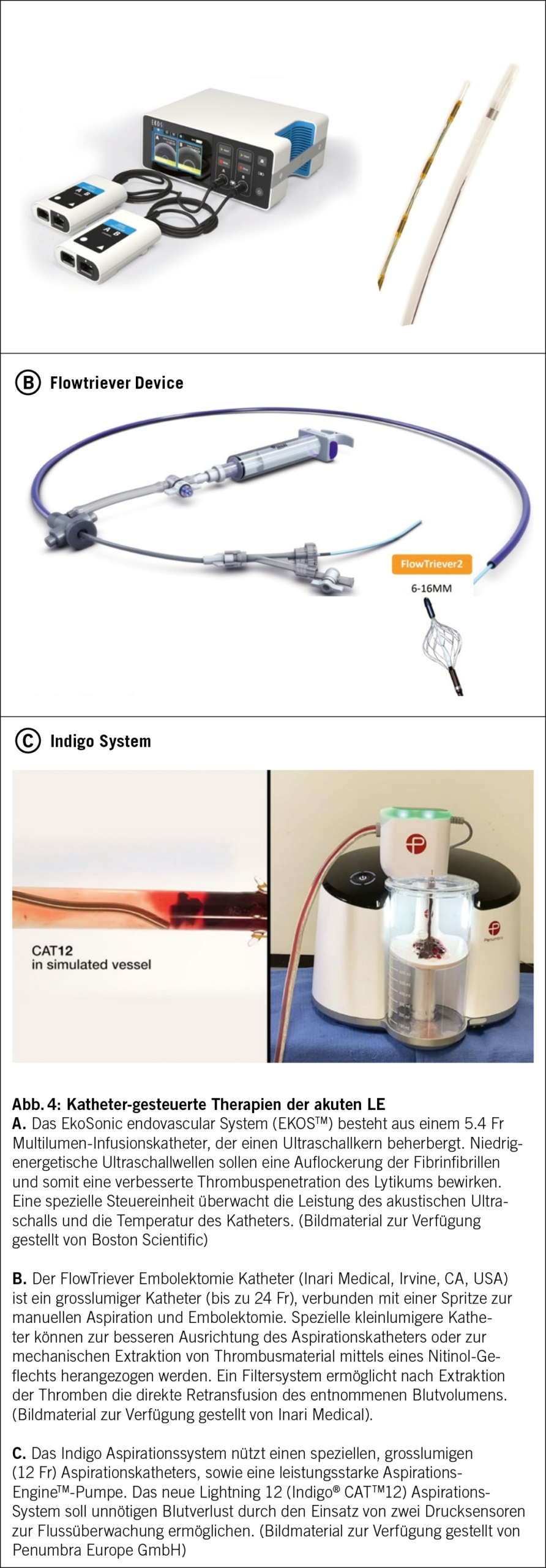
Die operative Therapie des Ovarialkarzinoms ist zentral und zielt auf eine vollständige Tumorentfernung ab, wobei die systematische Lymphadenektomie nach der LION-Studie bei unauffälligen Lymphknoten im CT und intraoperativ nicht mehr empfohlen wird. Ausser bei sehr frühen Karzinomen ist die adjuvante Chemotherapie Standard, meist in Form einer Kombination aus Carboplatin und Paclitaxel. Die Erhaltungstherapie mit Bevacizumab oder PARP-Inhibitoren wie Olaparib und Niraparib verlängert das progressionsfreie Überleben, insbesondere bei BRCA-mutierten und HRD-positiven Tumoren. Neoadjuvante Chemotherapie kann je nach Tumorausdehnung und Patientenzustand eine Alternative zur primären Operation sein. Bei Rezidiven werden erneute platinhaltige Chemotherapien oder Operationen individuell abgewogen. Bei einem platinsensitiven Rezidiv kann eine chirurgische Intervention im Kontext der rezidivierenden Erkrankung von Vorteil sein.
Surgical treatment is central to the management of ovarian cancer and aims for complete tumor resection. A systematic pelvic and paraaortic lymphadenectomy is no longer recommended for patients with normal lymph nodes based on the CT scan and intraoperative findinds, according to the LION study. Except for very early-stage cancers, adjuvant chemotherapy, typically a combination of carboplatin and paclitaxel, is standard. Maintenance therapy with Bevacizumab or PARP inhibitors like Olaparib and Niraparib extends progression-free survival, especially in BRCA-mutated and HRD-positive tumors. Neoadjuvant chemotherapy can be an alternative to primary surgery depending on tumor extent and patient condition. In cases of recurrence, repeat platinum-based chemotherapies or surgeries are considered on an individual basis. For platinum-sensitive recurrences, surgical intervention in the context of recurrent disease can be beneficial.
