Die Nuklearkardiologie, die SPECT und PET einsetzt, bietet wichtige Instrumente für die Diagnose von koronarer Herzerkrankung, Amyloidose und Sarkoidose. Sie ermöglicht eine präzise Beurteilung von Myokarddurchblütungsstörungen, Myokardablagerungen und Entzündungsprozessen. Die Fortschritte in der Bildgebung und Quantifizierung bieten Internisten und Kardiologen wertvolle Erkenntnisse für die Risikostratifizierung, Therapieplanung und Überwachung komplexer kardiovaskulärer Erkrankungen.
Nuclear cardiology, utilizing SPECT and PET, provides essential tools for diagnosing coronary artery disease, amyloidosis, and sarcoidosis. It enables precise assessment of perfusion abnormalities, myocardial deposits, and inflammatory processes. Advances in imaging and quantification offer internists and cardiologists valuable insights for risk stratification, therapy planning, and monitoring in complex cardiovascular conditions. Keywords: Nuklearkardiologie; PET Perfusion; Skelettszintigraphie; Inflammatorische Kardiomyopathie
Einführung
Die Nuklearkardiologie ist ein zentrales Feld der Herz-Kreislauf-Diagnostik und umfasst hochentwickelte Verfahren wie die Single-Photon-Emissions-Computertomographie (SPECT) und die Positronen-Emissions-Tomographie (PET). Diese ermöglichen die präzise Erkennung von Durchblutungsstörungen bei der koronaren Herzerkrankung (KHK), die nicht-invasive Diagnose der ATTR-Amyloidose und die Diagnose inflammatorischer Kardiomyopathien wie bei der kardialen Sarkoidose. Durch die Kombination funktioneller und anatomischer Informationen leisten diese Techniken einen entscheidenden Beitrag zu präziser Diagnostik, individualisierter Therapieplanung und Risikostratifizierung, was sie zu einem unverzichtbaren Bestandteil der kardiologischen Versorgung machen. Auf den folgenden Seiten finden Sie eine Übersicht über den aktuellen Stand der nuklearkardiologischen Methoden.
Koronare Herzerkrankung
Die KHK wird als pathologischer Prozess definiert, der durch die Ansammlung atherosklerotischer Plaques in den epikardialen Herzkranzarterien, unabhängig von deren obstruktivem oder nicht-obstruktivem Charakter, gekennzeichnet ist (1). Chronische Koronarsyndrome umfassen klinische Präsentationen, die durch strukturelle oder funktionelle Veränderungen der Koronararterien oder Mikrozirkulation eine Diskrepanz zwischen myokardialem Sauerstoffbedarf und Blutversorgung verursachen können. Dies führt zu ischämischen Durchblutungsstörungen, die sich typischerweise, aber nicht ausschliesslich, als Angina pectoris oder Dyspnoe äussern. Zwei etablierte Verfahren zur KHK-Diagnostik sind die Myokardsperfusion-SPECT und -PET. Beide ermöglichen die nicht-invasive Erkennung von Durchblutungsstörungen. SPECT verwendet Gamma-strahlende Radionuklide wie 99mTechnetium mit Tracern wie Sestamibi oder Tetrofosmin. PET nutzt β+-strahlende Radionuklide wie 13N-Ammoniak oder 82Rubidium, die kürzere Halbwertszeiten haben und den Einsatz von Zyklotronen oder Generatoren erfordern.
Ablauf
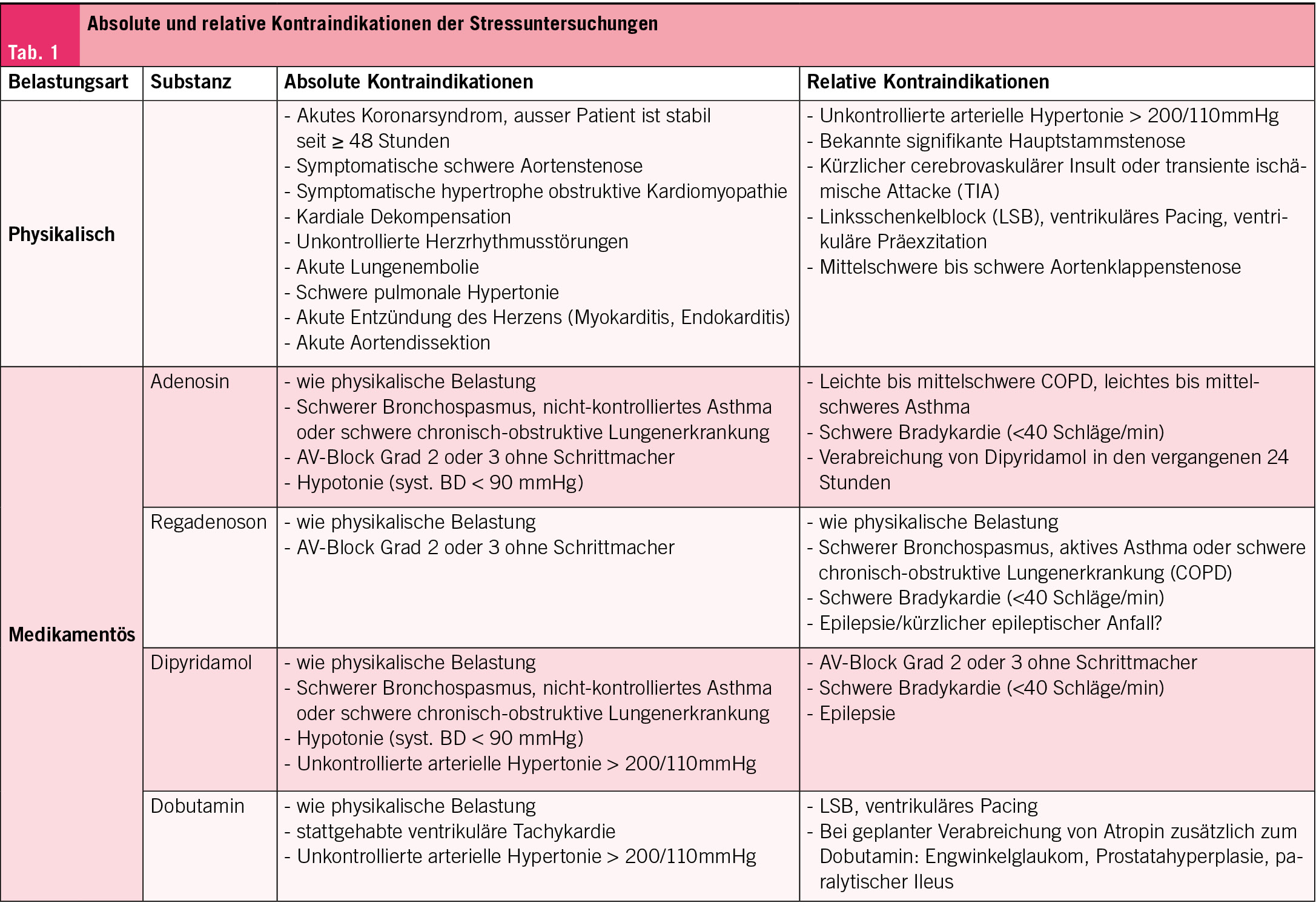
Zur Durchführung nuklearmedizinischer Myokardperfusionsuntersuchungen mittels SPECT oder PET müssen Patienten mindestens sechs Stunden nüchtern bleiben und koffeinhaltige Getränke wie Kaffee oder Tee mindestens zwölf Stunden meiden, da diese die Wirkung von Stressmitteln wie Regadenoson und Adenosin beeinträchtigen können. Myokardperfusionsuntersuchungen bestehen aus einer Ruhe- und einer Stressphase. Bei der PET-Myokardsperfusion wird die Ruheaufnahme typischerweise vor der Stressaufnahme durchgeführt, was durch die kurzen Halbwertszeiten der Radiotracer eine schnelle Testabfolge ermöglicht. Die Stressinduktion erfolgt fast ausschliesslich medikamentös, wobei die Wahl des Stressmittels von individuellen Kontraindikationen abhängt (Tab. 1). SPECT-Untersuchungen nutzen aufgrund der längeren Halbwertszeit von 99mTechnetium oft körperliche Belastung mittels Ergometrie. Hierbei ist ein Zeitabstand von mindestens zwei Stunden zwischen den Phasen erforderlich, wodurch flexible Protokolle (z. B. Ruhe-Stress, Stress-Ruhe oder «Stress-nur») möglich sind. Das «Stress-nur»-Protokoll reduziert die Untersuchungsdauer erheblich und verringert die Strahlenbelastung um über 50 % (2).
Beurteilung
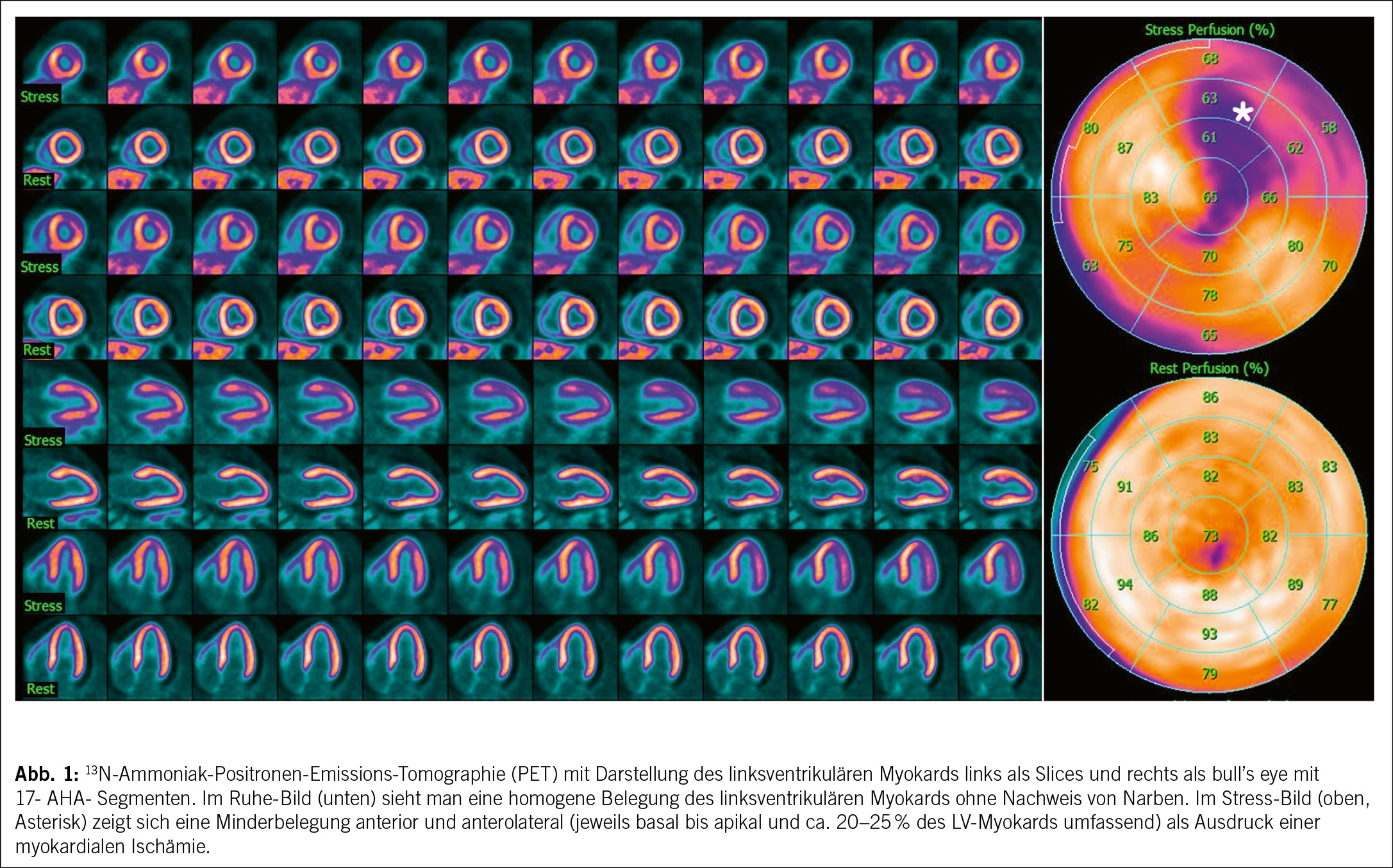
Ziel der Myokardperfusionsuntersuchungen ist es, physiologische und/oder verminderte Myokardperfusion im Sinne von Ischämien oder Narben zu diagnostizieren sowie deren Schweregrad zu bewerten. Narbengewebe, das durch den Verlust funktionsfähiger Myozyten entsteht, zeigt fixierte Perfusionsdefizite in Ruhe und unter Belastung, da keine Radionuklidanreicherung mehr erfolgt. Ischämisches Myokardgewebe, das in Ruhe noch ausreichend durchblutet ist, kann unter Stress (entweder medikamentös oder durch körperliche Belastung) ein relatives Perfusionsdefizit zeigen, wenn eine hämodynamisch relevante Stenose den Blutfluss begrenzt. PET- und SPECT-Untersuchungen erlauben nicht nur eine qualitative Beurteilung der Myokardperfusion, sondern – insbesondere bei der PET – auch eine absolute Quantifizierung des myokardialen Blutflusses (in Millilitern pro Minute pro Gramm Myokard) sowohl unter Stress als auch in Ruhe. Diese erfolgt
mittels dynamischer Akquisition während der Radiotracer-Anflutungsphase und ist bei der PET inzwischen Standard, während sie bei der SPECT nur in spezialisierten Zentren verfügbar ist.
Das Verhältnis des myokardialen Blutflusses unter Belastung und in Ruhe ist die myokardiale Flussreserve (MFR), die eine zentrale diagnostische und prognostische Rolle spielt. Sie spiegelt die Funktion der epikardialen Koronararterien sowie der Mikrozirkulation wider und erlaubt eine umfassende Bewertung der koronaren Gesundheit. Patienten mit einer normalen MFR (> 2) haben eine ausgezeichnete Prognose mit einer Event-Rate von weniger als 1 % pro Jahr. Zusätzlich, dank der Blutfluss-Quantifizierung, ist PET die einzige klinisch genutzte Modalität, die eine nicht-invasive Diagnose der mikrovaskulären Dysfunktion ermöglicht. Prognostisch entscheidend ist zudem die sogenannte Ischämie-Last: Ab einer Ischämie von 10-15 % gilt das kardiovaskuläre Risiko gemäss Leitlinien als erhöht, was therapeutische Konsequenzen nach sich ziehen kann. Ein weiterer Risikomarker ist der Kalzium-Score, der mithilfe einer nicht-kontrastverstärkten Low-Dose-CT zur Attenuierungskorrektur bestimmt wird. Solche CT-Scans werden immer bei PET durchgeführt und sind auch für SPECT-Untersuchungen empfohlen. Sie ermöglichen eine zusätzliche Ermittlung des Agatston-Scores zur Beurteilung der koronaren Kalziumlast und liefern wesentliche Informationen zur Risikostratifizierung, selbst bei fehlenden flusslimitierenden Koronarstenosen.
Die Wahl zwischen PET und SPECT richtet sich nach den individuellen Anforderungen der Diagnostik. PET bietet durch höhere räumliche und zeitliche Auflösung sowie die Möglichkeit der quantitativen Blutflussmessung deutliche Vorteile, insbesondere bei diffuser KHK oder mikrovaskulären Störungen. Zudem ist die Strahlenbelastung bei PET geringer (1–2 mSv) im Vergleich zur SPECT (ca. 5 mSv). Die SPECT hingegen punktet mit flexiblen Protokollen, der Möglichkeit einer physischen Belastung und modernen Kameras, die auch Patienten mit Platzangst mehr Komfort bieten. Beide Verfahren ergänzen sich in der nuklearmedizinischen Diagnostik und erlauben eine präzise Beurteilung von Ischämien und Narben sowie eine umfassende Risikobewertung. Ihre Kombination mit innovativen Technologien wie Low-Dose-CT und dynamischer Akquisition eröffnet weitere Möglichkeiten in der personalisierten Diagnostik und Therapieplanung bei Patienten mit koronarer Herzerkrankung (3) (Abb. 1).
Zukunft
Die nuklearkardiologische Bildgebung erlebt derzeit bedeutende Fortschritte, insbesondere in der Diagnostik der koronaren Herzerkrankung. Ein Meilenstein ist die Einführung des neuen PET-Radiotracers 18F-Flurpiridaz, der in zwei grossen klinischen Studien untersucht und kürzlich von der U.S. Food and Drug Administration (FDA) zugelassen wurde. Die Studienergebnisse belegen, dass 18F-Flurpiridaz eine überlegene diagnostische Genauigkeit im Vergleich zu herkömmlichen Radiotracern wie 99mTechnetium aufweist. Insbesondere bei der Detektion von koronaren Stenosen zeigt 18F-Flurpiridaz PET eine deutlich höhere Sensitivität und überlegene Bildqualität im Vergleich zur SPECT-Bildgebung. 18F-Flurpiridaz hat zudem eine längere Halbwertszeit als andere PET-Tracer, was die Verteilung und den Einsatz in mehr Instituten ermöglicht. Dank dieser Eigenschaften könnte der Tracer zukünftig auch bei Belastungstests mit physikalischer Belastung eingesetzt werden, was bisher mit anderen PET-Tracern nicht möglich war. Auch im Bereich der SPECT-Bildgebung gibt es bedeutende Fortschritte. Neue SPECT-Kameras bieten die Möglichkeit, dynamische Akquisitionen durchzuführen, was eine Quantifizierung des myokardialen Blutflusses erlaubt. Dies könnte die diagnostische Genauigkeit der SPECT in den kommenden Jahren erheblich steigern und die Rolle dieser Technologie weiter festigen.
Amyloidose
Die Amyloidose ist eine Erkrankung, bei der gestörter Proteinstoffwechsel zu Ablagerungen von Amyloidfibrillen im extrazellulären Raum führt, was die Funktion betroffener Organe beeinträchtigt. Bei kardialer Beteiligung erhöhen die Ablagerungen zwischen Myofibrillen die Myokardsteifigkeit und verursachen eine diastolische Dysfunktion. Die häufigsten Formen sind die AL-Amyloidose, verursacht durch Plasmazellmyelome, und die Transthyretin (ATTR)-Amyloidose, die durch Ablagerungen von Transthyretin entsteht, einem Protein aus der Leber. Die 99mTc-Skelettszintigraphie hat sich als nicht-invasive Methode mit hoher diagnostischer Genauigkeit zur Diagnose der ATTR-Amyloidose etabliert und kann heutzutage die invasive Endomyokardbiopsie ersetzen. Die Diagnostik erfolgt schrittweise: Bei Verdacht auf Amyloidose werden Serum-Leichtketten und Protein-Immunfixation in Serum und Urin durchgeführt. Ein positiver Befund erfordert hämatologische Abklärung. Bei negativem Ergebnis wird eine Skelettszintigraphie oder SPECT veranlasst, die eine Sensitivität von 92 % und eine Spezifität von 95 % für die ATTR-Amyloidose aufweist (4, 5).
