqu’ à un eGFR de 30 ml/min. Les effets positifs en termes de paramètres cardiovasculaires ou de protection rénale demeurent, bien que l’ effet d’ abaissement de l’ HbA1c soit réduit.
Il n’ est plus recommandé de commencer une thérapie avec la metformine et si une thérapie établie existe déjà, une réduction de la dose à 1000 mg par jour est raisonnable.
eGFR < 30ml/min
Les agonistes du GLP1 (si l’ IMC > 28 kg/m2) peuvent être administrés jusqu’ à un eGFR de 15 ml/min, et même jusqu’ à la dialyse. Les inhibiteurs de la DPP4 sont sûrs, mais n’ ont aucun effet positif sur la mortalité globale. Pour la linagliptine (Trajenta), aucun ajustement de dose n’ est nécessaire, même si une dialyse est nécessaire. Pour la sitagliptine, un ajustement de la dose est nécessaire. Seule la saxagliptine a montré des indices d’ une augmentation du taux d’ insuffisance cardiaque. La metformine doit être arrêtée.
3. Le patient souffre-t-il d’ une insuffisance cardiaque ou faut-il la prévenir ?
Un inhibiteur du SGLT2 et la metformine devraient être établis car celui-ci peut réduire de manière significative le risque cardiovasculaire. Il existe de solides preuves d’ une insuffisance cardiaque avec fraction d’ éjection réduite (HFREF), cependant, un effet positif sur l’ insuffisance cardiaque avec fracture d’ éjection préservée (HFPEF), en tant que forme la plus courante chez les patients atteints de diabète sucré de type 2, est pathophysiologiquement probable. Même si tous les facteurs de risque sont ajustés de manière optimale pour le diabète sucré de type 2, le risque d’ insuffisance cardiaque est élevé de 45 %.
4. Quel est l’ objectif de l’ HbA1c ?
Chez les jeunes patients sans lésions secondaires des organes, le taux d’ HbA1c doit être réduit à 6,5 %, tant qu’ il n’ y a pas d’ hypoglycémie. Si le taux d’ HbA1c baisse en dessous de 6,5 %, par exemple lors d’ un traitement par un agoniste du GLP1, la thérapie ne doit pas être réduite à cause de cela.
Les personnes âgées et les patients polymorbides qui prennent de l’ insuline ou des sulfonylurées ne doivent pas présenter d’ hypoglycémie et un taux d’ HbA1c de < 8 % est approprié.
Source : Recommandations suisses de la Société d’ endocrinologie et de diabète (SGED/SSED) pour le traitement du diabète de type 2 (2019). Elles seront accessibles prochainement sur le site www.sgedssed.ch
Clinique d’endocrinologie, de diabétologie et nutrition clinique
Hôpital universitaire de Zurich
Rämistrasse 100
8091 Zurich
Roger.Lehmann@usz.ch
UniversitätsSpital Zürich
Rämistrasse 100
8091 Zurich
matthias.ernst@usz.ch
RL: Participation à des Advisory Boards et honoraires de conférencier de Novo Nordisk, Sanofi, MSD, Boehringer Ingelheim, Servier et Astra Zeneca. ME: Frais de voyage et de congrès de Novo Nordisk, Eli Lilly et Ipsen.