Keywords: Ovarian/ primary peritoneal/ tubal carcinoma – surgery – chemotherapy – maintenance therapy – PARP inhibitor
Das Ovarialkarzinom stellt eine der schwerwiegendsten gynäkologischen Krebserkrankungen dar und erfordert eine sorgfältige und umfassende Behandlungsstrategie. Die operative Therapie ist ein zentraler Bestandteil der Behandlung und zielt darauf ab, das gesamte sichtbare Tumorgewebe zu entfernen, um die Prognose der Patientinnen zu verbessern. Ergänzend zur Chirurgie sind systemische Therapien wie Chemotherapie und moderne Erhaltungstherapien entscheidend, um das Überleben zu verlängern und Rückfälle zu verhindern. Aktuelle klinische Studien haben zu signifikanten Veränderungen in der Behandlungsstrategie geführt, insbesondere hinsichtlich der Rolle der systematischen Lymphadenektomie und der Anwendung von PARP-Inhibitoren. Dieser Text bietet einen Überblick über die wesentlichen Aspekte der operativen und systemischen Therapie bei Ovarialkarzinom.
(Zur besseren Lesbarkeit sind Ovarial-, Tuben- und primäres Peritonealkarzinom im Text unter dem Oberbegriff «Ovarialkarzinom» zusammengefasst).
Erstdiagnose eines Ovarialkarzinoms
Operative Therapie bei Erstdiagnose:
Die operative Therapie spielt eine zentrale Rolle bei der Behandlung des Ovarialkarzinoms, da eine makroskopisch tumorfreie Resektion wesentlich für die Prognose der Patientinnen ist (1). Das Standardverfahren umfasst eine mediane Längs-Laparotomie mit Hysterektomie und bilateraler Adnexektomie sowie mindestens eine infrakolische Omentektomie, Peritonealbiopsien und die Entfernung allen weiteren tumorverdächtigen Gewebes, um eine makroskopisch tumorfreie Resektion zu erzielen (S3-Leitlinie Ovar).