Ablauf
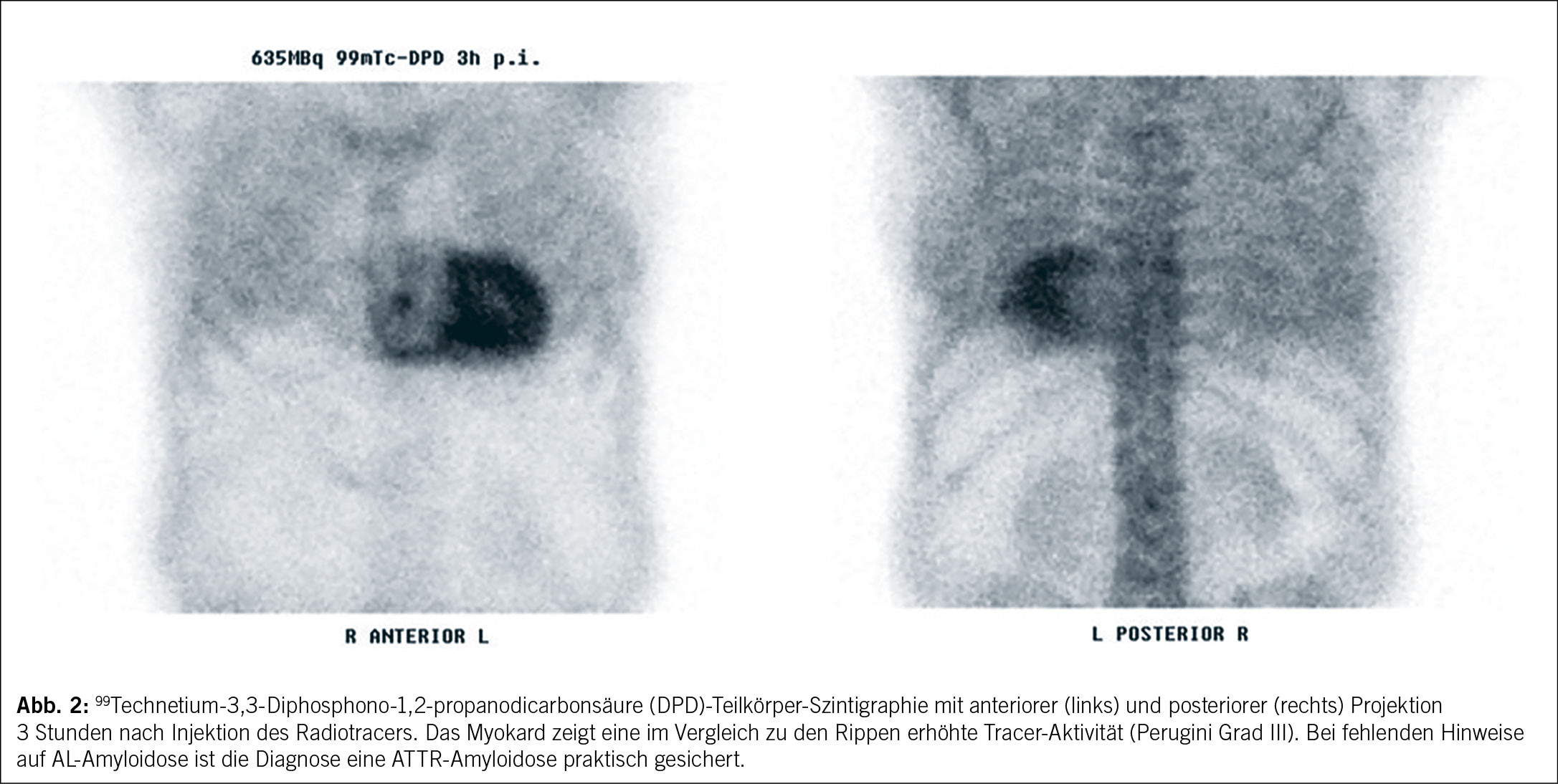
In der Skelettszintigraphie zur ATTR-Diagnostik werden knochenaffine Biphosphonate wie 3,3-Diphosphono-1,2-propanodicarbonsäure (DPD) verwendet. Diese Substanzen binden spezifisch an Amyloidfibrillen, wobei der Bindungsmechanismus nicht vollständig geklärt ist. Nach intravenöser Gabe des Radiotracers erfolgt eine Wartezeit von 3 Stunden, bevor biplanare oder Ganzkörperaufnahmen erstellt werden, um extrakardiale Ablagerungen zu detektieren.
Beurteilung
Bei Nachweis myokardialer Aktivität wird immer eine SPECT-Untersuchung zur dreidimensionalen Darstellung der Tracerverteilung ergänzt. Die semiquantitative Bewertung der Myokardaktivität erfolgt in der Regel anhand eines Gradings (Perugini-Score):
• Grad 0: keine myokardiale Aktivität
• Grad I: Myokard-Uptake < Rippen-Uptake
• Grad II: Myokard-Uptake = Rippen-Uptake
• Grad III: Myokard-Uptake > Rippen-Uptake
Bei Patienten, bei denen eine AL-Amyloidose ausgeschlossen wurde und die in der Skelettszintigraphie Grad-II- oder Grad-III-Scores aufweisen, gilt die Diagnose einer ATTR-Amyloidose als praktisch gesichert (Abb. 2).
Zukunft
Neue PET-Radiotracer wie 18F-Florbetapir, 18F-Florbetaben und 18F-Flutemetamol, ursprünglich für β-Amyloid im Gehirn entwickelt, zeigen grosses Potenzial zur kardialen Amyloidose-Diagnostik. Sie bieten durch längere Halbwertszeit und höhere räumliche Auflösung eine genauere Quantifizierung der Ablagerungen, sind jedoch noch nicht klinisch etabliert (6, 7). Ein weiterer Fortschritt ist 124I-Evuzamitid, ein unspezifisch an Amyloidfibrillen bindender Tracer, der sowohl ATTR- als auch AL-Amyloidose nachweisen kann und ebenfalls grosses Potenzial für die klinische Anwendung bietet (8).
Sarkoidose
Die Sarkoidose ist eine multisystemische entzündliche Erkrankung unbekannter Ursache, gekennzeichnet durch nicht-nekrotisierende Granulome in betroffenen Organen. Kardialer Befall wird oft bei Patienten mit extrakardialer Sarkoidose diagnostiziert, während in 20–25 % der Fälle eine isolierte kardiale Sarkoidose vorliegt (9). Häufige klinische Manifestationen sind höhergradige AV-Blockierungen, Herzinsuffizienz und ventrikuläre Arrhythmien (10). Die Diagnostik basiert auf klinischen Symptomen, histologischen Befunden und dem Ausschluss alternativer Diagnosen. Eine zentrale Rolle spielt die 18F-FDG-PET, besonders bei unklaren oder negativen MRT-Befunden, und ist laut Leitlinien als Zweitliniendiagnostik empfohlen (11).
Ablauf
Die 18F-FDG-PET unterscheidet entzündliches von normalem Myokard durch deren Stoffwechselaktivität: Normales Myokard nutzt bei kohlenhydratfreier Diät Fettsäuren, während entzündliches Gewebe Glukose bevorzugt. Eine kohlenhydratfreie Diät 24–72 Stunden vor der Untersuchung unterdrückt die FDG-Aufnahme im gesunden Myokard, wodurch pathologische FDG-Akkumulation in entzündeten Arealen sichtbar wird. Fettreiche Ernährung unterstützt diesen Effekt. Oft wird Heparin zur Fettsäurenfreisetzung zur Verstärkung der FDG-Aufnahme im entzündlichen Gewebe verabreicht. Nach intravenöser Gabe des Radiotracers 18F-FDG folgt eine 60-minütige Wartezeit, bevor die PET-Aufnahme erfolgt. Diese kann als Thorax/Abdomen- oder Ganzkörper-Scan durchgeführt werden.
Beurteilung
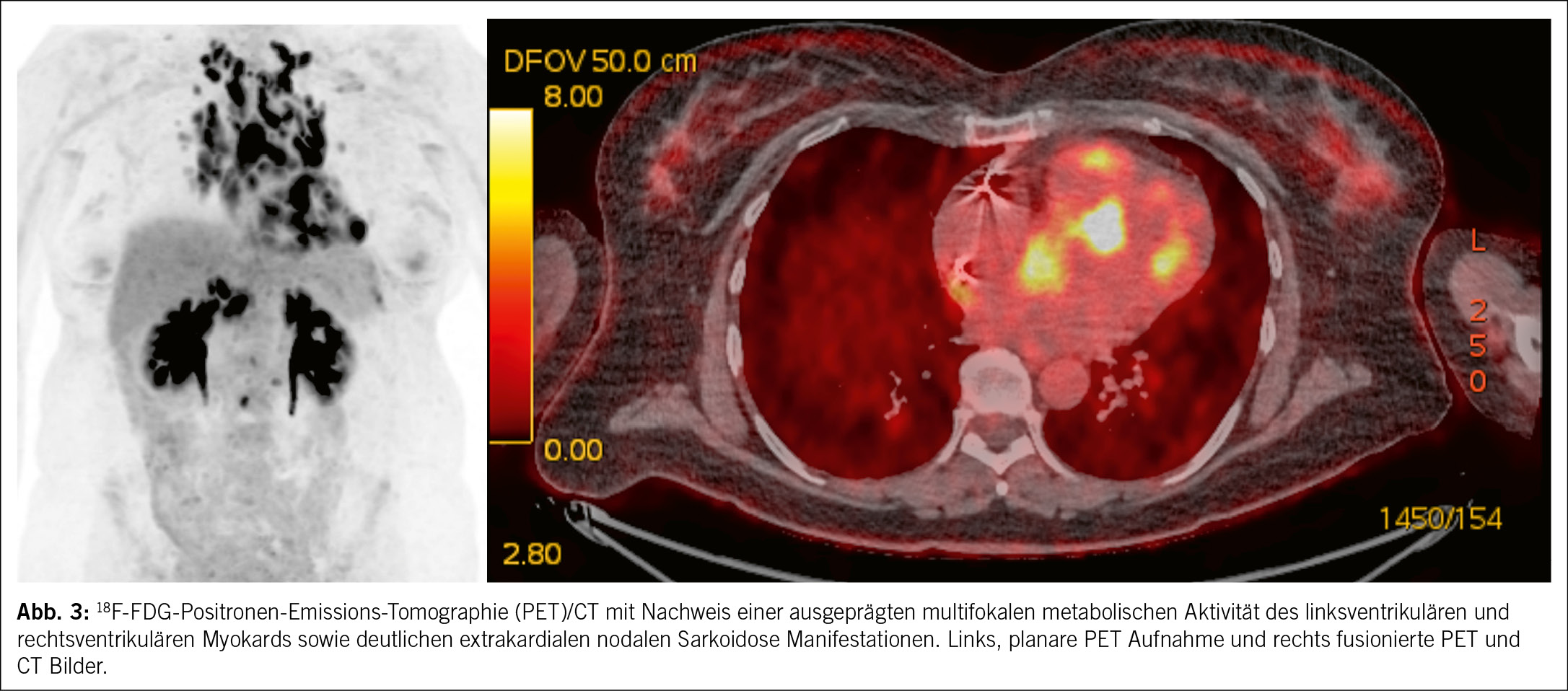
Die Interpretation kardialer Befunde erfordert eine sorgfältige Differenzierung zwischen physiologischer FDG-Aufnahme und pathologischen Entzündungsherden. Pathologische FDG-Akkumulation im Myokard zeigt typischerweise ein fleckförmiges (multifokales) Uptake-Muster, das auf aktive Entzündungen hinweist und weniger häufig ein diffuses Uptake-Muster, welches suspekt auf nicht supprimierte physiologische Glukoseaufnahme ist. Der standardisierte Uptake-Wert (SUV) hilft, die Intensität der FDG-Aufnahme zu quantifizieren und liefert Hinweise auf den Schweregrad der Entzündung. FDG-PET ist zudem nützlich für das Monitoring des Therapieansprechens und zur Planung von Follow-up-Untersuchungen, um die Krankheitsaktivität und das Risiko für Ereignisse zu bewerten. Darüber hinaus dient die 18F-FDG-PET als Basis für die Risikostratifizierung, indem sie hilft, das Ausmass der Myokardentzündung zu quantifizieren und den potenziellen Krankheitsverlauf einzuschätzen. Ein Vorteil der 18F-FDG-PET gegenüber der MRT ist die Möglichkeit, neben der myokardialen auch die extrakardiale Entzündungsaktivität zu erfassen. Extrakardiale FDG-Anreicherungen, beispielsweise in Lymphknoten oder anderen Organen, können eine gezielte Biopsie unterstützen, insbesondere, wenn histologische Nachweise erforderlich sind (Abb. 3).
Zukunft
Fibroblast activation protein Inhibitor (FAPI) ist ein Inhibitor des Proteins alpha, welches von aktiven Fibroblasten produziert wird. Die Entwicklung von FAPIs, die mit Radionukliden wie 68Ga oder 18F markiert sind, eröffnet neue Möglichkeiten für die Sarkoidose Bildgebung welche bereits in verschiedenen Zentren zu Forschungszwecken im Einsatz und dient dem Nachweis von Gewebeaktivität im Rahmen fibrotischer Prozesse, wie sie nach aktiven Entzündungsreaktionen häufig auftreten. Ein potentieller Anwendungsbereich dieses Radiotracers ist die Risikostratifizierung und Verlaufskontrolle von kardialer Sarkoidose-Patienten unter immunsuppressiver Therapie, in welchen oft noch eine chronische Fibroblastenaktivität als Hinweis auf ein Remodeling dargestellt werden kann, während mittels 18F-FDG-PET bereits keine Entzündungsaktivität mehr nachgewiesen werden kann (12, 13).
Copyright
Aerzteverlag medinfo AG
Dr. med. Pascal S. Heiniger
Klinik für Nuklearmedizin
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
PD Dr. med. et Dr. sc. med. Andreas A. Giannopoulos
Klinik für Nuklearmedizin
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
Die Autoren haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
◆ Präzise Diagnostik für die KHK: SPECT und PET bieten eine detaillierte Analyse von Durchblutungsstörungen und liefern wichtige prognostische Parameter wie die Ischämie-Last und die myokardiale Flussreserve.
◆ Nicht-invasive Amyloidose Diagnostik durch Skelettszintigraphie:
Die Skelettszintigraphie ermöglicht eine präzise Diagnostik der ATTR-Amyloidose, während neue quantitative Ansätze die diagnostische Genauigkeit weiter verbessern.
◆ Kardiale Sarkoidose Detektion: Mittels 18F-FDG, erlaubt die PET-Bildgebung die genaue Identifikation von Myokardentzündungen und extrakardialen Manifestationen, unterstützt die gezielte Biopsie-Planung und bietet wertvolle Informationen zur Therapieüberwachung.
1. Vrints C, Andreotti F, Koskinas KC, Rossello X, Adamo M, Ainslie J, et al. 2024 ESC Guidelines for the management of chronic coronary syndromes: Developed by the task force for the management of chronic coronary syndromes of the European Society of Cardiology (ESC) Endorsed by the European Association for Cardio-Thoracic Surgery (EACTS). European Heart Journal. 2024;45(36):3415-537.
2. Garefa C, Sager DF, Heiniger PS, Markendorf S, Albertini T, Jurisic S, et al. Duration of adenosine-induced myocardial hyperaemia: insights from quantitative 13N-ammonia positron emission tomography myocardial perfusion imaging. Eur Heart J Cardiovasc Imaging. 2024;25(10):1367-73.
3. Chang SM, Nabi F, Xu J, Peterson LE, Achari A, Pratt CM, Mahmarian JJ. The coronary artery calcium score and stress myocardial perfusion imaging provide independent and complementary prediction of cardiac risk. J Am Coll Cardiol. 2009;54(20):1872-82.
4. Maurer MS, Bokhari S, Damy T, Dorbala S, Drachman BM, Fontana M, et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Heart Fail. 2019;12(9):e006075.
5. Treglia G, Glaudemans A, Bertagna F, Hazenberg BPC, Erba PA, Giubbini R, et al. Diagnostic accuracy of bone scintigraphy in the assessment of cardiac transthyretin-related amyloidosis: a bivariate meta-analysis. Eur J Nucl Med Mol Imaging. 2018;45(11):1945-55.
6. Sperry BW, Bock A, DiFilippo FP, Donnelly JP, Hanna M, Jaber WA. Pilot Study of F18-Florbetapir in the Early Evaluation of Cardiac Amyloidosis. Front Cardiovasc Med. 2021;8:693194.
7. Genovesi D, Vergaro G, Giorgetti A, Marzullo P, Scipioni M, Santarelli MF, et al. [18F]-Florbetaben PET/CT for Differential Diagnosis Among Cardiac Immunoglobulin Light Chain, Transthyretin Amyloidosis, and Mimicking Conditions. JACC Cardiovasc Imaging. 2021;14(1):246-55.
8. Wall JS, Martin EB, Lands R, Ramchandren R, Stuckey A, Heidel RE, et al. Cardiac Amyloid Detection by PET/CT Imaging of Iodine ((124)I) Evuzamitide ((124)I-p5+14): A Phase 1/2 Study. JACC Cardiovasc Imaging. 2023;16(11):1433-48.
9. Kawai H, Sarai M, Kato Y, Naruse H, Watanabe A, Matsuyama T, et al. Diagnosis of isolated cardiac sarcoidosis based on new guidelines. ESC Heart Fail. 2020;7(5):2662-71.
10. Ekström K, Lehtonen J, Nordenswan HK, Mäyränpää MI, Räisänen-Sokolowski A, Kandolin R, et al. Sudden death in cardiac sarcoidosis: an analysis of nationwide clinical and cause-of-death registries. Eur Heart J. 2019;40(37):3121-8.
11. Cheng RK, Kittleson MM, Beavers CJ, Birnie DH, Blankstein R, Bravo PE, et al. Diagnosis and Management of Cardiac Sarcoidosis: A Scientific Statement From the American Heart Association. Circulation. 2024;149(21):e1197-e216.