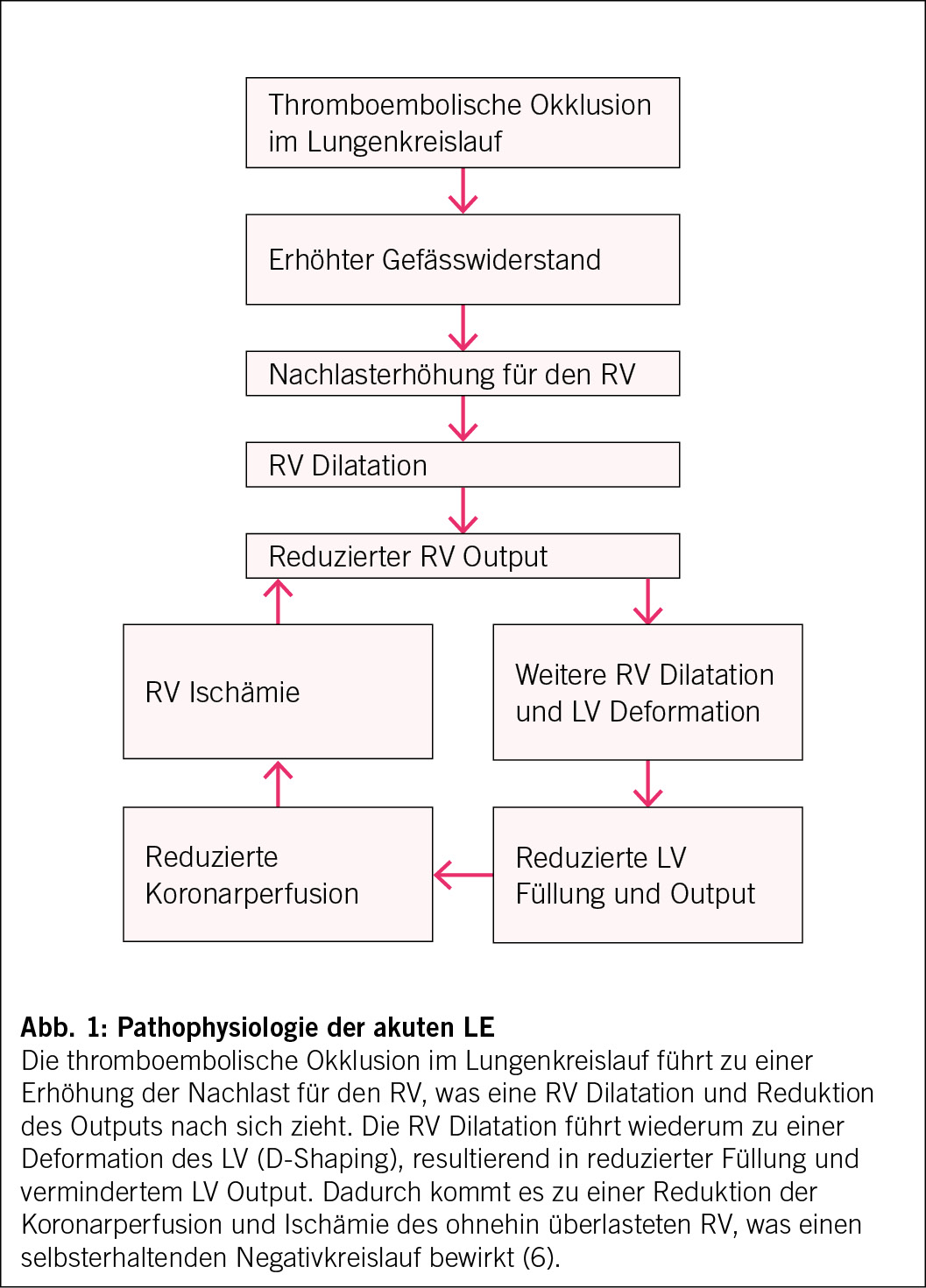
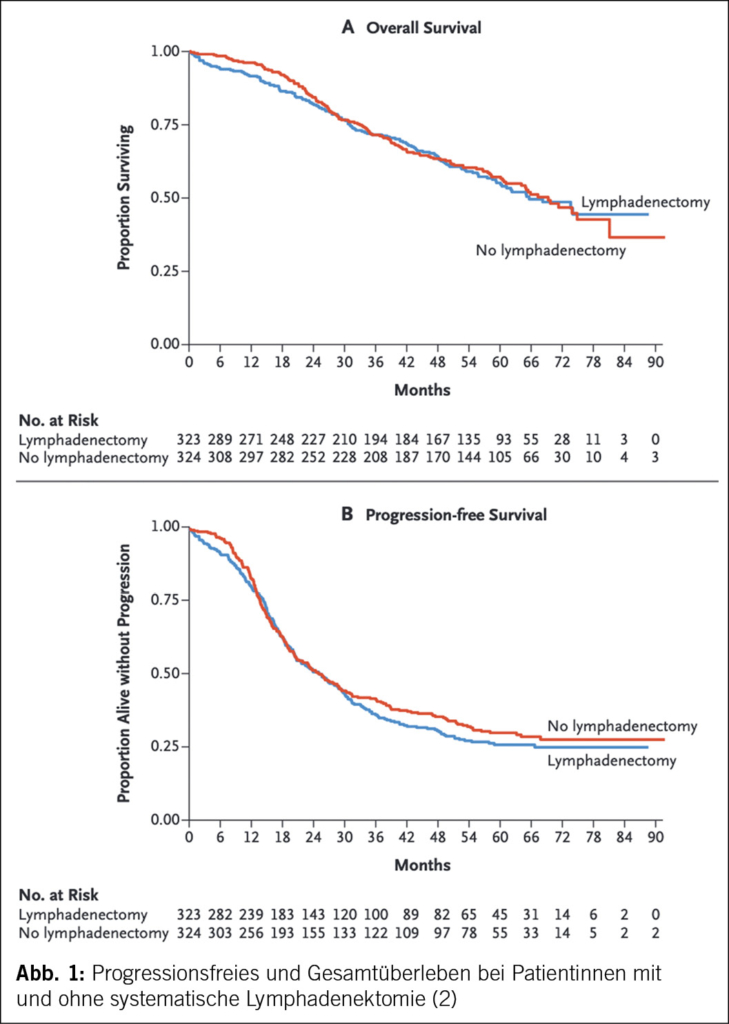
Bis 2017 war die systematische pelvine und paraaortale Lymphadenektomie ein fester Bestandteil der operativen Therapie des Ovarialkarzinoms. Die LION-Studie («Lymphadenectomy in Ovarian Neoplasms») führte jedoch zu einem Paradigmenwechsel ((2), S3-Leitlinie). Trotz der Tatsache, dass bei 55.7 % der Patientinnen mit Lymphadenektomie mikroskopische Lymphknotenmetastasen nachgewiesen wurden, gab es keinen Unterschied im Gesamt-(OS) oder progressionsfreien Überleben (PFS) (Abb. 1). Die Morbidität und die perioperative Mortalität waren in der Lymphadenektomie-Gruppe signifikant höher (2). Diese Ergebnisse haben dazu geführt, dass von einer systematischen Lymphadenektomie bei fortgeschrittenem high-grade serösen Ovarialkarzinom und unauffälligen Lymphknoten abgeraten wird (S3-Leitlinie).
Es ist wichtig zu betonen, dass bei der Diagnose eines fortgeschrittenen Ovarialkarzinoms üblicherweise eine adjuvante platinbasierte Chemotherapie indiziert ist, unabhängig von einem möglichen Tumorbefall der Lymphknoten ((1),S3-Leitlinie). Bei klinisch früh eingestuften Ovarialkarzinomen (FIGO I-IIA) hängt die Indikation für eine adjuvante Chemotherapie jedoch vom Nachweis positiver pelviner und/oder paraaortaler Lymphknoten ab. Bis zu 30 % dieser Patientinnen haben okkulte Lymphknotenmetastasen, was eine Höherklassifikation zu einem FIGO-Stadium III und damit eine Indikation für eine adjuvante Chemotherapie bedeutet (3). Daher bleibt die systematische pelvine und paraaortale Lymphadenektomie bei diesen Patientinnen empfohlen (Schmalfeldt et al. 2018, S3-Leitlinie).