12. Siebermair J, Kessler L, Kupusovic J, Rassaf T, Rischpler C. Cardiac fibroblast activation detected by (68)Gallium-FAPI-46 positron emission tomography-magnetic resonance imaging as a sign of chronic activity in cardiac sarcoidosis. Eur Heart J Case Rep. 2022;6(1):ytac005.
13. Solanki R, Singh H, Mehta V, Panda P, Singhal M, Sood A, Mittal BR. Potential application of (68)Ga-FAPI PET/CT for diagnosing cardiac sarcoidosis. J Nucl Cardiol. 2024;36:101835.
Die operative Therapie des Ovarialkarzinoms ist zentral und zielt auf eine vollständige Tumorentfernung ab, wobei die systematische Lymphadenektomie nach der LION-Studie bei unauffälligen Lymphknoten im CT und intraoperativ nicht mehr empfohlen wird. Ausser bei sehr frühen Karzinomen ist die adjuvante Chemotherapie Standard, meist in Form einer Kombination aus Carboplatin und Paclitaxel. Die Erhaltungstherapie mit Bevacizumab oder PARP-Inhibitoren wie Olaparib und Niraparib verlängert das progressionsfreie Überleben, insbesondere bei BRCA-mutierten und HRD-positiven Tumoren. Neoadjuvante Chemotherapie kann je nach Tumorausdehnung und Patientenzustand eine Alternative zur primären Operation sein. Bei Rezidiven werden erneute platinhaltige Chemotherapien oder Operationen individuell abgewogen. Bei einem platinsensitiven Rezidiv kann eine chirurgische Intervention im Kontext der rezidivierenden Erkrankung von Vorteil sein.
Surgical treatment is central to the management of ovarian cancer and aims for complete tumor resection. A systematic pelvic and paraaortic lymphadenectomy is no longer recommended for patients with normal lymph nodes based on the CT scan and intraoperative findinds, according to the LION study. Except for very early-stage cancers, adjuvant chemotherapy, typically a combination of carboplatin and paclitaxel, is standard. Maintenance therapy with Bevacizumab or PARP inhibitors like Olaparib and Niraparib extends progression-free survival, especially in BRCA-mutated and HRD-positive tumors. Neoadjuvant chemotherapy can be an alternative to primary surgery depending on tumor extent and patient condition. In cases of recurrence, repeat platinum-based chemotherapies or surgeries are considered on an individual basis. For platinum-sensitive recurrences, surgical intervention in the context of recurrent disease can be beneficial. Keywords: Ovarian/ primary peritoneal/ tubal carcinoma – surgery – chemotherapy – maintenance therapy – PARP inhibitor
Das Ovarialkarzinom stellt eine der schwerwiegendsten gynäkologischen Krebserkrankungen dar und erfordert eine sorgfältige und umfassende Behandlungsstrategie. Die operative Therapie ist ein zentraler Bestandteil der Behandlung und zielt darauf ab, das gesamte sichtbare Tumorgewebe zu entfernen, um die Prognose der Patientinnen zu verbessern. Ergänzend zur Chirurgie sind systemische Therapien wie Chemotherapie und moderne Erhaltungstherapien entscheidend, um das Überleben zu verlängern und Rückfälle zu verhindern. Aktuelle klinische Studien haben zu signifikanten Veränderungen in der Behandlungsstrategie geführt, insbesondere hinsichtlich der Rolle der systematischen Lymphadenektomie und der Anwendung von PARP-Inhibitoren. Dieser Text bietet einen Überblick über die wesentlichen Aspekte der operativen und systemischen Therapie bei Ovarialkarzinom.
(Zur besseren Lesbarkeit sind Ovarial-, Tuben- und primäres Peritonealkarzinom im Text unter dem Oberbegriff «Ovarialkarzinom» zusammengefasst).
Erstdiagnose eines Ovarialkarzinoms
Operative Therapie bei Erstdiagnose:
Die operative Therapie spielt eine zentrale Rolle bei der Behandlung des Ovarialkarzinoms, da eine makroskopisch tumorfreie Resektion wesentlich für die Prognose der Patientinnen ist (1). Das Standardverfahren umfasst eine mediane Längs-Laparotomie mit Hysterektomie und bilateraler Adnexektomie sowie mindestens eine infrakolische Omentektomie, Peritonealbiopsien und die Entfernung allen weiteren tumorverdächtigen Gewebes, um eine makroskopisch tumorfreie Resektion zu erzielen (S3-Leitlinie Ovar).
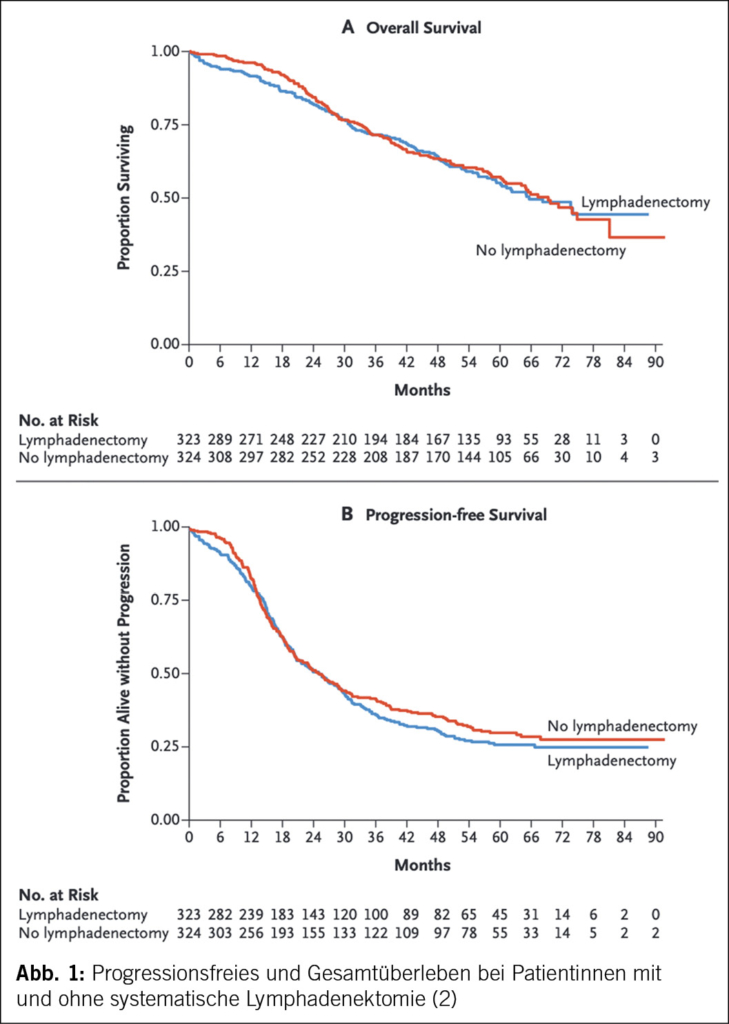
Bis 2017 war die systematische pelvine und paraaortale Lymphadenektomie ein fester Bestandteil der operativen Therapie des Ovarialkarzinoms. Die LION-Studie («Lymphadenectomy in Ovarian Neoplasms») führte jedoch zu einem Paradigmenwechsel ((2), S3-Leitlinie). Trotz der Tatsache, dass bei 55.7 % der Patientinnen mit Lymphadenektomie mikroskopische Lymphknotenmetastasen nachgewiesen wurden, gab es keinen Unterschied im Gesamt-(OS) oder progressionsfreien Überleben (PFS) (Abb. 1). Die Morbidität und die perioperative Mortalität waren in der Lymphadenektomie-Gruppe signifikant höher (2). Diese Ergebnisse haben dazu geführt, dass von einer systematischen Lymphadenektomie bei fortgeschrittenem high-grade serösen Ovarialkarzinom und unauffälligen Lymphknoten abgeraten wird (S3-Leitlinie).
Es ist wichtig zu betonen, dass bei der Diagnose eines fortgeschrittenen Ovarialkarzinoms üblicherweise eine adjuvante platinbasierte Chemotherapie indiziert ist, unabhängig von einem möglichen Tumorbefall der Lymphknoten ((1),S3-Leitlinie). Bei klinisch früh eingestuften Ovarialkarzinomen (FIGO I-IIA) hängt die Indikation für eine adjuvante Chemotherapie jedoch vom Nachweis positiver pelviner und/oder paraaortaler Lymphknoten ab. Bis zu 30 % dieser Patientinnen haben okkulte Lymphknotenmetastasen, was eine Höherklassifikation zu einem FIGO-Stadium III und damit eine Indikation für eine adjuvante Chemotherapie bedeutet (3). Daher bleibt die systematische pelvine und paraaortale Lymphadenektomie bei diesen Patientinnen empfohlen (Schmalfeldt et al. 2018, S3-Leitlinie).
Neoadjuvante Chemotherapie
Die grösste bisher veröffentlichte Studie zur neoadjuvanten Chemotherapie (NACT) zeigte keinen Unterschied im OS zwischen der Gruppe mit neoadjuvanter Chemotherapie für 3 Zyklen und Intervall-Debulking versus Patientinnen mit Primär-Debulking (PDS) und adjuvanter Kombinationstherapie für 6 Zyklen (HR 0,98; 90 %-KI 0,84–1,13; p= 0,01) (4). Eine Metaanalyse (5) ergab jedoch einen Vorteil für das primäre Debulking bei Patientinnen im FIGO-Stadium IIIC mit einer maximalen Tumorgrösse von < 5 cm (6). Auch retrospektive Studien zeigten teilweise einen Vorteil der PDS (7, 8).
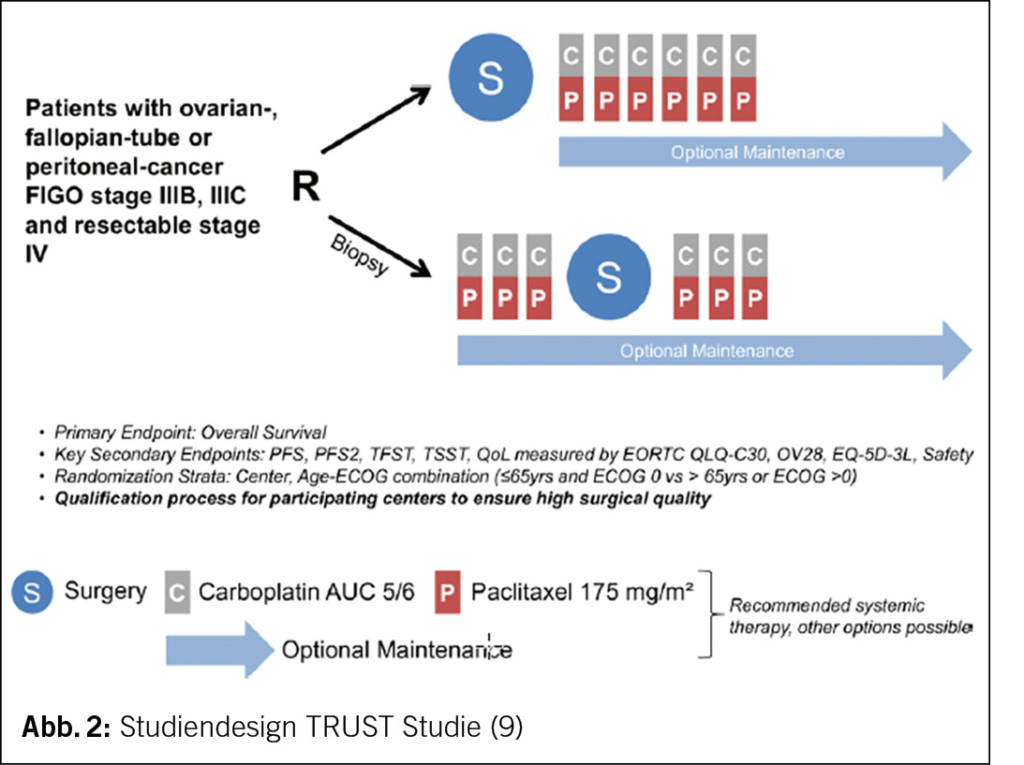
Die randomisierte, multizentrische TRUST-Studie («Trial of Radical Upfront Surgical Therapy in advanced ovarian cancer») untersucht das OS von Frauen mit epithelialem Ovarial-, Tuben- oder primärem Peritonealkarzinom FIGO IIIB-IVB bei primärer zytoreduktiver Operation versus neoadjuvanter Chemotherapie und Intervall-Debulking nach 3 Zyklen mit Carboplatin und Paclitaxel an Zentren mit hoher operativer Expertise (Abb. 2) (9). Ergebnisse werden dieses Jahr erwartet (9).
Adjuvante Systemtherapie
Frühes Ovarialkarzinom
Patientinnen mit frühem Ovarialkarzinom im Stadium FIGO IA G1 benötigen keine adjuvante Chemotherapie; in den Stadien IA G2 und IB G1/2 kann eine platinhaltige Chemotherapie diskutiert werden (3, 10). In den Stadien IC oder IA/ B G3 sollte eine platinhaltige (Mono-)Therapie gegeben werden (Verbesserung des 5-Jahres-OS von 75 % auf 82 % (3, 10).
Fortgeschrittenes Ovarialkarzinom
Seit den frühen 2000er Jahren hat sich die Kombination aus Carboplatin und Paclitaxel durchgesetzt (11). Die ICON-8-Studie konnte keine signifikanten Unterschiede zwischen verschiedenen Dosisdichten von Paclitaxel nachweisen, weshalb die dreiwöchentliche Kombination aus Carboplatin und Paclitaxel als Standard beibehalten wurde (12). Diese wird gegebenenfalls mit einer Erhaltungstherapie kombiniert.
Erhaltungstherapie
Antiangiogenetische Therapie
Bevacizumab, ein Angiogenesehemmer, wird in Kombination mit Chemotherapie und als Erhaltungstherapie eingesetzt. Phase-III-Studien wie GOG-0218 und ICON-7 haben gezeigt, dass Bevacizumab das PFS signifikant verlängert, besonders in Hochrisikogruppen (FIGO III und IV) (13, 14). In der Primärtherapie wird Bevacizumab zunächst mit Chemotherapie kombiniert und anschliessend als Erhaltungstherapie fortgeführt.
PARP-Inhibitoren in der Erhaltungstherapie bei Erstlinien- und Rezidivtherapie
PARP-Inhibitoren (PARPi) sind orale Medikamente, die die Reparatur von DNA-Einzelstrangbrüchen hemmen und dadurch Doppelstrangbrüche verursachen. Karzinomzellen, die nicht über die homologe Rekombinationsreparatur verfügen und somit eine homologe Rekombinations-Defizienz (HRD) aufweisen, können diese Brüche nicht richtig reparieren, was zu Chromosomenveränderungen und schliesslich zum Zelltod führt (15). Tests auf BRCA-Mutationen und HRD-Status sind zum Standard geworden, um Patientinnen zu identifizieren, die von PARPi profitieren können. Olaparib kann bei BRCA1/2-mutierten Ovarialkarzinomen eingesetzt werden (16) und die Kombination mit Bevacizumab ist bei HRD-positiven Tumoren möglich (17). Niraparib kann als Monotherapie verwendet werden (18). Veliparib und Rucaparib zeigten Vorteile in verschiedenen Studien (19, 20), sind jedoch in Europa noch nicht zugelassen.
Bei der Erhaltungstherapie mit einem PARPi bei einem Rezidiv war ein OS-Vorteil schwerer nachzuweisen, was teilweise auf PARPi-Crossover und eine lange Überlebenszeit nach Progression zurückzuführen sein könnte. Die Wahl des Medikaments sollte nach Nebenwirkungsprofil und Patientinnen-Präferenz erfolgen, da vergleichende Studien fehlen. Patientinnen, die unter PARPi progredient sind, haben meist nur geringen Nutzen von einer erneuten PARPi-Erhaltungstherapie (21). Es gibt bislang keine Daten zu einer gleichzeitigen Erhaltungstherapie mit Bevacizumab und Olaparib bei einem Rezidiv.
Bei Patientinnen mit platinsensiblem Rezidiv eines BRCA-mutierten high-grade Ovarialkarzinoms nach zwei oder mehr platinhaltigen Vortherapien kann eine Monotherapie mit Rucaparib eine Option sein (22).
Rezidiv
Operation beim Rezidiv
Ein erheblicher Anteil der Patientinnen mit fortgeschrittenem Ovarialkarzinom entwickelt ein Rezidiv. Die DESKTOP III-Studie definierte prädiktive Parameter, um geeignete Patientinnen für eine erneute Operation zu identifizieren. Diese beinhalten Patientinnen mit einem ersten platin-sensitiven Rezidiv, einem ECOG-Performance-Status von 0, Aszites ≤ 500 ml und einer makroskopischen Komplettresektion bei der Erstoperation (du Bois et al. 2020).
Chemotherapie beim Rezidiv
Bei Rezidiven des epithelialen Ovarialkarzinoms sollte bei Patientinnen zunächst evaluiert werden, ob sie für eine platinhaltige Therapie geeignet sind (früher «platinsensibel» oder «platinresistent»). Bei frühem Rezidiv (< 6 Monate nach Abschluss der adjuvanten Systemtherapie) haben Mono-Chemotherapien mit Topotecan, Gemcitabin, Paclitaxel oder pegyliertes liposomales Doxorubicin bessere Verträglichkeit und vergleichbare Effektivität gezeigt (23). Die Optimierung der Lebensqualität ist besonders wichtig (24). Mirvetuximab Soravtansin zeigte in der Phase-III-Studie MI-RASOL einen signifikanten PFS- und OS-Vorteil sowie ein besseres Sicherheitsprofil im Vergleich zur Chemotherapie (25).
Bei einem Rezidiv > 6 Monate nach der letzten Platintherapie wird in der Regel eine erneute platinhaltige Kombinationschemotherapie durchgeführt. Vor Beginn der Rezidivtherapie sollte die Möglichkeit einer Rezidivoperation geprüft werden. Bevorzugtes Regime beim Rezidiv sind Kombinationen aus Carboplatin und pegyliertem liposomalem Doxorubicin oder Carboplatin und Gemcitabin (26).
Antiangiogenetische Therapie beim Rezidiv
Bevacizumab kann in der Rezidivtherapie bei Patientinnen, die bisher kein Bevacizumab erhalten haben, in Kombination mit einer Monochemotherapie das PFS signifikant verlängern (27). Es kann auch off-label zur Reduktion der Aszitesbildung beitragen (27).
Spezielle Situationen
Low-grade seröses Ovarialkarzinom
Für Patientinnen im FIGO-Stadium IC bis IIA wird eine Monotherapie mit Carboplatin empfohlen, während ab Stadium IIB eine Kombinationstherapie aus Carboplatin und Paclitaxel eingesetzt werden sollte (Ansprechrate unter 25 %) (24). In retrospektiven Studien konnte gezeigt werden, dass eine endokrine Erhaltungstherapie das PFS verdoppeln kann (28). Die MATAO-Studie untersucht den Effekt von Letrozol versus Placebo nach Chemotherapie bei hormonrezeptorpositiven Patientinnen prospektiv (29).
Die Behandlung mit dem MEK-Inhibitor Trametinib zeigte in einer Phase-II/III-Studie ein signifikant längeres PFS als die Standardtherapie (HR 0.48, p<0.0001) und bietet eine neue Behandlungsoption für Patienten mit Rezidiv eines low-grade serösen Karzinoms (30).
Die Wirksamkeit von Bevacizumab bei low-grade serösen Ovarialkarzinomen ist unklar (14).
Die erste Phase-III-Studie zur HIPEC bei Ovarialkarzinom-Patientinnen nach neoadjuvanter Chemotherapie zeigte eine signifikante Verbesserung des rückfallfreien Überlebens (HR 0.66, p=0.003) und des OS im Vergleich zur Standardtherapie, jedoch mit ähnlichen Raten schwerer Nebenwirkungen. Die Studie wirft jedoch erhebliche methodische Fragen auf. Aktuell wird HIPEC nicht als Standardtherapie empfohlen und sollte nur in kontrollierten Studien verwendet werden (31).
Abkürzungen AGO Arbeitsgemeinschaft Gynäkologische Onkologie ECOG Eastern European Cooperative Oncology Group FIGO Fédération Internationale de la Gynécologie et d’Obstétrique HIPEC Hypertherme intraperitoneale Chemotherapie HRD Homologe Rekombinations-Defizienz MEK Mitogen-aktivierte Proteinkinase NACT Neoadjuvante Chemotherapie OS Gesamtüberleben PARP Poly(ADP-ribose)-Polymerasen PARPi PARP-Inhibitoren PDS primary debulking surgery, primäre Debulking-Operation PFS progressionsfreies Überleben
Die Autorenschaft hat keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
Grundsatz der operativen Therapie ist das Ziel der tumorfreien Resektion. Diese verbessert die Prognose der Erkrankung.
Eine systematische pelvine und paraaortale Lymphadenektomie sollte bei Patientinnen mit fortgeschrittenem high-grade serösen Ovarialkarzinom (FIGO IIB-IV) und unauffälligen Lymphknoten nicht durchgeführt werden (LION-Studie).
Eine Platin-haltige Kombinations-Chemotherapie ist in den meisten Fällen der Standard der adjuvanten Therapie.
Als Erhaltungstherapie nach Abschluss der adjuvanten Chemotherapie und Therapieansprechen kommen bei BRCA-Mutation oder HRD-Positivität PARP-Inhibitoren in Frage.
Bei Patientinnen mit einem Platin-sensitiven Erstrezidiv eines Ovarialkarzinoms und einem positiven AGO-Score (ECOG Performance Status von 0, Aszites von ≤ 500 ml und eine makroskopische
Komplettresektion) sollte beim Rezidiv eine erneute Operation in Erwägung gezogen werden (DESKTOP III-Studie).
1. du Bois, A., et al., Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: a combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: by the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d’Investigateurs Nationaux Pour les Etudes des Cancers de l’Ovaire (GINECO). Cancer, 2009. 115(6): p. 1234-44.
2. Harter, P., et al., A Randomized Trial of Lymphadenectomy in Patients with Advanced Ovarian Neoplasms. N Engl J Med, 2019. 380(9): p. 822-832.
3. Trimbos, B., et al., Surgical staging and treatment of early ovarian cancer: long-term analysis from a randomized trial. J Natl Cancer Inst, 2010. 102(13): p. 982-7.
4. Vergote, I., et al., Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N Engl J Med, 2010. 363(10): p. 943-53.
5. Kehoe, S., et al., Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): an open-label, randomised, controlled, non-inferiority trial. Lancet, 2015. 386(9990): p. 249-57.
6. Vergote, I., et al., Neoadjuvant chemotherapy versus debulking surgery in advanced tubo-ovarian cancers: pooled analysis of individual patient data from the EORTC 55971 and CHORUS trials. Lancet Oncol, 2018. 19(12): p. 1680-1687.
7. Sorensen, S.M., et al., Residual tumor and primary debulking surgery vs interval debulking surgery in stage IV epithelial ovarian cancer. Acta Obstet Gynecol Scand, 2022. 101(3): p. 334-343.
8. Rauh-Hain, J.A., et al., Primary debulking surgery versus neoadjuvant chemotherapy in stage IV ovarian cancer. Ann Surg Oncol, 2012. 19(3): p. 959-65.
9. Reuss, A., et al., TRUST: Trial of Radical Upfront Surgical Therapy in advanced ovarian cancer (ENGOT ov33/AGO-OVAR OP7). Int J Gynecol Cancer, 2019. 29(8): p. 1327-1331.
10. Trimbos, J.B., et al., Impact of adjuvant chemotherapy and surgical staging in early-stage ovarian carcinoma: European Organisation for Research and Treatment of Cancer-Adjuvant ChemoTherapy in Ovarian Neoplasm trial. J Natl Cancer Inst, 2003. 95(2): p. 113-25.
11. du Bois, A., et al., A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J Natl Cancer Inst, 2003. 95(17): p. 1320-9.
12. Blagden, S.P., et al., Weekly platinum-based chemotherapy versus 3-weekly platinum-based chemotherapy for newly diagnosed ovarian cancer (ICON8): quality-of-life results of a phase 3, randomised, controlled trial. Lancet Oncol, 2020. 21(7): p. 969-977.
13. Tewari, K.S., et al., Final Overall Survival of a Randomized Trial of Bevacizumab for Primary Treatment of Ovarian Cancer. J Clin Oncol, 2019. 37(26): p. 2317-2328.
14. Oza, A.M., et al., Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): overall survival results of a phase 3 randomised trial. The Lancet Oncology, 2015. 16(8): p. 928-936.
15. Lord, C.J. and A. Ashworth, PARP inhibitors: Synthetic lethality in the clinic. Science, 2017. 355(6330): p. 1152-1158.
16. Moore, K., et al., Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med, 2018. 379(26): p. 2495-2505.
17. Ray-Coquard, I., et al., Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N Engl J Med, 2019. 381(25): p. 2416-2428.
18. Gonzalez-Martin, A., et al., Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med, 2019. 381(25): p. 2391-2402.
19. Coleman, R.L., et al., Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N Engl J Med, 2019. 381(25): p. 2403-2415.
20. Monk, B.J., et al., A Randomized, Phase III Trial to Evaluate Rucaparib Monotherapy as Maintenance Treatment in Patients With Newly Diagnosed Ovarian Cancer (ATHENA-MONO/GOG-3020/ENGOT-ov45). J Clin Oncol, 2022. 40(34): p. 3952-3964.
21. Pujade-Lauraine, E., et al., Maintenance olaparib rechallenge in patients with platinum-sensitive relapsed ovarian cancer previously treated with a PARP inhibitor (OReO/ENGOT-ov38): a phase IIIb trial. Ann Oncol, 2023. 34(12): p. 1152-1164.
22. Kristeleit, R., et al., Rucaparib versus standard-of-care chemotherapy in patients with relapsed ovarian cancer and a deleterious BRCA1 or BRCA2 mutation (ARIEL4): an international, open-label, randomised, phase 3 trial. Lancet Oncol, 2022. 23(4): p. 465-478.
23. Gordon, A.N., et al., Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol, 2001. 19(14): p. 3312-22.
24. Colombo, N., et al., ESMO-ESGO consensus conference recommendations on ovarian cancer: pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease†. Ann Oncol, 2019. 30(5): p. 672-705.
25. Moore, K.N., et al., Phase III MIRASOL (GOG 3045/ENGOT-ov55) study: Initial report of mirvetuximab soravtansine vs. investigator’s choice of chemotherapy in platinum-resistant, advanced high-grade epithelial ovarian, primary peritoneal, or fallopian tube cancers with high folate receptor-alpha expression. Journal of Clinical Oncology, 2023. 41(17_suppl): p. LBA5507-LBA5507.
26. Wagner, U., et al., Final overall survival results of phase III GCIG CALYPSO trial of pegylated liposomal doxorubicin and carboplatin vs paclitaxel and carboplatin in platinum-sensitive ovarian cancer patients. Br J Cancer, 2012. 107(4): p. 588-91.
27. Pujade-Lauraine, E., et al., Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. J Clin Oncol, 2014. 32(13): p. 1302-8.
28. Gershenson, D.M., et al., Hormonal Maintenance Therapy for Women With Low-Grade Serous Cancer of the Ovary or Peritoneum. J Clin Oncol, 2017. 35(10): p. 1103-1111.
29. Heinzelmann-Schwarz, V.A., et al., ENGOT-ov54/Swiss-GO-2/MATAO including LOGOS (Low-Grade Ovarian cancer Sub-study): MAintenance Therapy with Aromatase inhibitor in epithelial Ovarian cancer—A randomized, double-blinded, placebo-controlled, multicenter phase III Trial. Journal of Clinical Oncology, 2021. 39(15_suppl): p. TPS5598-TPS5598.
30. Gershenson, D.M., et al., Trametinib versus standard of care in patients with recurrent low-grade serous ovarian cancer (GOG 281/LOGS): an international, randomised, open-label, multicentre, phase 2/3 trial. Lancet, 2022. 399(10324): p. 541-553.
31. van Driel, W.J., et al., Hyperthermic Intraperitoneal Chemotherapy in Ovarian Cancer. N Engl J Med, 2018. 378(3): p. 230-240.
Separat im Text gelistet:
1. Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF): S3-Leitlinie Diagnostik, Therapie und Nachsorge maligner Ovarialtumoren, Langversion 4.0, 2020, AWMF-Registernummer: 032/035OL, https://www.leitlinienprogramm-onkologie.de/leitlinien/ovarialkarzinom/, (abgerufen am: 30.06.2024).
2. Schmalfeldt B et al. – für die Kommission Ovar der AGO: Wie ist die Evidenz für die Lym-phonodektomie beim frühen Ovarialkarzinom? Der Frauenarzt 2018;59(10):751-753.
3. du Bois A et al.: Randomized controlled phase III study evaluating the impact of secondary cytoreductive surgery in recurrent ovarian cancer: The final analysis of AGO DESKTOP III/ENGOT ov20. ASCO 2020, Abstr. #6000.
Les enjeux éthiques des demandes de suicide assisté venant des personnes atteintes de démence sont complexes. Il s’agit avant tout de pouvoir adresser leurs préoccupations et explorer leurs motivations et attitudes face à la vie et à la mort. Une demande de suicide assisté peut être exprimée en anticipation du déclin cognitif, mais la réalisation nécessite la capacité de discernement. Si souhaité, l’élaboration d’un projet de soins anticipé permet de discuter des alternatives en cas de complications et des préférences y relatives. Une approche interdisciplinaire de soins palliatifs gériatriques est recommandée afin d’offrir des soins concordant avec les objectifs des personnes et de les respecter dans leurs décisions de fin de vie.
The ethical issues surrounding requests for assisted suicide from people with dementia are complex. First and foremost, we need to address their concerns and explore their motivations and attitudes towards life and death. A request for assisted suicide may be expressed in anticipation of cognitive decline, but its realization requires decision-making capacity. If desired, advance care planning can help discuss and determine alternatives in the event of complications and related preferences. An interdisciplinary approach to geriatric palliative care is recommended in order to provide goal-concordant care and to respect the person’s end-of-life decisions. Keywords: Dementia, assisted suicide, decision-making capacity, advance care planning
Introduction
Dans le présent article, nous proposons d’aborder quelques enjeux éthiques actuels du suicide assisté chez les personnes atteintes de démence. Comme souvent face à une maladie grave et incurable, les préférences des personnes confrontées à un diagnostic de démence sont très variables et dépendent de leurs valeurs et attitudes face à la vie et à la mort. Certains expriment initialement ne pas s’imaginer vivre avec des troubles cognitifs mais montrent plus tard des signes de plaisir à vivre. D’autres présentent des signes de souffrance existentielle, de perte de sens ou d’une crainte face au déclin cognitif, pouvant motiver une demande de suicide assisté dès l’ annonce du diagnostic. Il s’agit aussi, pour certaines personnes comme Gunter Sachs, de garder le contrôle de leur vie et de ne pas subir une fin de vie qu’elles ne jugent pas digne selon leur échelle de valeur (cf. encadré).
Pour les professionnelles de santé, l’expression d’un désir de mort, voire d’une demande d’assistance au suicide, est un défi qui nécessite des compétences communicationnelles, d’écoute active et de respect des valeurs d’autrui (2). Les soignants ont une obligation déontologique d’essayer de comprendre leurs patients en tant que personnes, et de considérer leurs souffrances et ses potentielles causes. Une évaluation pluriprofessionnelle et interdisciplinaire permet de mieux comprendre les enjeux qui découlent de chaque situation, en impliquant des spécialistes médecins, infirmiers, psychologues, accompagnants spirituels des domaines de la gériatrie, de la psycho-gériatrie et des soins palliatifs, mais aussi des éthiciens et juristes parfois.
Comme le montre le cas de Gunter Sachs, un désir de mort au début d’une démence est souvent motivé par la crainte d’un avenir sombre: les craintes d’être totalement dépendant, de perdre son identité et sa personnalité, d’être un fardeau pour les proches, de souffrir de douleurs ou d’autres symptômes insupportables, et finalement les craintes de ne plus pouvoir contrôler sa vie et de ne plus pouvoir mettre fin à ses jours à cause d’une perte de la capacité de discernement (3). Une revue systématique de la littérature a dévoilé que la démence en soi n’est pas un facteur de risque suicidaire, mais que certains sous-groupes peuvent présenter un risque accru, par exemple dans des situations de patients plus jeunes, en cas de comorbidités, de dépression, de démence sémantique, ou dans la phase initiale après l’annonce du diagnostic (4).
Cadre juridique et déontologique
En Suisse, l’assistance au suicide n’est pas considérée comme une infraction pénale à condition que la personne qui assiste n’ait pas de mobile égoïste (art. 155 Code Pénal suisse, CP). L’euthanasie est cependant interdite (art. 114 CP), contrairement à un nombre croissant d’autres pays (Pays-Bas, Belgique, Luxembourg, Canada, Australie, Espagne, Portugal). Dans le cas du suicide assisté la personne désirant mourir garde le contrôle ultime de l’acte: c’est elle qui doit boire la substance létale ou démarrer la perfusion. L’assistance au suicide est uniquement permise si le suicide est volontaire et si la personne a encore sa capacité de discernement par rapport à cet acte. Pour assurer la nature volontaire de l’acte, il est nécessaire d’exclure toutes pressions, manipulations, tromperies ou contraintes intérieures ou extérieures. La capacité de discernement se décline en 4 sous-capacités: la compréhension des informations données de manière compréhensible, l’appréciation de sa propre situation et des conséquences de l’acte, le raisonnement selon la balance des arguments, et finalement le choix personnel exprimé (5). Cependant, l’évaluation de cette capacité est complexe quand elle concerne le suicide assisté et la littérature montre qu’il y a beaucoup d’incertitude et de diversité dans ces évaluations médicales (6).
La directive de l’Académie Suisse des Sciences Médicales « Attitude face à la fin de vie et à la mort », adoptée comme déontologie par la FMH, émet des conditions supplémentaires (7): le médecin doit attester que la personne ait une souffrance insupportable objectivée par un diagnostic ou un pronostic. De plus, les alternatives au suicide assisté doivent avoir été expliquées, discutées et proposées. Selon cette directive, le médecin ne peut apporter une assistance au suicide si le désir de suicide « constitue un symptôme actuel d’un trouble psychique » (7).
Alternatives et enjeux éthiques
Sur le plan éthique, un enjeu majeur est le respect de l’autonomie et de la dignité de la personne atteinte de démence, et des décisions de fin de vie qui en découlent (8). Afin de pouvoir prendre une décision éclairée, la personne capable de discernement doit avant tout être informée de manière complète et objective sur le pronostic de la maladie, les conséquences de celle-ci sur la santé et la fin de vie, les options de projet de soins et les risques et bénéfices de ces options.
Les patients doivent aussi être informés des différentes options légales permettant de soulager la souffrance et d’accompagner la fin de vie, en particulier des soins palliatifs. Comme toutes mesures de soins, les mesures prolongeant la vie et prescrites dans le cadre d’une démence (p.ex. des antibiotiques), ne doivent être entamées que si elles sont alignées avec la volonté autonome de la personne. Afin de respecter au mieux cette autonomie selon l’échelle de valeurs de l’individu, il est primordial de pouvoir engager ces discussions dans la phase précoce de la maladie (« autonomie relationnelle »). Il est recommandé que le patient exprime ses souhaits, valeurs et objectifs de soins en amont avec ses proches, ses médecins et ses soignants, idéalement dans le cadre d’un Projet de Soins Anticipé (ProSA), accompagné par un professionnel qualifié qui anime et documente ces entretiens (9, 10). Dans ce processus, le patient a aussi l’occasion de nommer une personne de confiance comme représentant thérapeutique et de rédiger des directives anticipées. Ces dernières permettent de se déterminer sur les situations dans lesquelles, en cas d’incapacité de discernement, la personne souhaiterait renoncer aux mesures de soins. On ne peut pas diriger son futur soi incapable de discernement à se suicider (comme montré dans le film « Still Alice »), et si on pouvait le faire ceci serait un suicide non volontaire qui devrait être empêché plutôt qu‘assisté.
Le ProSA permet également d’explorer d’autres options possibles, comme celles relatives à l’alimentation et à l’hydratation. Tant qu’une personne garde sa capacité de discernement, un arrêt volontaire de manger et de boire est une décision de fin de vie légitime qui permet de raccourcir sa vie (11). Cependant, il est éthiquement controversé qu’une personne puisse exiger en amont de ne plus recevoir de boissons et de nourriture en cas de démence avancée, surtout si elle montre des signes qu’elle souhaite boire et manger – une offre d’entraide humaine soutenue par le principe bioéthique de bienfaisance (12, 13). La prise hydrique et alimentaire orale n’est pas un traitement médical mais un geste d‘assistance interpersonnelle, tout comme la protection contre le froid. Leur arrêt ne peut pas être prescrit en amont car ceci équivaudrait à un suicide assisté non volontaire (en l‘absence d‘autodétermination), ce qui n’est ni légal ni éthiquement bien-fondé (13, 14).