Neoadjuvante Chemotherapie
Die grösste bisher veröffentlichte Studie zur neoadjuvanten Chemotherapie (NACT) zeigte keinen Unterschied im OS zwischen der Gruppe mit neoadjuvanter Chemotherapie für 3 Zyklen und Intervall-Debulking versus Patientinnen mit Primär-Debulking (PDS) und adjuvanter Kombinationstherapie für 6 Zyklen (HR 0,98; 90 %-KI 0,84–1,13; p= 0,01) (4). Eine Metaanalyse (5) ergab jedoch einen Vorteil für das primäre Debulking bei Patientinnen im FIGO-Stadium IIIC mit einer maximalen Tumorgrösse von < 5 cm (6). Auch retrospektive Studien zeigten teilweise einen Vorteil der PDS (7, 8).
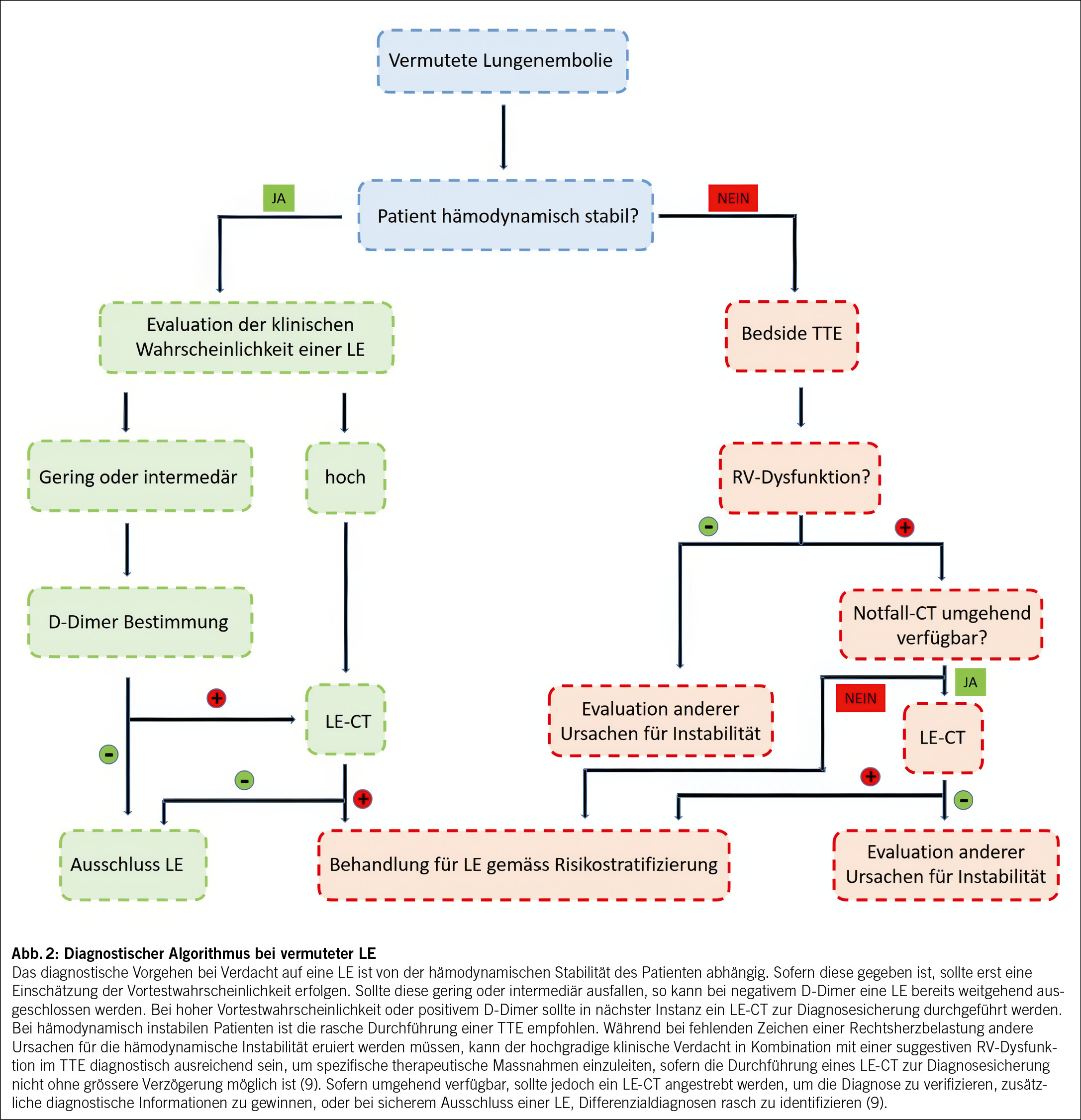
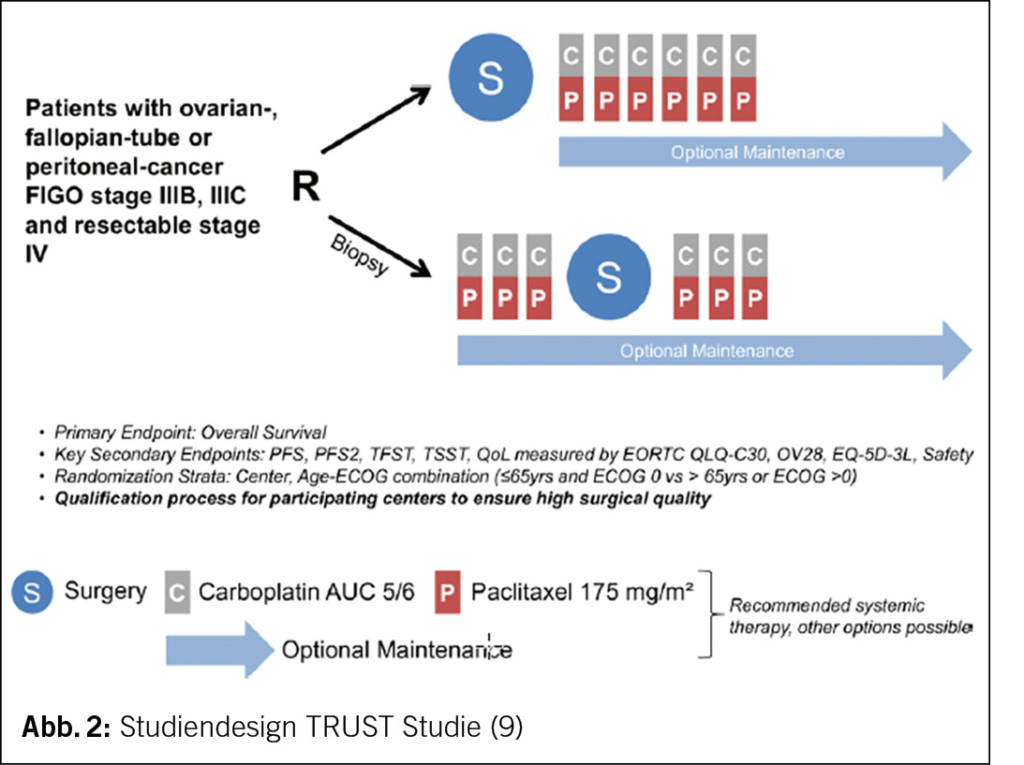
Die randomisierte, multizentrische TRUST-Studie («Trial of Radical Upfront Surgical Therapy in advanced ovarian cancer») untersucht das OS von Frauen mit epithelialem Ovarial-, Tuben- oder primärem Peritonealkarzinom FIGO IIIB-IVB bei primärer zytoreduktiver Operation versus neoadjuvanter Chemotherapie und Intervall-Debulking nach 3 Zyklen mit Carboplatin und Paclitaxel an Zentren mit hoher operativer Expertise (Abb. 2) (9). Ergebnisse werden dieses Jahr erwartet (9).
Adjuvante Systemtherapie
Frühes Ovarialkarzinom