La démence présumée de Gunter Sachs n’a pas été diagnostiquée et il était connu pour avoir des épisodes dépressifs. Il aurait été mérité d’être accompagné par une équipe médico-soignante et soutenue par une culture de communication ouverte dans une société qui s’engage à respecter les décisions de fin de vie de toutes les personnes, quelles qu’elles soient.
Copyright
Aerzteverlag medinfo AG
Dre Rachel Rutz Voumard
Unité d’éthique clinique,
Institut des Humanités en Médecine, CHUV-UNIL
Dre Eve Rubli Truchard
Chaire de soins palliatifs gériatriques
Service de gériatrie et de réadaptation gériatrique
Centre hospitalier universitaire vaudois (CHUV)
Avenue Pierre-Decker 9
1011 Lausanne
Pr Ralf Jox
– Unité d’éthique clinique, Institut des Humanités en Médecine, CHUV-UNIL
– Chaire de soins palliatifs gériatriques, Service de soins palliatifs et de support CHUV-UNIL,
Les auteurs n’ont pas déclaré de conflit d’intérêts en rapport avec cet article.
Les personnes atteintes de démence peuvent exprimer une souffrance existentielle, une perte de sens ou une crainte face au risque de déclin cognitif, pouvant motiver un désir de mort précoce à l’annonce du diagnostic.
Il est primordial d’écouter leurs préoccupations, les informer de la trajectoire de la maladie, discuter des options de soins et anticiper le projet de soins.
Consciente des alternatives possibles, une personne atteinte de démence est plus à même de prendre des décisions existentielles concernant sa vie et sa mort.
1. Der Abschiedsbrief von Gunter Sachs. Frankfurter Allgemeine Zeitung, 8.5.2011, https://www.faz.net/aktuell/gesellschaft/menschen/wortlaut-der-abschiedsbrief-von-gunter-sachs-1637779.html (accédé le 19.8.2024)
2. Fachgesellschaft für Palliative Geriatrie (FGPG), Grundsatzpapier „Sterbewünsche in der palliativen Geriatrie“. FGPG Berlin 2023, https://www.fgpg.eu/wp-content/uploads/2023/05/GSP-05_Sterbewuensche-Druck.pdf (accédé le 19.8.2024)
3. van Rickstal R, De Vleminck A, Chambaere K, Van den Block L. People with young-onset dementia and their family caregivers discussing euthanasia: A qualitative analysis of their considerations. Patient Educ Couns 2023; Oct:115:107882. doi: 10.1016/j.pec.2023.107882
4. Schmid J, Jox R, Gauthier S, Belleville S, Racine E, Schüle C, Turecki G, Richard-Devantoy S. Suicide and assisted dying in dementia: what we know and what we need to know. A narrative literature review. Int Psychogeriatr 2017;29(8):1247-59.
5. Appelbaum PS. Assessment of Patients’ Competence to Consent to Medical Treatment. New Engl J Med 2007 ;357:1834-40.
6. Mangino DR, Nicolini ME, De Vries RG, Kim SYH. Euthanasia and Assisted Suicide of Persons With Dementia in the Netherlands. Am J Geriatr Psychiatry 2020;28(4):466-77.
7. Académie Suisse des Sciences Médicales. Directives médico-éthiques: Attitude fae à la fin de vie et à la mort. Version de 2018, révisée en 2021. Bâle 2022. https://www.samw.ch/fr/Publications/Directives.html (accédé le 19.8.2024)
8. Académie Suisse des Sciences Médicales. Directives médico-éthiques: Prise en charge et traitements des personnes atteintes de démences, ASSM, Bâle 2018.
9. Bosisio F, Sterie AC, Rubli Truchard E, Jox RJ. Implementing advace care planning in early dementia care: results and insights from a pilot interventional trial. BMC Geriatr 2021;21:573.
10. Bosisio F, Jox RJ, Jones L, Rubli Truchard E. Planning ahead with dementia: what roles can advance care planning play? A review on opportunities and challenges. Swiss Med Wkly 2018:148w14706.
11. Jox RJ, Black I, Borasio GD, Anneser J. Voluntary stopping of eating and drinking: is medical support ethically justified? BMC Med 2017;15(1):186.
12. Largent EA, Lowers J, Pope TM, Quill TE, Wynia MK. When people facing dementia choose to hasten death: the landscape of current ethical, legal, medical, and social considerations in the United States. Hastings Cent Rep 2024 ;54 Suppl 1:S11-21.
13. Jox RJ. Ethische Fragen im Zusammenhang mit der Ernährung von Menschen mit Demenz. Zeitschrift für medizinische Ethik 2022;68:49-61.
14. Commission nationale d’éthique pour la médecine humaine, Considérations éthiques sur le nouveau droit de la protection de l’adulte, tenant compte en particulier de la démence, prise de position n°17/2011dans l’article en ligne sous www.medinfo-verlag.ch
Von der Auseinandersetzung mit dem eigenen ökologischen Fussabdruck über neue Erkenntnisse zu Kardiomyopathien bis hin zu optimierten Behandlungsstrategien für die akute Lungenembolie – in dieser Ausgabe präsentieren wir Ihnen drei Beiträge, die die Vielschichtigkeit der modernen Kardiologie beleuchten:
Der erste Artikel widmet sich einem Thema, das längst überfällig ist: Nachhaltigkeit in der Kardiologie. Der Gesundheitssektor verursacht weltweit 4–5 % der gesamten Treibhausgasemissionen – ein Anteil, der in Zeiten der Klimakrise nicht länger ignoriert werden kann. Der Artikel bietet einen umfassenden Überblick über die Zusammenhänge zwischen Medizin und Klimawandel, zeigt die grössten Emissionsquellen in der Kardiologie auf und diskutiert innovative Lösungen, um den ökologischen Fussabdruck zu reduzieren. Dabei wird deutlich: Jede Massnahme zählt – von energieeffizienten Geräten über nachhaltige Materialien bis hin zu klimafreundlichen Transportkonzepten.
Der zweite Beitrag dieser Ausgabe beschäftigt sich mit den häufigsten Kardiomyopathie-Formen und stellt die aktuellen pathophysiologischen Erkenntnisse, diagnostischen Verfahren und therapeutischen Konzepte vor. Neben der hypertrophen, der dilatativen und der arrhythmogenen rechtsventrikulären Kardiomyopathie wird auch eine neue, von der Europäischen Gesellschaft für Kardiologie definierte Entität – die nicht-dilatierte linksventrikuläre Kardiomyopathie – vorgestellt. Die Kardiomyopathien sind ein vielschichtiges Krankheitsbild, dessen Management sich, wie in vielen anderen medizinischen Fachgebieten, zunehmend individualisiert.
Abgerundet wird die Ausgabe durch einen Beitrag zur akuten Lungenembolie, einer der häufigsten Ursachen für kardiovaskuläre Morbidität und Mortalität. Die Autoren erläutern, wie entscheidend eine frühzeitige Diagnostik und Risikostratifizierung für das Patientenmanagement sind. Von der Antikoagulation im ambulanten Bereich bis hin zu lebensrettenden Reperfusionsmassnahmen im Rahmen spezialisierter Lungenembolie-Teams – der Artikel zeigt, wie sich interdisziplinäre und strukturierte Vorgehensweisen in der klinischen Praxis
bewähren.
Diese drei Artikel verdeutlichen, wie dynamisch sich die Kardiologie weiterentwickelt – sei es in Richtung Nachhaltigkeit, durch präzisere Diagnosen oder optimierte Therapieansätze. Wir wünschen Ihnen eine inspirierende Lektüre und freuen uns auf angeregte Diskussionen zu diesen wichtigen Themen.
Kardiomyopathien sind Herzmuskelerkrankungen mit strukturellen und funktionellen Anomalien des Herzmuskels. In dieser Übersicht werden pathophysiologische Aspekte, die klinische Manifestation, die Diagnosestellung, die Risikostratifizierung und die neusten therapeutischen Konzepte der drei häufigsten Kardiomyopathieformen – hypertrophe Kardiomyopathie (HCM), dilatative Kardiomyopathie (DCM) und arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC) – aufgezeigt. Zusätzlich berichten wir über die in den Richtlinien für das Management von Kardiomyopathien der Europäischen Gesellschaft für Kardiologie (ESC) von 2023 neu definierte Entität der nicht-dilatierten linksventrikulären Kardiomyopathie (NDLVC).
Cardiomyopathies are usually hereditary conditions of the heart muscle including structural and functional abnormalities. In this overview we cover pathophysiological aspects, clinical manifestations, diagnostics, risk stratification and the newest therapeutic concepts of the three most common forms of cardiomyopathy: hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and arrhythmogenic cardiomyopathy (ARVC). Furthermore, we report on the newly described for of non-dilated left ventricular cardiomyopathy (NDLVC).
Key Words: hypertrophic cardiomyopathy, dilated cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, non-dilated left ventricular cardiomyopathy.
Einleitung
Kardiomyopathien sind Herzmuskelerkrankungen, bei denen der Herzmuskel strukturell und funktionell auffällig ist, ohne dass eine erkennbare Grunderkrankung vorliegt (1).
Obwohl viele dieser Erkrankungen genetisch bedingt sind, ist eine rein genetische Klassifikation in der klinischen Praxis weder sinnvoll noch möglich (2). Es ist wichtig zu erkennen, dass verschiedene Phänotypen von Kardiomyopathien innerhalb einer Familie koexistieren können und dass der Progress der Erkrankung beim einzelnen Patienten von einem Kardiomyopathie-Phänotyp zu einem anderen führen kann (3). Daher werden die Kardiomyopathien weiterhin nach morphologischen und funktionellen Kriterien eingeteilt, wobei es zu Überschneidungen zwischen den verschiedenen Formen kommen kann (2). Es ist daher nicht möglich, ein einziges Klassifikationsschema für alle möglichen Ursachen und Krankheitsbilder zu erstellen. So wurde in den ESC Guidelines 2023 die bestehende klinische Klassifikation aktualisiert, um neue phänotypische Beschreibungen aufzunehmen und die Terminologie zu vereinfachen, während sie gleichzeitig einen konzeptionellen Rahmen für Diagnose und Behandlung bietet (3). Es wird jedoch empfohlen, bei der Nomenklatur und Diagnose der Erkrankung nach dem bei der Präsentation vorherrschenden kardialen Phänotyp vorzugehen (3).
In diesem Artikel werden die drei häufigsten Formen der Kardiomyopathien beschrieben: die hypertrophe Kardiomyopathie (HCM), die dilatative Kardiomyopathie (DCM) und die arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC) sowie die neu definierte Entität der nicht-dilatierten linksventrikulären Kardiomyopathie (NDLVC).
Hypertrophe Kardiomyopathie (HCM)
Definition
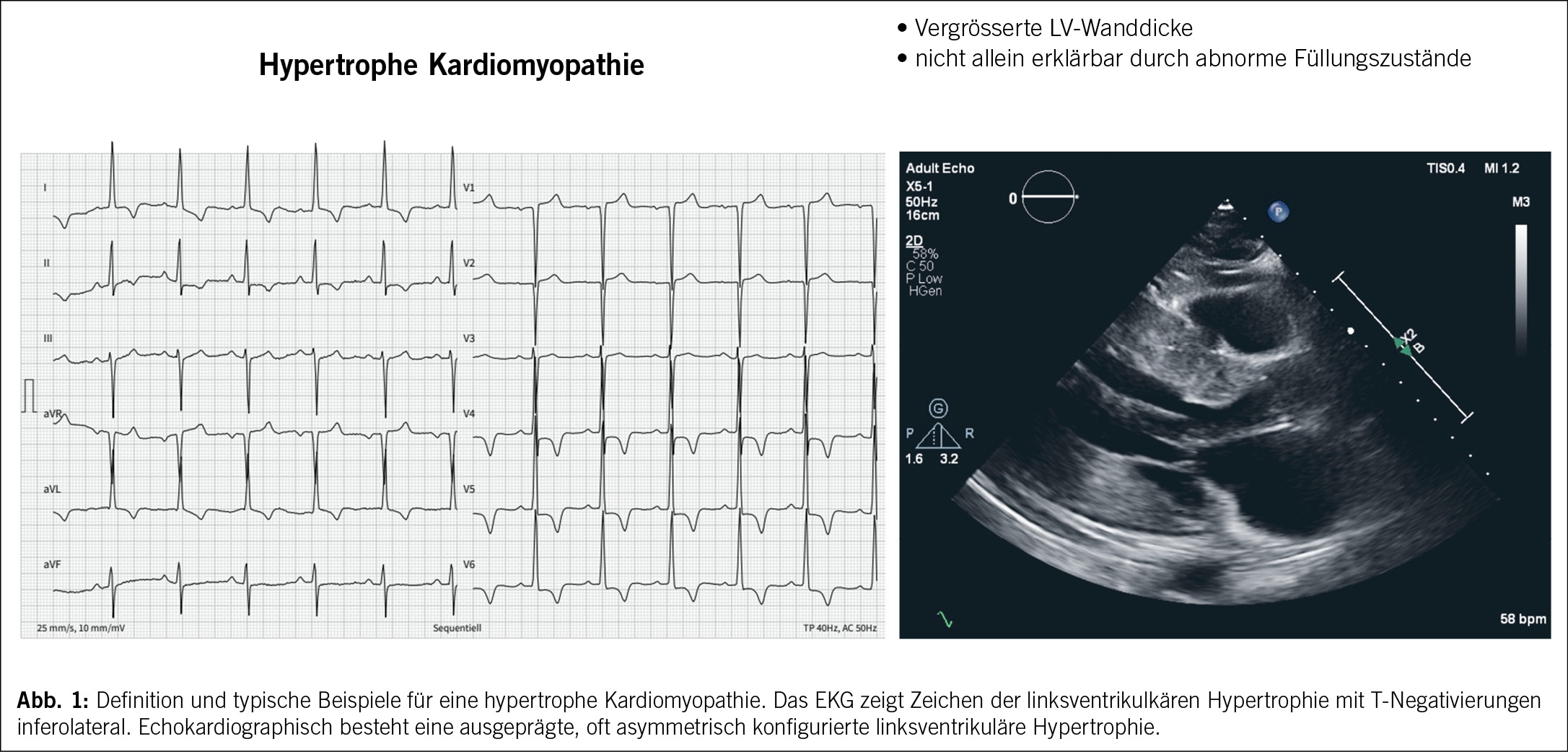
Die HCM ist definiert als das Vorliegen einer erhöhten linksventrikulären Wanddicke (mit oder ohne rechtsventrikulärer Hypertrophie) oder Masse, die nicht allein durch abnormale linksventrikuläre Füllungszustände erklärt werden kann (3). (Abb. 1)
Die HCM ist eine relativ häufige genetisch bedingte Herzerkrankung, die in der Regel autosomal dominant vererbt wird und weltweit in allen Bevölkerungsgruppen vorkommt (1).
Pathogenese
Die Annahme einer morphologischen Krankheitsdefinition impliziert eine Reihe unterschiedlicher Ätiologien, die zu einer hypertrophen Kardiomyopathie führen können. Ca. 60 % aller Fälle sind auf Varianten in Genen für kardiale Sarkomerproteine, insbesondere in der schweren β-Myosinkette (MYH7) und dem Myosin-bindenden Protein C (MYPBC3) zurückzuführen (1). Erbliche Stoffwechselstörungen wie Morbus Fabry, infiltrative Speichererkrankungen wie Amyloidose, neuromuskuläre und endokrine Pathologien, Mitochondriopathien und genetische Syndrome verursachen etwa 5-10 % der Fälle. Für einige von ihnen gibt es kurative Optionen (4).
Die klassische sarkomerische Form der HCM ist typischerweise eine monogenetische Erkrankung. Das typische Erscheinungsbild mit diastolischer Dysfunktion und ventrikulären Arrhythmien wird durch die Dysfunktion des sarkomerischen Proteins und das dadurch verursachte adverse Remodelling verursacht. Auf zellulärer Ebene sind vor allem Veränderungen der Aktin-Myosin-Interaktionen und der Ca2+-Sensitivität zu verzeichnen. Für das Verständnis der aktuellen Therapieoptionen ist hier vor allem relevant, dass HCM-verursachende Genvarianten zu vermehrten kardialen Aktin-Myosin-Querbrücken, welche wiederum für hyperdynamische Kontraktionen und gesteigerten Energieverbrauch verantwortlich sind (1, 5, 6).
Klinik
Das klinische Bild der HCM kann sehr unterschiedlich sein. Es kann als Zufallsbefund bei einer Vorsorgeuntersuchung entdeckt werden oder sich als Auftreten neuer Symptome wie Dyspnoe, Angina pectoris, Palpitationen und Synkopen oder sich im schlimmsten Fall primär durch einen Herzstillstand zeigen (5).
Diagnose
Bei Erwachsenen kann die klinische Diagnose einer HCM durch eine maximale enddiastolische Wanddicke von ≥ 15 mm in einem oder mehreren linksventrikulären Segmenten bei Fehlen anderer Ursachen der Hypertrophie, gemessen mittels Echokardiographie, Magnetresonanztomographie des Herzens oder Computertomographie definiert werden (1).
Wie bei allen Kardiomyopathien berichten die Patienten sehr unterschiedliche Beschwerden wie Dyspnoe, Thoraxschmerzen, Palpitationen, Synkopen oder Leistungsintoleranz, viele Patienten werden jedoch auch als Zufallsbefund diagnostiziert (3). Ein wichtiger Teil der Anamnese ist die Analyse des Stammbaums über drei Generationen bezüglich Herzerkrankungen, plötzlichem Herztod oder unklaren Unfällen, welche auf Synkopen hinführen könnten. Das 12-Kanal-EKG ist häufig pathologisch und kann daher ein hilfreiches Instrument sein, da Zeichen einer linksventrikulären Hypertrophie, ST-, T- sowie Q-Wellenanomalien nachgewiesen werden können. Ebenso wichtig ist das 24-Stunden-EKG, mit dem das Vorliegen nicht anhaltenden ventrikulärer Tachykardien dokumentiert wird und das Risiko eines plötzlichen Herztodes berechnet werden kann. Zusätzlich kann Vorhofflimmern erkannt und damit das Schlaganfallrisiko bestimmt werden (7).
Die Echokardiographie ist das wichtigste diagnostische Mittel bei der Abklärung einer HCM. Sie kann die Diagnose stellen, die Ursache der Symptome identifizieren und ist somit nützlich für das Therapiemanagement und schliesslich für die Risikostratifizierung des plötzlichen Herztodes (7).