Patientinnen mit frühem Ovarialkarzinom im Stadium FIGO IA G1 benötigen keine adjuvante Chemotherapie; in den Stadien IA G2 und IB G1/2 kann eine platinhaltige Chemotherapie diskutiert werden (3, 10). In den Stadien IC oder IA/ B G3 sollte eine platinhaltige (Mono-)Therapie gegeben werden (Verbesserung des 5-Jahres-OS von 75 % auf 82 % (3, 10).
Fortgeschrittenes Ovarialkarzinom
Seit den frühen 2000er Jahren hat sich die Kombination aus Carboplatin und Paclitaxel durchgesetzt (11). Die ICON-8-Studie konnte keine signifikanten Unterschiede zwischen verschiedenen Dosisdichten von Paclitaxel nachweisen, weshalb die dreiwöchentliche Kombination aus Carboplatin und Paclitaxel als Standard beibehalten wurde (12). Diese wird gegebenenfalls mit einer Erhaltungstherapie kombiniert.
Erhaltungstherapie
Antiangiogenetische Therapie
Bevacizumab, ein Angiogenesehemmer, wird in Kombination mit Chemotherapie und als Erhaltungstherapie eingesetzt. Phase-III-Studien wie GOG-0218 und ICON-7 haben gezeigt, dass Bevacizumab das PFS signifikant verlängert, besonders in Hochrisikogruppen (FIGO III und IV) (13, 14). In der Primärtherapie wird Bevacizumab zunächst mit Chemotherapie kombiniert und anschliessend als Erhaltungstherapie fortgeführt.
PARP-Inhibitoren in der Erhaltungstherapie bei Erstlinien- und Rezidivtherapie
PARP-Inhibitoren (PARPi) sind orale Medikamente, die die Reparatur von DNA-Einzelstrangbrüchen hemmen und dadurch Doppelstrangbrüche verursachen. Karzinomzellen, die nicht über die homologe Rekombinationsreparatur verfügen und somit eine homologe Rekombinations-Defizienz (HRD) aufweisen, können diese Brüche nicht richtig reparieren, was zu Chromosomenveränderungen und schliesslich zum Zelltod führt (15). Tests auf BRCA-Mutationen und HRD-Status sind zum Standard geworden, um Patientinnen zu identifizieren, die von PARPi profitieren können. Olaparib kann bei BRCA1/2-mutierten Ovarialkarzinomen eingesetzt werden (16) und die Kombination mit Bevacizumab ist bei HRD-positiven Tumoren möglich (17). Niraparib kann als Monotherapie verwendet werden (18). Veliparib und Rucaparib zeigten Vorteile in verschiedenen Studien (19, 20), sind jedoch in Europa noch nicht zugelassen.
Bei der Erhaltungstherapie mit einem PARPi bei einem Rezidiv war ein OS-Vorteil schwerer nachzuweisen, was teilweise auf PARPi-Crossover und eine lange Überlebenszeit nach Progression zurückzuführen sein könnte. Die Wahl des Medikaments sollte nach Nebenwirkungsprofil und Patientinnen-Präferenz erfolgen, da vergleichende Studien fehlen. Patientinnen, die unter PARPi progredient sind, haben meist nur geringen Nutzen von einer erneuten PARPi-Erhaltungstherapie (21). Es gibt bislang keine Daten zu einer gleichzeitigen Erhaltungstherapie mit Bevacizumab und Olaparib bei einem Rezidiv.
Bei Patientinnen mit platinsensiblem Rezidiv eines BRCA-mutierten high-grade Ovarialkarzinoms nach zwei oder mehr platinhaltigen Vortherapien kann eine Monotherapie mit Rucaparib eine Option sein (22).
Rezidiv
Operation beim Rezidiv
Ein erheblicher Anteil der Patientinnen mit fortgeschrittenem Ovarialkarzinom entwickelt ein Rezidiv. Die DESKTOP III-Studie definierte prädiktive Parameter, um geeignete Patientinnen für eine erneute Operation zu identifizieren. Diese beinhalten Patientinnen mit einem ersten platin-sensitiven Rezidiv, einem ECOG-Performance-Status von 0, Aszites ≤ 500 ml und einer makroskopischen Komplettresektion bei der Erstoperation (du Bois et al. 2020).
Chemotherapie beim Rezidiv
Bei Rezidiven des epithelialen Ovarialkarzinoms sollte bei Patientinnen zunächst evaluiert werden, ob sie für eine platinhaltige Therapie geeignet sind (früher «platinsensibel» oder «platinresistent»). Bei frühem Rezidiv (< 6 Monate nach Abschluss der adjuvanten Systemtherapie) haben Mono-Chemotherapien mit Topotecan, Gemcitabin, Paclitaxel oder pegyliertes liposomales Doxorubicin bessere Verträglichkeit und vergleichbare Effektivität gezeigt (23). Die Optimierung der Lebensqualität ist besonders wichtig (24). Mirvetuximab Soravtansin zeigte in der Phase-III-Studie MI-RASOL einen signifikanten PFS- und OS-Vorteil sowie ein besseres Sicherheitsprofil im Vergleich zur Chemotherapie (25).
Bei einem Rezidiv > 6 Monate nach der letzten Platintherapie wird in der Regel eine erneute platinhaltige Kombinationschemotherapie durchgeführt. Vor Beginn der Rezidivtherapie sollte die Möglichkeit einer Rezidivoperation geprüft werden. Bevorzugtes Regime beim Rezidiv sind Kombinationen aus Carboplatin und pegyliertem liposomalem Doxorubicin oder Carboplatin und Gemcitabin (26).
Antiangiogenetische Therapie beim Rezidiv
Bevacizumab kann in der Rezidivtherapie bei Patientinnen, die bisher kein Bevacizumab erhalten haben, in Kombination mit einer Monochemotherapie das PFS signifikant verlängern (27). Es kann auch off-label zur Reduktion der Aszitesbildung beitragen (27).
Spezielle Situationen
Low-grade seröses Ovarialkarzinom
Für Patientinnen im FIGO-Stadium IC bis IIA wird eine Monotherapie mit Carboplatin empfohlen, während ab Stadium IIB eine Kombinationstherapie aus Carboplatin und Paclitaxel eingesetzt werden sollte (Ansprechrate unter 25 %) (24). In retrospektiven Studien konnte gezeigt werden, dass eine endokrine Erhaltungstherapie das PFS verdoppeln kann (28). Die MATAO-Studie untersucht den Effekt von Letrozol versus Placebo nach Chemotherapie bei hormonrezeptorpositiven Patientinnen prospektiv (29).