Risikostratifizierung
Da die HCM als eine relevante Ursache für den plötzlichen Herztod vor allem bei jungen Menschen gilt, ist es ein Ziel, das Risiko für das Auftreten maligner Arrhythmien abzuschätzen und ein geeignetes Risikomodell zur Abschätzung der Häufigkeit zukünftiger kardialer Ereignisse zu finden, um diese durch die Implantation eines ICDs zu verhindern.
Die Empfehlung, die Überlebenden von SCD mit einem ICD zu versorgen, wurde bereits vor Jahren ausgesprochen; die Debatte über die Primärprophylaxe einer ICD-Implantation in der HCM-Population ist jedoch eine schwierigere Frage. Im Jahr 2014 haben O’Mahony et al. ein Risikoprädiktionsmodell, das nun von der ESC zur Stratifizierung des individuellen Risikos für SCD bei HCM und zur Unterstützung der Entscheidung für eine ICD-Implantation dient, vorgestellt (8). Da das Risiko für tödliche Arrhythmien über einen langen Zeitraum bestehen bleiben kann, ist es wichtig, das Risiko des Patienten regelmässig neu zu bewerten (1).
Management
Das Ziel der HCM-Behandlung besteht darin, die Symptome der Patienten (Angina Pectoris, Herzinsuffizienz, Synkopen, Palpitationen usw.) zu lindern und ein Fortschreiten der Krankheit sowie schwerwiegende kardiovaskuläre Komplikationen und den plötzlichen Herztod zu verhindern (1).
Das Hauptmerkmal, das bei HCM-Patienten zu Symptomen führt, ist das Vorhandensein einer linksventrikulären Ausflusstraktobstruktion, allgemein definiert als ein Doppler-Spitzengradient im linksventrikulären Ausflusstrakt (LVOT) von ≥ 30 mmHg (5).
Die Erstlinientherapie in dieser Situation umfasst nicht-vasodilatierende β-Blocker und bei Unverträglichkeit oder Unwirksamkeit Nicht-Dihydropyridin-Kalziumkanalblocker. Zusätzlich kann bei schlechtem klinischem Ansprechen das Antiarrhythmikum Disopyramid verabreicht werden (1).
Kürzlich wurde eine neue vielversprechende Therapie mit dem ersten kardialen Myosin-Inhibitor zugelassen. Mavacamten ist ein reversibler kardialer Myosin-Adenosintriphosphatase (ATPase)-Hemmer, der die Bildung von Aktin-Myosin-Kreuzbrücken reduziert und dadurch die Kontraktilität verringert und den Energiehaushalt des Herzmuskels verbessert. In der kürzlich veröffentlichten klinischen Studie zur Bewertung von Mavacamten bei Erwachsenen mit symptomatischer obstruktiver hypertropher Kardiomyopathie (EXPLORER-HCM), reduzierte Mavacamten den Gradienten des linksventrikulären Ausflusstrakts (LVOT) und verbesserte die körperliche Leistungsfähigkeit im Vergleich zu Placebo bei Patienten mit HCM und symptomatischer LVOT (NYHA II-III und EF >55%); 27% der Patienten, die Mavacamten erhielten, hatten eine Reduzierung des LVOT-Gradienten auf <30 mmHg und verbesserten sich in die NYHA-Klasse I (9). Diese Therapie hat bereits Eingang in die aktuellen Richtlinien gefunden (10).
Für Patienten mit persistierenden Symptomen trotz medikamentöser Therapie stehen invasive Optionen wie die Septum-Myektomie oder die Septum-Alkohol-Ablation in spezialisierten Zentren zur Verfügung. Die Therapie von HCM-Patienten mit Herzinsuffizienz (LVEF < 50 %) richtet sich nach den etablierten Leitlinien zur Herzinsuffizienz (1).
Vorhofflimmern ist die häufigste persistierende Arrhythmie bei HCM-Patienten, die bei etwa 20% der Patienten vorkommt und mit einer hohen Inzidenz thromboembolischer Ereignisse assoziiert ist. Eine orale Antikoagulation sollte daher bei jedem Patienten mit HCM und paroxysmalem, persistierendem oder permanentem Vorhofflimmern empfohlen werden, unabhängig vom CHA2DS2Vasc-Score, es sei denn, es besteht eine strenge Kontraindikation (3, 11).
Erwachsene Patienten mit HCM können auch eine atherosklerotische Koronararterienerkrankung (KHK) entwickeln. Die Berichte über die Prävalenz der KHK bei HCM variieren, aber bei bis zu 20 % der HCM-Patienten wurde eine koexistierende KHK festgestellt. Der Befund einer schweren KHK bei HCM-Patienten ist im Vergleich zu Patienten ohne KHK oder mit leichter bis mittelschwerer KHK mit einem reduzierten Gesamtüberleben, einem reduzierten Überleben ohne SCD assoziiert. Dieser Befund kann daher als zusätzlicher prognostischer Faktor bei der Beurteilung von Patienten mit HCM herangezogen werden (12).
Dilatative Kardiomyopathie
Definition
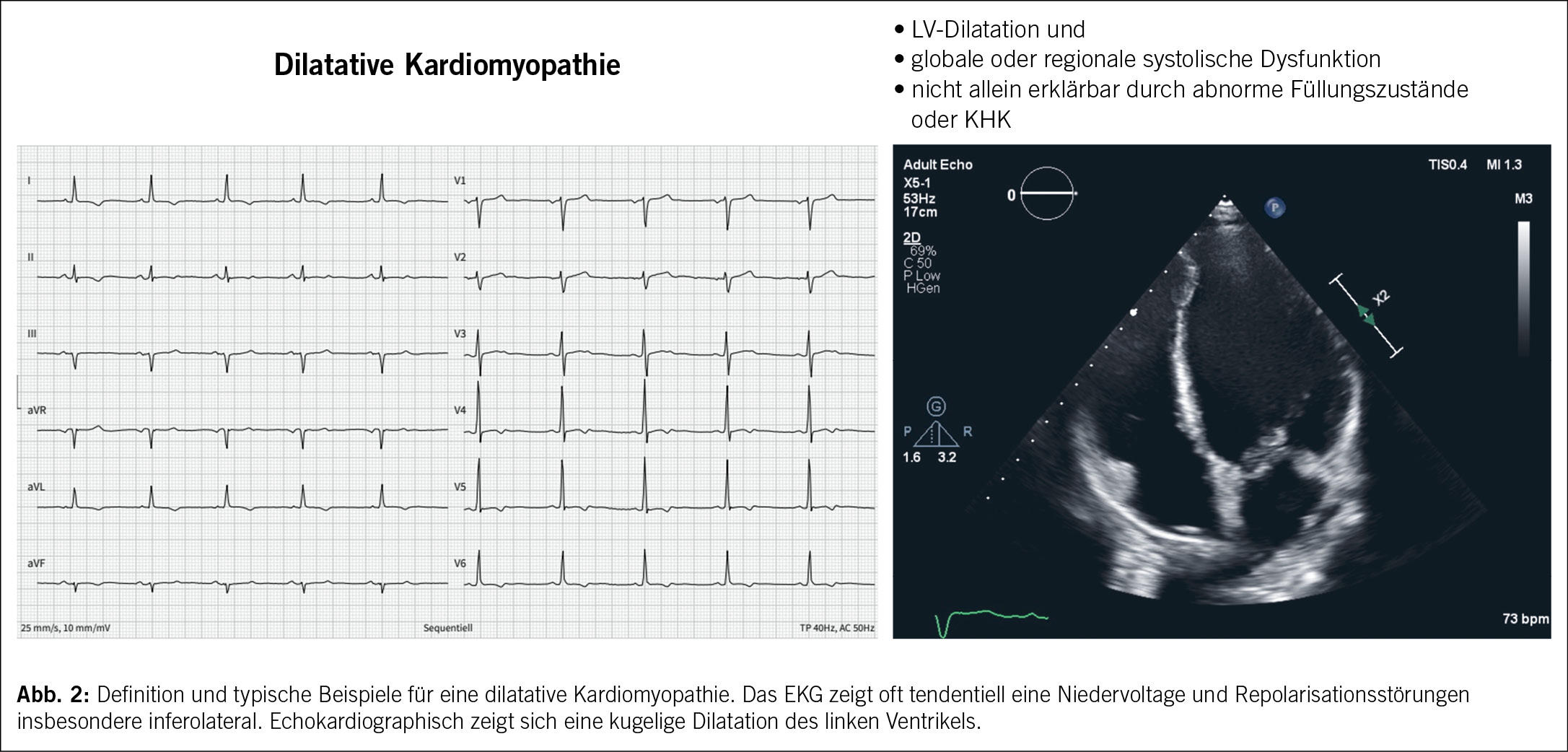
Die dilatative Kardiomyopathie (DCM) ist definiert als linksventrikuläre oder biventrikuläre Dilatation sowie systolische Dysfunktion bei Fehlen einer ischämischen, hypertensiven oder kongenitalen Herzerkrankung (1). Die ESC klassifiziert die Kardiomyopathien als familiär oder nicht-familiär, je nachdem, ob ein genetisches Substrat vorliegt oder nicht (3). (Abb. 2)
Pathogenese
Es ist bekannt, dass DCM durch eine Reihe von Prozessen verursacht werden kann. Diese wird in der klinischen Praxis gerne als «nicht-ischämische Kardiomyopathie» bezeichnet. Diese Bezeichnung entspricht jedoch nicht dem heutigen Kenntnisstand, da DCM eine Familie von Krankheiten darstellt, die durch komplexe Wechselwirkungen zwischen Umwelt und genetischer Prädisposition gekennzeichnet sind (13). Selbst in Fällen mit scheinbar eindeutiger Ursache muss zusätzlich von einer genetischen Ursache ausgegangen werden. Patienten mit alkoholbedingter DCM weisen z.B. im Vergleich zu gesunden Probanden und Bevölkerungskontrollen signifikant häufiger seltene, proteinalterierende genetische Varianten auf (14). Bei bis zu 50% der Patienten mit DCM kann eine pathogene Genvariante gefunden werden (15). Auch wenn das Wissen um die genetische Grundlage der Erkrankung in der klinischen Praxis «akademisch» erscheinen mag, kann es für den Patienten von grosser Bedeutung sein, nicht nur, um ein Kaskadenscreening bei Familienmitgliedern anzubieten, sondern auch für die Risikoabschätzung von Patienten, da das Risiko für SCD bei DCM-Patienten mit einer desmosomalen oder Lamin A/C-Genvariante deutlich erhöht ist (15). Eine Besonderheit der DCM ist das breite Spektrum möglicher Ätiologien, die einen gemeinsamen pathogenetischen Weg zur Herzschädigung implizieren, unabhängig davon, ob es sich um eine umweltbedingte oder genetische Ursache handelt, die eine Entzündung auslöst und eine Infiltration von Immunzellen verursacht, um das geschädigte Myokard zu reparieren (1).
Klinik
Die klinischen Merkmale eines Patienten mit DCM hängen hauptsächlich mit Symptomen der Herzinsuffizienz zusammen, d. h. Dyspnoe, Müdigkeit und Leistungsintoleranz, Beinödeme, pulmonale Stauung oder durch Herzrhythmusstörungen, wie Herzklopfen, Schwindel oder Synkopen. Das Ausmass dieser Symptome hängt von der Schwere der links- oder biventrikulären Dysfunktion ab, und das Auftreten kann akut, subakut oder chronisch sein und unterschiedliche Ursachen haben (16). Zusätzlich können Befunde auftreten, die Hinweise auf die genetische Grunderkrankung liefern können, wie z.B. Muskelbeschwerden (3).
Diagnostik
Die diagnostischen Kriterien basieren auf der kardialen Bildgebung, in erster Linie auf der Echokardiographie zum Nachweis eines enddiastolischen linksventrikulären Volumens oder Durchmesser > 2 SD vom Normbereich, korrigiert für Alter und Körperoberfläche sowie einer Ejektionsfraktion < 50%. Eine Koronarangiographie kann zum Ausschluss einer begleitenden koronaren Herzkrankheit durchgeführt werden. Ein MRT des Herzens kann die Dilatation bestätigen und ist entscheidend für die Beurteilung von Ödemen/Fibrosen durch spätes Gadolinium-Enhancement, das stark auf eine Entzündung hinweist und somit für die prognostische Stratifizierung nützlich ist (17). Die Myokardbiopsie kann bei Verdacht auf eine Speicherkrankheit, bei infiltrativen Prozessen und bei möglicher entzündlicher Kardiomyopathie eingesetzt werden (1).
Das breite Spektrum unterschiedlicher Ursachen erfordert aufwendige Untersuchungen, um das phänotypische Bild der DCM von der tatsächlichen Pathologie zu unterscheiden, da die Identifizierung einer spezifischen Ätiologie eine krankheitsspezifische Behandlung ermöglichen oder auf die Notwendigkeit eines Familienscreenings hinweisen und Informationen über die Prognose liefern kann (1).
Eine detaillierte Familienanamnese (bis zur dritten Generation) und eine Anamnese der Noxenexposition, Blutuntersuchung auf Eisenspeicher, Nierenfunktion, Elektrolyte, Kreatinkinase, CRP, Schilddrüsenfunktion und 12-Kanal-EKG ist unerlässlich (1).
Risikostratifizierung
Die Vorhersage von SCD ist ein schwieriger Aspekt der klinischen Versorgung von Patienten mit DCM. Implantierbare Kardioverter-Defibrillatoren (ICD) sind wirksam bei der Behandlung lebensbedrohlicher ventrikulärer Arrhythmien und bei der Prävention des plötzlichen Herztods, sind aber auch mit Komplikationen verbunden, insbesondere bei jungen Patienten, die im Laufe ihres Lebens die ICDs mehrmals ausgetauscht werden müssen (3). In der Sekundärprävention ist entsprechend die Empfehlung zur ICD-Implantation unumstritten (3).
In der Primärprävention besteht bei symptomatischen Patienten mit einer LVEF von ≤ 35% eine Klasse IIa-Indikation zur ICD-Implantation (3). Bei Patienten mit einer LVEF von > 35% müssen aufgrund der vielfältigen Ätiologie der Erkrankung zusätzliche Risikofaktoren in Betracht gezogen werden (3). Daher ist es notwendig, nach Anzeichen für Formen der Krankheit zu suchen, die ein höheres arrhythmogenes Potenzial aufweisen. Red Flags sind genetische Erkrankungen, insbesondere hochgradig arrhythmogene Formen wie Laminopathien, schwere ventrikuläre Arrhythmien, plötzlicher Herztod in der Familienanamnese, Skelettmuskelbeteiligung, EKG-Anomalien und eine posterolaterale Akinesie in der Echokardiographie. In diesen Fällen oder bei eindeutiger Familienanamnese wird eine genetische Untersuchung empfohlen, um Varianten mit erhöhtem Risiko zu erkennen (18).
Verschiedene Studien weisen darauf hin, dass der Genotyp eine Rolle für das SCD-Risiko spielt, wobei Patienten mit krankheitsverursachenden Varianten in PLN, DSP, LMNA, FLNC, TMEM43 und RBM20 unabhängig von der LVEF eine signifikant höhere Rate an schweren Herzrhythmusstörungen aufweisen als Patienten mit anderen Ursachen für DCM unabhängig von der LVEF (3). Somit sollte beim Nachweis solcher Varianten eine ICD-Implantation in einem früheren Stadium evaluiert werden.
Die zunehmende Durchführbarkeit von Gentests in Verbindung mit der Verfügbarkeit diagnostischer Untersuchungen für DCM-Patienten könnte es in Zukunft ermöglichen, detailliertere Risikostratifizierungsschemata zu definieren (16).
Management
Neben der möglichen ätiologiebasierten Therapie ist die Standardtherapie der symptomatischen DCM mit reduzierter LVEF gemäss den aktuellen ESC-Leitlinien für Herzinsuffizienz wie folgt: Angiotensin-Converting-Enzym-Inhibitoren (ACE-I) oder Angiotensin-Rezeptor-Blocker (ARB), in Verbindung mit β-Blockern, Mineralcorticoid-Antagonisten (MRA) und in ausgewählten Fällen Vasodilatatoren (19). In letzter Zeit wurden die Angiotensin-Rezeptor-Neprilysin Inhibitoren (ARNI) und Ivabradin in die Liste der wirksamen Behandlungen für diejenigen, die nicht auf eine optimale medizinische Therapie ansprechen, hinzugefügt. Bei anhaltenden Symptomen (NYHA ≥ II) trotz optimaler Therapie, linksventrikulärer systolischen Dysfunktion (LVEF ≤ 35 %) und ventrikulären Asynchronie (QRS-Dauer ≥ 130 ms) ist eine kardiale Resynchronisationstherapie (CRT) indiziert (19).