Die Behandlung mit dem MEK-Inhibitor Trametinib zeigte in einer Phase-II/III-Studie ein signifikant längeres PFS als die Standardtherapie (HR 0.48, p<0.0001) und bietet eine neue Behandlungsoption für Patienten mit Rezidiv eines low-grade serösen Karzinoms (30).
Die Wirksamkeit von Bevacizumab bei low-grade serösen Ovarialkarzinomen ist unklar (14).
Hypertherme intraperitoneale Chemotherapie (HIPEC)
Die erste Phase-III-Studie zur HIPEC bei Ovarialkarzinom-Patientinnen nach neoadjuvanter Chemotherapie zeigte eine signifikante Verbesserung des rückfallfreien Überlebens (HR 0.66, p=0.003) und des OS im Vergleich zur Standardtherapie, jedoch mit ähnlichen Raten schwerer Nebenwirkungen. Die Studie wirft jedoch erhebliche methodische Fragen auf. Aktuell wird HIPEC nicht als Standardtherapie empfohlen und sollte nur in kontrollierten Studien verwendet werden (31).
Abkürzungen
AGO Arbeitsgemeinschaft Gynäkologische Onkologie
ECOG Eastern European Cooperative Oncology Group
FIGO Fédération Internationale de la Gynécologie et d’Obstétrique
HIPEC Hypertherme intraperitoneale Chemotherapie
HRD Homologe Rekombinations-Defizienz
MEK Mitogen-aktivierte Proteinkinase
NACT Neoadjuvante Chemotherapie
OS Gesamtüberleben
PARP Poly(ADP-ribose)-Polymerasen
PARPi PARP-Inhibitoren
PDS primary debulking surgery, primäre Debulking-Operation
PFS progressionsfreies Überleben
Copyright Aerzteverlag medinfo AG
Zweitabdruck aus info@gynäkologie 06/2024
Luzerner Kantonsspital
Frauenklinik
Spitalstrasse
6000 Luzern 16
Luzerner Kantonsspital
Frauenklinik
Spitalstrasse
6000 Luzern 16
Luzerner Kantonsspital
Frauenklinik
Spitalstrasse
6000 Luzern 16
Die Autorenschaft hat keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
1. du Bois, A., et al., Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: a combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: by the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d’Investigateurs Nationaux Pour les Etudes des Cancers de l’Ovaire (GINECO). Cancer, 2009. 115(6): p. 1234-44.
2. Harter, P., et al., A Randomized Trial of Lymphadenectomy in Patients with Advanced Ovarian Neoplasms. N Engl J Med, 2019. 380(9): p. 822-832.
3. Trimbos, B., et al., Surgical staging and treatment of early ovarian cancer: long-term analysis from a randomized trial. J Natl Cancer Inst, 2010. 102(13): p. 982-7.
4. Vergote, I., et al., Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N Engl J Med, 2010. 363(10): p. 943-53.
5. Kehoe, S., et al., Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): an open-label, randomised, controlled, non-inferiority trial. Lancet, 2015. 386(9990): p. 249-57.
6. Vergote, I., et al., Neoadjuvant chemotherapy versus debulking surgery in advanced tubo-ovarian cancers: pooled analysis of individual patient data from the EORTC 55971 and CHORUS trials. Lancet Oncol, 2018. 19(12): p. 1680-1687.
7. Sorensen, S.M., et al., Residual tumor and primary debulking surgery vs interval debulking surgery in stage IV epithelial ovarian cancer. Acta Obstet Gynecol Scand, 2022. 101(3): p. 334-343.
8. Rauh-Hain, J.A., et al., Primary debulking surgery versus neoadjuvant chemotherapy in stage IV ovarian cancer. Ann Surg Oncol, 2012. 19(3): p. 959-65.
9. Reuss, A., et al., TRUST: Trial of Radical Upfront Surgical Therapy in advanced ovarian cancer (ENGOT ov33/AGO-OVAR OP7). Int J Gynecol Cancer, 2019. 29(8): p. 1327-1331.
10. Trimbos, J.B., et al., Impact of adjuvant chemotherapy and surgical staging in early-stage ovarian carcinoma: European Organisation for Research and Treatment of Cancer-Adjuvant ChemoTherapy in Ovarian Neoplasm trial. J Natl Cancer Inst, 2003. 95(2): p. 113-25.
11. du Bois, A., et al., A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J Natl Cancer Inst, 2003. 95(17): p. 1320-9.
12. Blagden, S.P., et al., Weekly platinum-based chemotherapy versus 3-weekly platinum-based chemotherapy for newly diagnosed ovarian cancer (ICON8): quality-of-life results of a phase 3, randomised, controlled trial. Lancet Oncol, 2020. 21(7): p. 969-977.
13. Tewari, K.S., et al., Final Overall Survival of a Randomized Trial of Bevacizumab for Primary Treatment of Ovarian Cancer. J Clin Oncol, 2019. 37(26): p. 2317-2328.
14. Oza, A.M., et al., Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): overall survival results of a phase 3 randomised trial. The Lancet Oncology, 2015. 16(8): p. 928-936.
15. Lord, C.J. and A. Ashworth, PARP inhibitors: Synthetic lethality in the clinic. Science, 2017. 355(6330): p. 1152-1158.
16. Moore, K., et al., Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med, 2018. 379(26): p. 2495-2505.
17. Ray-Coquard, I., et al., Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N Engl J Med, 2019. 381(25): p. 2416-2428.
18. Gonzalez-Martin, A., et al., Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med, 2019. 381(25): p. 2391-2402.
19. Coleman, R.L., et al., Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N Engl J Med, 2019. 381(25): p. 2403-2415.
20. Monk, B.J., et al., A Randomized, Phase III Trial to Evaluate Rucaparib Monotherapy as Maintenance Treatment in Patients With Newly Diagnosed Ovarian Cancer (ATHENA-MONO/GOG-3020/ENGOT-ov45). J Clin Oncol, 2022. 40(34): p. 3952-3964.
21. Pujade-Lauraine, E., et al., Maintenance olaparib rechallenge in patients with platinum-sensitive relapsed ovarian cancer previously treated with a PARP inhibitor (OReO/ENGOT-ov38): a phase IIIb trial. Ann Oncol, 2023. 34(12): p. 1152-1164.
22. Kristeleit, R., et al., Rucaparib versus standard-of-care chemotherapy in patients with relapsed ovarian cancer and a deleterious BRCA1 or BRCA2 mutation (ARIEL4): an international, open-label, randomised, phase 3 trial. Lancet Oncol, 2022. 23(4): p. 465-478.
23. Gordon, A.N., et al., Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol, 2001. 19(14): p. 3312-22.
24. Colombo, N., et al., ESMO-ESGO consensus conference recommendations on ovarian cancer: pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease†. Ann Oncol, 2019. 30(5): p. 672-705.
25. Moore, K.N., et al., Phase III MIRASOL (GOG 3045/ENGOT-ov55) study: Initial report of mirvetuximab soravtansine vs. investigator’s choice of chemotherapy in platinum-resistant, advanced high-grade epithelial ovarian, primary peritoneal, or fallopian tube cancers with high folate receptor-alpha expression. Journal of Clinical Oncology, 2023. 41(17_suppl): p. LBA5507-LBA5507.
26. Wagner, U., et al., Final overall survival results of phase III GCIG CALYPSO trial of pegylated liposomal doxorubicin and carboplatin vs paclitaxel and carboplatin in platinum-sensitive ovarian cancer patients. Br J Cancer, 2012. 107(4): p. 588-91.
27. Pujade-Lauraine, E., et al., Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. J Clin Oncol, 2014. 32(13): p. 1302-8.
28. Gershenson, D.M., et al., Hormonal Maintenance Therapy for Women With Low-Grade Serous Cancer of the Ovary or Peritoneum. J Clin Oncol, 2017. 35(10): p. 1103-1111.
29. Heinzelmann-Schwarz, V.A., et al., ENGOT-ov54/Swiss-GO-2/MATAO including LOGOS (Low-Grade Ovarian cancer Sub-study): MAintenance Therapy with Aromatase inhibitor in epithelial Ovarian cancer—A randomized, double-blinded, placebo-controlled, multicenter phase III Trial. Journal of Clinical Oncology, 2021. 39(15_suppl): p. TPS5598-TPS5598.
30. Gershenson, D.M., et al., Trametinib versus standard of care in patients with recurrent low-grade serous ovarian cancer (GOG 281/LOGS): an international, randomised, open-label, multicentre, phase 2/3 trial. Lancet, 2022. 399(10324): p. 541-553.
31. van Driel, W.J., et al., Hyperthermic Intraperitoneal Chemotherapy in Ovarian Cancer. N Engl J Med, 2018. 378(3): p. 230-240.
Separat im Text gelistet:
1. Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF): S3-Leitlinie Diagnostik, Therapie und Nachsorge maligner Ovarialtumoren, Langversion 4.0, 2020, AWMF-Registernummer: 032/035OL, https://www.leitlinienprogramm-onkologie.de/leitlinien/ovarialkarzinom/, (abgerufen am: 30.06.2024).
2. Schmalfeldt B et al. – für die Kommission Ovar der AGO: Wie ist die Evidenz für die Lym-phonodektomie beim frühen Ovarialkarzinom? Der Frauenarzt 2018;59(10):751-753.
3. du Bois A et al.: Randomized controlled phase III study evaluating the impact of secondary cytoreductive surgery in recurrent ovarian cancer: The final analysis of AGO DESKTOP III/ENGOT ov20. ASCO 2020, Abstr. #6000.