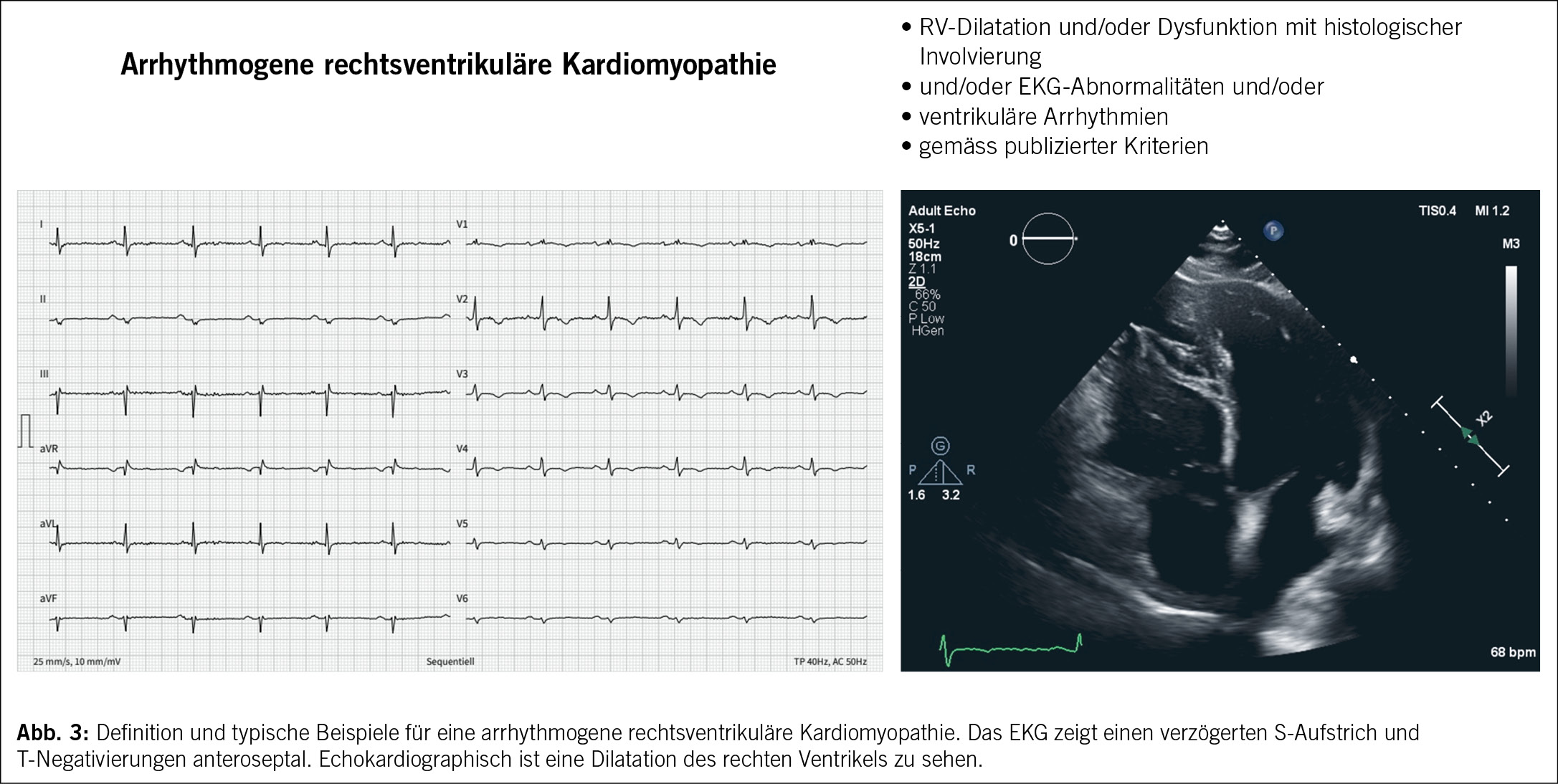
Die arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC) wird als eine dominant genetisch determinierte Herzmuskelerkrankung definiert. Die Pathologie ist durch einen fettig-fibrösen Ersatz des rechtsventrikulären (und linksventrikulären) Myokards charakterisiert (20). Klinisch kommt es zu ausgeprägten ventrikulären Arrhythmien und eine Beeinträchtigung der systolischen Funktion des Ventrikels (20). (Abb. 3)
Nomenklatur
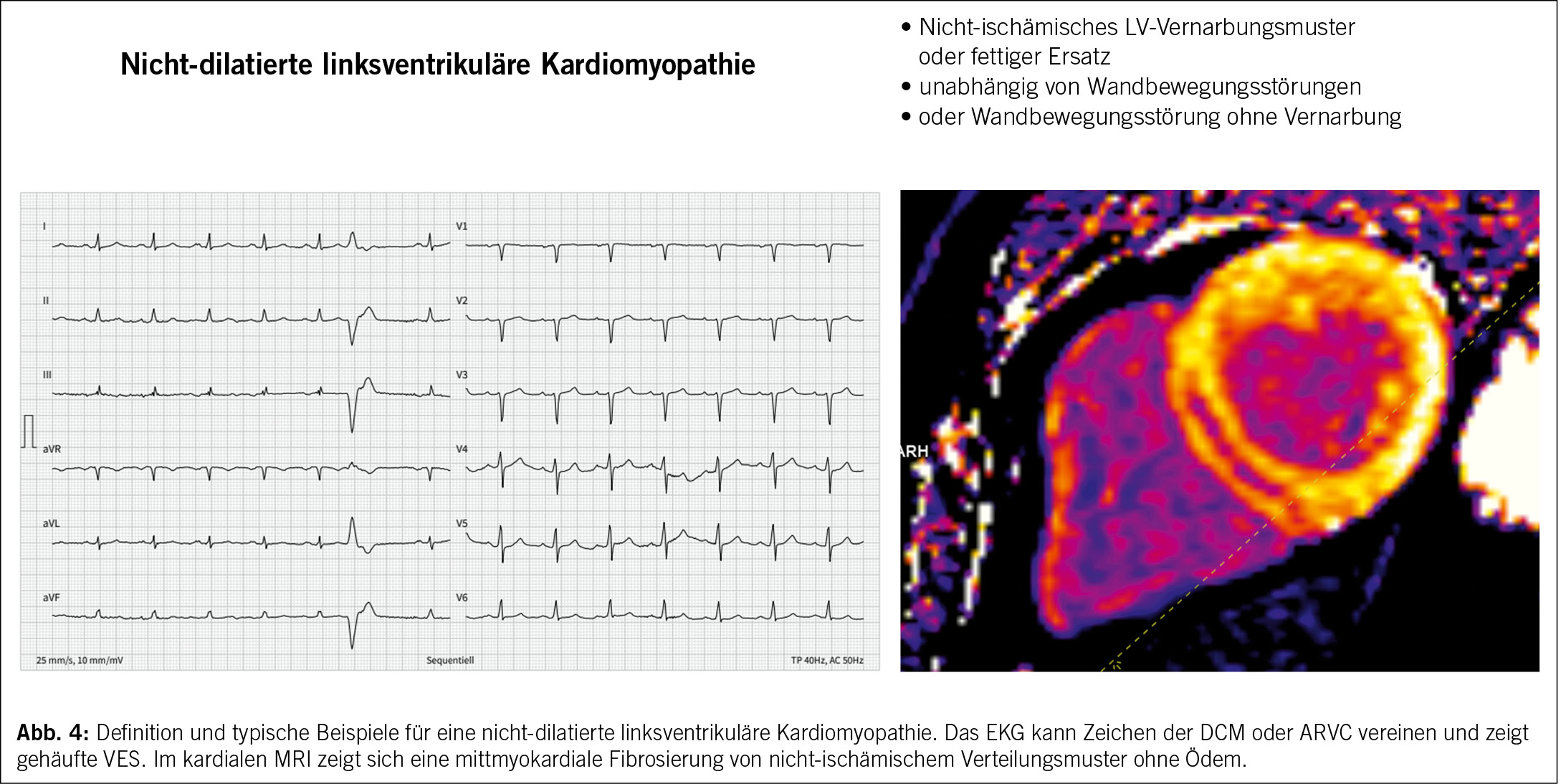
Bezüglich der korrekten Nomenklatur für die Erkrankung besteht eine jahrelange Diskussion. Aktuell werden dominant zwei Termini parallel verwendet: (i) die «arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC)», welcher die Dominanz der rechtsventriuklären Veränderungen betont und in den aktuellen ESC-Richtlinien verwendet wird (3). Linksventrikuläre Formen sollen somit als NDLVC diagnostiziert werden. (ii) Die «arrhythmogene Kardiomyopathie (ACM)», welche die biventrikulären, rechts- und linksventrikulären Formen unterstreicht und in den neuesten Diagnosekriterien verwendet wird (21). Wir haben uns für diesen Artikel für ARVC als Terminus entschieden.
Pathogenese
Die Pathogenese der ARVC beruht typischerweise auf Varianten in Genen, welche hauptsächlich für Proteine des kardialen Desmosoms, aber auch der Kernhülle, des Natriumkanals oder des Sarkomers kodieren (20). Die Desmosomen sind für die Zell-Zell-Adhäsion verantwortlich und wichtige Mediatoren der intra- und interzelluläre Signalwege. Der Verlust der Adhäsion zwischen den Myozyten führt dazu, dass sie sich voneinander lösen und es dann allmählich zum Absterben der Herzmuskelzellen kommt. Somit wird dies durch fibröses oder faserig-fettiges Gewebe ersetzt, welches oft durch Entzündungen vermittelt wird. Dieses gilt als wesentlicher Mechanismus der Kardiomyopathie (22, 23).
Der Trigger ist die körperliche Belastung, die diesen Adhäsionsdefekt verschlimmert, wobei der rechte Ventrikel stärker betroffen ist, da seine Wand dünner ist als die des linken Ventrikels (1). Die Myokardatrophie ist ein fortlaufender Prozess, welcher vom Epikard beginnt und sich in Richtung Endokard ausbreitet und schliesslich transmural wird. Somit kommt es zu einer progressiven Ausdünnung der Wände und schliesslich zur Aneurysmabildung (1). So wird bei Patienten, die kompetitiven Sport betreiben, ein früheres Einsetzen von Symptomen sowie ein höheres Risiko für ventrikuläre Arrhythmien beobachtet (24). In einigen Fällen verläuft dieser Prozess jedoch nicht kontinuierlich, sondern in Form von periodischen akuten Schüben einer ansonsten stabilen Erkrankung, die eine Myokarditis imitieren, was eine wichtige Rolle bei der Pathogenese des Phänotyps spielen könnte (1, 20). So zeigen eine Grosszahl der autoptisch untersuchten Herzen mit ARVC ein entzündliches Infiltrat (20).
Klinik
Die Symptomatik der ARVC kann stark variieren, und zwar von asymptomatischen Familienmitgliedern mit verborgenen strukturellen Anomalien und ohne Arrhythmien bis hin zu symptomatischen Patienten, die einen plötzlichen Herztod erleiden oder die sich aufgrund einer therapierefraktären Herzinsuffizienz einer Herztransplantation unterziehen müssen (1, 25). Die häufigste klinische Präsentation ist zur Abklärung unspezifischer kardialer Symptome wie z.B. Palpitationen, Dyspnoe oder Synkopen, oder aber, die Patienten werden wegen Arrhythmien oder zum Familienscreening vorstellig (26). Leider kommt es immer wieder vor, dass bislang komplett unauffällige Patienten sich erstmals mit einem plötzlichen Herztod manifestieren (20).
Diagnose
Die Diagnose erfolgt durch verschiedene Modalitäten und wurde unter anderem durch die 2010 Task Force Kriterien (27) definiert, welche zuletzt durch die Padua-Kriterien ersetzt wurden, die auf verschiedenen Aspekten wie strukturellen Veränderungen, Depolarisations-/Repolarisationsstörungen, Arrhythmien und Familienanamnese beruhen (27). Um eine ARVC diagnostizieren zu können, müssen mindestens zwei unterschiedliche diagnostische Modalitäten eingesetzt werden. Dabei ist zu berücksichtigen, dass elektrische Veränderungen vor sichtbaren strukturellen Modifikationen auftreten können (28). Um die sichere Diagnose einer ARVC stellen zu können, müssen zwei Hauptkriterien oder ein Hauptkriterium und zwei sekundäre Kriterien oder vier sekundäre Kriterien aus verschiedenen Kategorien erfüllt werden. Für eine Borderline-Diagnose genügen ein Hauptkriterium und ein Nebenkriterium oder drei Kriterien aus verschiedenen Kategorien und für eine mögliche Diagnose ein Hauptkriterium oder zwei Nebenkriterien aus verschiedenen Kategorien (27).
Natürlicher Verlauf der Erkrankung
Der Verlauf der ARVC ist, wie bei allen Kardiomyopathien, höchst variabel und reicht von relativ oligosymptomatischen Patienten zu Patienten mit anhaltenden ventrikulären Arrhythmien, plötzlichem Herztod und Herzinsuffizienz. (25, 26, 29). Hier ist speziell zu erwähnen, dass der plötzliche Herztod als Endpunkt in allen Beobachtungsstudien relativ selten auftrat, obwohl durchschnittlich ein Risiko für anhaltende ventrikulären Arrhythmien von 3.7 – 10.6%/Jahr angegeben wird. (3) Dies hängt speziell damit zusammen, dass ARVC-Patienten öfters an anhaltenden ventrikulären Arrhythmien leiden, welche hämodynamisch gut toleriert werden und entsprechend nicht zu einem plötzlichen Herztod führen (3, 25).
Risikostratifizierung
Die grösste Herausforderung bei der Behandlung der ARVC ist nach wie vor die Risikostratifizierung für den plötzlichen Herztod und somit die Indikationsstellung für die ICD-Implantation. Das Hauptproblem bei der Indikationsstellung für einen ICD ist zwischen Komplikationen, fehlendem Nutzen und geretteten Leben abzuwägen. Basierend auf Daten zur jährlichen Mortalität in Verbindung mit Risikofaktoren teilt der Konsensalgorithmus der International Task Force die Patienten in drei verschiedene Risikokategorien ein (29). Das geschätzte Risiko für schwere arrhythmische Ereignisse liegt in der Hochrisikokategorie bei > 10 %/Jahr, in der Zwischenrisikokategorie zwischen 1 und 10 %/Jahr und in der Niedrigrisikokategorie bei < 1 %/Jahr. Kürzlich wurde in einer retrospektiven Studie mit ARVC-Patienten aus fünf Registern ein neues Modell zur Vorhersage des 5-Jahres-Risikos für ventrikuläre Arrhythmien bei ARVC vorgestellt, das vielversprechende, wenn auch nicht unumstrittene Ergebnisse liefert (31). Dieses Prognosemodell (ARVC Risk Calculator, www.arvcrisk.com) umfasst sieben Variablen: Geschlecht, Alter, kürzlich aufgetretene kardiale Synkopen (<6 Monate), nicht anhaltende ventrikuläre Tachykardien, 24-Stunden-VES (ventrikulkäre Extrasystolen)-Anzahl, Anzahl der Ableitungen mit T-Wellen-Inversion in den präkordialen und inferioren Ableitungen und rechtsventrikuläre Ejektionsfraktion (31). In der Originalpublikation wurde dieser Score jedoch nicht extern validiert. Mehrere Studien untersuchten die Leistungsfähigkeit des ARVC-Risikorechners in externen, unabhängigen Kohorten und zeigten, dass das Modell grundsätzlich Patienten mit einem erhöhten Risiko für ventrikuläre Arrhythmien gut identifiziert (32, 33). Es ist jedoch zu bemerken, dass der Rechner das Risiko für anhaltende ventrikuläre Arrhythmien – und nicht SCD – prognostiziert und dass das Risiko insgesamt, speziell aber bei Patienten mit niedrigem bis mittlerem Risiko, tendenziell überschätzt und die Aussagekraft bei Patienten Varianten in anderen Genen als PKP2 oder fehlenden pathogenen Varianten und nicht rechtsdominanten Formen der Erkrankung deutlich eingeschränkt ist (23, 33). Bislang wurde kein Schwellenwert validiert. Die ICD-Indikation sollte somit eine gemeinsame Entscheidung bleiben, die nicht nur auf dem Ergebnis des Scores, sondern auch auf der Gesamtbeurteilung des Patienten beruht (23).
Management
Das wichtigste klinische Ziel bei der Behandlung von ARVC ist die Verhinderung von SCD. Die derzeitigen therapeutischen Optionen umfassen Lebensstiländerungen, pharmakologische Optionen (β-Blocker und Antiarrhythmika), Katheterablation, ICD-Implantation und Herztransplantation (1).
Da körperliche Aktivität ein wichtiger Faktor ist, der die phänotypische Manifestation der Erkrankung favorisieren und damit lebensbedrohliche ventrikuläre Arrhythmien begünstigen kann, empfehlen die aktuellen Leitlinien die Abstinenz von Leistungssport für alle betroffenen Patienten und für Patienten mit einem hohen Risiko aufgrund einer pathogenen Variante (3). Leichte bis mittelschwere körperliche Aktivität können je nach Situation in einem gewissen Ausmass empfohlen werden – je intensiver die Aktivität, desto seltener sollte sie ausgeübt werden, im Sinne einer partizipativen Entscheidungsfindung (34).
Die Indikation für den Einsatz von β-Blockern bei ARVC beruht auf ihrer nachgewiesenen Wirksamkeit zur Reduktion der Inzidenz schwerer ventrikulärer Arrhythmien und in der Behandlung der Herzinsuffizienz (35). Daher sollte eine β-Blocker-Therapie bei allen Patienten mit eindeutiger Diagnose einer ARVC, speziell mit ventrikulären Arrhythmien (auch VES) empfohlen werden (3). Die vorliegende Evidenz zeigt, dass die Therapie mit Antiarrhythmika (Amiodaron und Sotalol) keinen ausreichenden Schutz vor plötzlichem Herztod bietet (35). Ferner sollte eine antiarrhythmische Therapie in Betracht gezogen werden, um die Arrhythmielast bei symptomatischen Patienten mit häufigen vorzeitigen ventrikulären Kontraktionen und nicht anhaltenden ventrikulären Tachykardien zu reduzieren (29).
Patienten mit ARVC, die eine rechts- oder biventrikuläre Herzinsuffizienz entwickeln – laut Literatur bis zu 13 % –, werden gemäss den ESC-Leitlinien 2016 mit Angiotensin-Converting-Enzym-Hemmern und Diuretika behandelt. Eine Herztransplantation kann die letzte Option bei schwerer Herzinsuffizienz sein, die auf die Standardtherapie nicht anspricht, oder bei unkontrollierbaren ventrikulären Tachyarrhythmien (25, 29). Zusätzlich sollte bei Patienten mit wiederholten appropriaten ICD-Therapien trotz β-Blockern eine Katheterablation der Kammertachykardien in Erwägung gezogen werden (3).
Die bisherige Definition von DCM und ARVC hat zur Folge, dass frühe und intermediäre Formen genetischer und erworbener Erkrankungen nicht genügend abgebildet wurden und somit das Risiko besteht, dass sie übersehen werden. Unter dieser Form werden Entitäten subsummiert, die früher z.B. als DCM ohne Dilatation, arrhythmogene linksventrikuläre Kardiomyopathie oder arrhythmogene DCM bezeichnet wurden. Bei dieser Form soll ein spezielles Augenmerk auf eine multiparametrische Abklärung, insbesondere auch die genetische Abklärung, geworfen werden, da die spezifische Ätiologie die klinische Behandlung beeinflusst. (Abb. 4)
Abkürzungen AAD Antiarrhythmische Medikamente ACE-I Angiotensin-Converting-Enzym-Inhibitoren ACM Arrhythmogene Kardiomyopathie ARB Angiotensin-Rezeptor-Blocker ARNI Angiotensin-Rezeptor-Neprilysin Inhibitoren ARVC Arrhythmogenic right ventricular cardiomyopathy CRT kardiale Resynchronisationstherapie DCM Dilatative Kardiomyopathie ESC Europäische Gesellschaft für Kardiologie HCM Hypertrophe Kardiomyopathie ICD Implantable Cardioverter Defibrillator LVEF linksventrikuläre Ejektionsfraktion LVOT Linksventrikulärer Ausflusstrakt MRA Mineralcorticoid-Antagonisten MYBPC3 Myosin-bindendes Protein C MYH7 schwere β-Myosinkette 7 NDLVC Nicht-dilatierte linksventrikuläre Kardiomyopathie NYHA New York Heart Association SCD Plötzlicher Herztod VA Ventrikuläre Arrhythmien VES Ventrikuläre Extrasystolen
Universitäres Zentrum für Hypertonie
Universitätsspital Basel
Petersgraben 4
4031 Basel
Annina.Vischer@usb.ch
PD Dr. A. Vischer hat Honorare von Bristol Myers Squibb, Amarin, Servier, Medtronic, Vifor und NovoNordisk erhalten.
◆ Kardiomyopathien werden primär nach ihren Phänotypen eingeteilt und behandelt.
◆ In der Regel handelt es sich um genetische Erkrankungen mit entsprechenden Implikationen für die ganze Familie
◆ Die Risikostratifizierung beruht auf verschiedenen Faktoren, die LVEF allein ist in der Regel nicht ausreichend
◆ Für die hypertrophe Kardiomyopathie gibt es neu spezifische Therapieansätze.
1. Schulz LP, Vischer AS. Cardiomyopathies in the Clinical Practice – an Overview. Praxis (Bern 1994). 2022;111(11):623-31.
2. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29(2):270-6.
3. Arbelo E, Protonotarios A, Gimeno JR, Arbustini E, Barriales-Villa R, Basso C, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023;44(37):3503-626.
4. Miles C, Fanton Z, Tome M, Behr ER. Inherited cardiomyopathies. Bmj. 2019;365:l1570.
5. Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733-79.
6. Zampieri M, Berteotti M, Ferrantini C, Tassetti L, Gabriele M, Tomberli B, et al. Pathophysiology and Treatment of Hypertrophic Cardiomyopathy: New Perspectives. Curr Heart Fail Rep. 2021;18(4):169-79.
7. Pantazis A, Vischer AS, Perez-Tome MC, Castelletti S. Diagnosis and management of hypertrophic cardiomyopathy. Echo Res Pract. 2015;2(1):R45-53.
8. O‘Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J. 2014;35(30):2010-20.
9. Olivotto I, Oreziak A, Barriales-Villa R, Abraham TP, Masri A, Garcia-Pavia P, Saberi S, Lakdawala NK, Wheeler MT, Owens A, Kubanek M, Wojakowski W, Jensen MK, Gimeno-Blanes J, Afshar K, Myers J, Hegde SM, Solomon SD, Sehnert AJ, Zhang D, Li W, Bhattacharya M, Edelberg JM, Waldman CB, Lester SJ, Wang A, Ho CY, Jacoby D; EXPLORER-HCM study investigators. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020 Sep 12;396(10253):759-769. doi: 10.1016/S0140-6736(20)31792-X. Epub 2020 Aug 29. Erratum in: Lancet. 2020 Sep 12;396(10253):758. doi: 10.1016/S0140-6736(20)31872-9.
10. Ommen SR, Ho CY, Asif IM, Balaji S, Burke MA, Day SM, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation. 2024;149(23):e1239-e311.
11. Guttmann OP, Rahman MS, O‘Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart. 2014;100(6):465-72.
12. Sorajja P, Ommen SR, Nishimura RA, Gersh BJ, Berger PB, Tajik AJ. Adverse prognosis of patients with hypertrophic cardiomyopathy who have epicardial coronary artery disease. Circulation. 2003;108(19):2342-8.
13. Merlo M, Cannatà A, Gobbo M, Stolfo D, Elliott PM, Sinagra G. Evolving concepts in dilated cardiomyopathy. Eur J Heart Fail. 2018;20(2):228-39.
14. Ware JS, Amor-Salamanca A, Tayal U, Govind R, Serrano I, Salazar-Mendiguchía J, et al. Genetic Etiology for Alcohol-Induced Cardiac Toxicity. J Am Coll Cardiol. 2018;71(20):2293-302.
15. Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J Am Coll Cardiol. 2019;74(11):1480-90.
16. Schultheiss HP, Fairweather D, Caforio ALP, Escher F, Hershberger RE, Lipshultz SE, et al. Dilated cardiomyopathy. Nat Rev Dis Primers. 2019;5(1):32.
17. Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Böhm M, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37(23):1850-8.
18. Paldino A, De Angelis G, Merlo M, Gigli M, Dal Ferro M, Severini GM, et al. Genetics of Dilated Cardiomyopathy: Clinical Implications. Curr Cardiol Rep. 2018;20(10):83.
19. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021;42(36):3599-726.
20. Corrado D, Basso C, Judge DP. Arrhythmogenic Cardiomyopathy. Circ Res. 2017;121(7):784-802.
21. Corrado D, Anastasakis A, Basso C, Bauce B, Blomström-Lundqvist C, Bucciarelli-Ducci C, et al. Proposed diagnostic criteria for arrhythmogenic cardiomyopathy: European Task Force consensus report. Int J Cardiol. 2024;395:131447.
22. Lin YN, Ibrahim A, Marbán E, Cingolani E. Pathogenesis of arrhythmogenic cardiomyopathy: role of inflammation. Basic Res Cardiol. 2021;116(1):39.
23. Gandjbakhch E, Redheuil A, Pousset F, Charron P, Frank R. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: JACC State-of-the-Art Review. J Am Coll Cardiol. 2018;72(7):784-804.
24. Ruwald AC, Marcus F, Estes NA, 3rd, Link M, McNitt S, Polonsky B, et al. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2015;36(27):1735-43.
25. Vischer AS, Castelletti S, Syrris P, McKenna WJ, Pantazis A. Heart failure in patients with arrhythmogenic right ventricular cardiomyopathy: Genetic characteristics. Int J Cardiol. 2019;286:99-103.
26. Vischer AS, Castelletti S, Syrris P, Bastiaenen R, Miles C, Akdis D, et al. Risk score for the exclusion of arrhythmic events in arrhythmogenic right ventricular cardiomyopathy at first presentation. Int J Cardiol. 2019;290:100-5.
27. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010;31(7):806-14.
28. Akdis D, Brunckhorst C, Duru F, Saguner AM. Arrhythmogenic Cardiomyopathy: Electrical and Structural Phenotypes. Arrhythm Electrophysiol Rev. 2016;5(2):90-101.
29. Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ Cardiovasc Genet. 2015;8(3):437-46.
30. Corrado D, Wichter T, Link MS, Hauer R, Marchlinski F, Anastasakis A, et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an international task force consensus statement. Eur Heart J. 2015;36(46):3227-37.
31. Cadrin-Tourigny J, Bosman LP, Nozza A, Wang W, Tadros R, Bhonsale A, et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2019;40(23):1850-8.
32. Jordà P, Bosman LP, Gasperetti A, Mazzanti A, Gourraud JB, Davies B, et al. Arrhythmic risk prediction in arrhythmogenic right ventricular cardiomyopathy: external validation of the arrhythmogenic right ventricular cardiomyopathy risk calculator. Eur Heart J. 2022;43(32):3041-52.
33. Protonotarios A, Bariani R, Cappelletto C, Pavlou M, García-García A, Cipriani A, et al. Importance of genotype for risk stratification in arrhythmogenic right ventricular cardiomyopathy using the 2019 ARVC risk calculator. Eur Heart J. 2022;43(32):3053-67.
34. Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm. 2019;16(11):e373-e407.
35. Cappelletto C, Gregorio C, Barbati G, Romani S, De Luca A, Merlo M, et al. Antiarrhythmic therapy and risk of cumulative ventricular arrhythmias in arrhythmogenic right ventricle cardiomyopathy. Int J Cardiol. 2021;334:58-64.
Die akute Lungenembolie zählt zu den häufigsten Ursachen für kardiovaskuläre Morbidität und Mortalität. Je nach Thrombuslast kann die Obstruktion im pulmonalen Kreislauf von leichter Rechtsherzbelastung bis hin zum Rechtsherzversagen und kardiogenen Schock führen. Eine rasche diagnostische Abklärung und Risikostratifizierung ist essenziell, um ehestmöglich eine adäquate Therapie einleiten zu können. Diese kann von einer Antikoagulation im ambulanten Setting bis hin zu kreislaufstabilisierenden Massnahmen und notfallmässiger Reperfusion durch ein multidisziplinäres, spezialisiertes Lungenembolie-Team reichen.
Epidemiologische Aspekte und Pathophysiologie der akuten Lungenembolie
Die akute Lungenembolie (LE) zählt zu den häufigsten Ursachen für kardiovaskuläre Morbidität mit einer jährlichen Inzidenz von 1 in 1000 Personen und einer Mortalität von bis zu 30 % in Hochrisikopopulationen (1-5). Die Ursache für die hohen Mortalitätsraten liegt hierbei in der akuten rechtsventrikulären Belastung mit resultierender Rechtsherzinsuffizienz bis hin zum kardiogenen Schock und Kreislaufstillstand (6).
Anatomisch ist der rechte Ventrikel (RV) muskelschwach, dünnwandig und dehnbar und ist durch eine Crescendo-Form an den dickwandigen starken linken Ventrikel (LV) angepasst (7). Dem RV nachgeschaltet befindet sich mit dem pulmonalen Kreislauf ein Niedrigdrucksystem, woraus sich eine deutlich geringere Nachlast ergibt, als dies für den LV mit dem nachgeschalteten Körperkreislauf der Fall ist. Ein physiologisch erhöhtes Herzzeitvolumen (z. B. unter körperlicher Belastung) kompensiert der pulmonale Kreislauf durch eine Rekrutierung der Mikrozirkulation. Die Anpassungsmechanismen des RV an eine akute Nachlasterhöhung sind jedoch deutlich schlechter ausgeprägt als beim LV. Durch seine Dehnbarkeit, kann er zwar grosse Blutvolumina fassen, jedoch ist das Schlagvolumen gegen einen erhöhten Widerstand stark eingeschränkt. Dies resultiert in einer paradoxen Verschiebung des interventrikulären Septums in Richtung des LV («D-Shaping» des LV Cavums), mit dadurch verminderter Füllung des LV, resultierender Output-Reduktion und reduzierter Perfusion der Koronararterien (Abb. 1). Dadurch, sowie durch den erhöhten Wandstress kommt es zu verminderter Perfusion des RV (8). Zusätzlich kommt es durch die RV-Erweiterung zu einer Erweiterung des Trikuspidalklappen-Annulus und als Konsequenz zu einer Trikuspidalinsuffizienz, wodurch der rechtsventrikuläre Vorwärtsfluss zusätzlich negativ beeinflusst wird (7).
Diagnostik und Risikostratifizierung der akuten Lungenembolie
Die akute LE kann sich mit einem variablen klinischen Erscheinungsbild in Hinblick auf Symptomintensität und Dauer präsentieren. Zu den häufigen Symptomen zählen Dyspnoe, Thoraxschmerzen, Präsynkopen oder Synkopen, sowie Husten und Hämoptysen. Weitere objektivierbare Faktoren im Notfallsetting umfassen eine Hypoxie, Hypokapnie, sowie typische EKG-Veränderungen (Zeichen einer Rechtsherzbelastung, SIQIIITIII Typ, Rechtsschenkelblock) (9).
Bei klinischem Verdacht auf eine akute LE ist das weitere Vorgehen massgeblich von der hämodynamischen Stabilität des Patienten abhängig (Abb. 2). Basierend auf der medizinischen Vorgeschichte und der klinischen Präsentation kann bei hämodynamischer Stabilität die Vortestwahrscheinlichkeit für das Vorliegen einer LE erhoben werden. Zur verbesserten Quantifizierung stehen hierzu Tools wie der Wells Score oder der Geneva Score zur Verfügung. Bei geringer oder intermediärer Vortestwahrscheinlichkeit kann die Bestimmung des D-Dimers eine LE bereits weitestgehend ausschliessen (9). Bei hoher Wahrscheinlichkeit oder altersadaptiert erhöhtem D-Dimer sollte eine weitere Abklärung mittels pulmonaler Angio-Computertomographie (LE-CT) erfolgen, in welcher sowohl das Ausmass der LE selbst, als auch eine RV Dilatation (RV/LV ratio > 0.9) als Zeichen der Rechtsherzbelastung dargestellt werden können (10). Bei hämodynamisch instabilen Patienten kommt im primären Assessment der transthorakalen Echokardiographie eine besondere Rolle in der Beurteilung einer möglichen RV-Dilatation und Dysfunktion zu (11). Sollten diese vorliegen, kann bei umgehender Verfügbarkeit eines LE-CT dieses zur Bestätigung erfolgen, oder auch unmittelbar eine Reperfusionstherapie neben der hämodynamischen Stabilisierung erfolgen, um eine unnötige Verzögerung zu vermeiden (9).
Die akute LE mit ausgeprägter RV Belastung und hämodynamischer Instabilität wird gemäss Richtlinien der Europäischen Gesellschaft für Kardiologie (ESC – European Society of Cardiology) als Hochrisiko – LE klassifiziert (9) und ist mit einer Mortalität zwischen 14 % und 29.8 % assoziiert (5). Am anderen Ende der Risikoskala stehen Niedrigrisiko – LE Patienten, welche weder Zeichen einer Rechtsherzbelastung in der Bildgebung, noch erhöhte kardiale Biomarker aufweisen. Zwischen den beiden extremen Risikogruppen, unterscheidet man bei symptomatischen Patienten zwischen intermediär-niedrigem und intermediär-hohem Risiko, je nachdem ob positive kardiale Biomarker oder ein Bildbefund einer RV Dysfunktion vorliegen (intermediär-niedrig), oder auch beide Befunde positiv ausfallen (intermediär-hohes Risiko) (9). Neben dieser Ampeleinteilung wird zur weiteren, objektivierten Risikostratifizierung auch der Pulmonary Embolism Severity Index (PESI) eingesetzt. Eine rasche Risikostratifizierung ist massgebend für das weitere Management hinsichtlich Monitoring und Therapie. (Abb. 3)
Therapie der akuten Lungenembolie
Die Initialtherapie der akuten LE umfasst die Korrektur der Hypoxämie mittels O2-Gabe bei Patienten mit einer Sauerstoffsättigung < 90 %. Die Spontanatmung sollte unbedingt erhalten bleiben, und nur bei zunehmender respiratorischer Erschöpfung oder auch insuffizienter spontaner Ventilation sollte eine mechanische Beatmungsunterstützung in Erwägung gezogen werden. Dabei ist es jedoch wichtig zu berücksichtigen, dass eine Beatmung ohne gewährleistete pulmonale Reperfusion keine Korrektur der Hypoxämie erwirken kann (9). Tatsächlich kann eine orotracheale Intubation und Allgemeinnarkose zu einer weiteren hämodynamischen Verschlechterung durch eine medikamentöse oder auch mechanische Überlastung des RV führen (9, 12–14).
Eine sofortige Antikoagulation mit unfraktioniertem (UFH) oder niedermolekularem (LMWH) Heparin ist bereits bei Patienten mit intermediärer oder hoher Wahrscheinlichkeit für das Vorliegen einer LE indiziert, bis weitere Testresultate zur Verfügung stehen. Der Einsatz oraler Antikoagulanzien sollte erst nach Abschluss der Riskostratifizierung und finalem weiteren Therapieentscheid erfolgen. Dabei stellen direkte orale Antikoagulanzien (NOAK), aufgrund eines deutlich verbesserten Nutzen-Risiko Profils im Vergleich zu den traditionellen Vitamin-K-Antagonisten, die Therapie der Wahl für die weiterführende Antikoagulation nach stattgehabter LE dar (9). Die Dauer der Antikoagulation sollte dabei mindestens 3 Monate umfassen; je nach Ursache kann jedoch auch eine langfristige, dauerhafte medikamentöse Therapie zur Prophylaxe erneuter Thrombosen / Embolien empfohlen werden (15).
Eine systemische Thrombolyse, zusätzlich zur Antikoagulation, sollte nur bei ausgewählten Hochrisikopatienten, in welchen eine rasche Auflösung des thrombotischen Materials zur Entlastung des RV angestrebt wird, erwogen werden. Metaanalysen konnten dabei eine signifikante Reduktion der Mortalität und etwaiger LE Rezidive nach dem Einsatz einer systemischen Thrombolyse nachweisen, allerdings zum Preis eines deutlich erhöhten Blutungsrisikos (16, 17). Absolute und relative Kontraindikationen für eine Lysetherapie können Tab. 1 entnommen werden.
Die Langzeit-Überlegenheit der systemischen Thrombolyse im Hinblick auf funktionelles Outcome und Entwicklung einer chronisch thromboembolischen pulmonalen Hypertonie ist aktuell nicht gesichert. Neuere Methoden umfassen die Katheter-gesteuerte Lyse, sowie die chirurgische oder minimal-invasive Katheter-gestützte Embolektomie (9). Abb. 4 illustriert die aktuell meist-verwendeten Kathetersysteme, welche an spezialisierten Lungenemboliezentren zur minimal-invasiven Behandlung der akuten LE eingesetzt werden (18-20).
Management von schwangeren Patientinnen mit akuter Lungenembolie
Das Risiko für Thromboembolien ist in der Schwangerschaft bis zu fünffach erhöht. Pathomechanistisch sind hierbei alle Komponenten der Virchow’schen Trias involviert, durch Hyperkoagulabilität, reduzierten venösen Fluss bis hin zur Stase und endotheliale Schädigung aufgrund einer venösen Hypertension oder auch im Rahmen der Entbindung. Besondere Vorsicht ist im Rahmen der Schwangerschaft hinsichtlich des Einsatzes strahlungsintensiver Untersuchungsmethoden, sowie auch den gegebenen Therapielimitationen geboten, um einen raschen Behandlungserfolg bei möglichst geringem Risiko für Mutter und Kind zu gewährleisten (21).
Der von der ESC empfohlene Diagnose- und Therapiealgorithmus ist in Abb. 5 zusammengefasst. Bei hämodynamisch stabilen Patientinnen sollte aufgrund der verlässlicheren Pharmakokinetik und Sicherheitsaspekten eine Antikoagulation mit LMWH ehestmöglich eingeleitet werden, wobei auch UFH eingesetzt werden kann (9). NOAKs sind hingegen in der Schwangerschaft kontraindiziert und auch Vitamin-K-Antagonisten mit teratogenen Effekten assoziiert. Vorsicht ist beim Einsatz von LMWH auch hinsichtlich spinaler und epiduraler Punktionen geboten, wobei die Antikoagulation nicht innerhalb der ersten 4 h nach Entfernung eines Epiduralkatheters begonnen und ein solcher auch nicht innerhalb 24 h Stunden nach letzter LMWH-Applikation eingelegt werden sollte (9).
Diagnostisches Mittel der Wahl ist ein Kompressionsultraschall zur Suche nach einer tiefen Venenthrombose (TVT) der unteren Extremität (9). Etwa 30 % offenbar isolierter LEs sind mit einer stummen TVT assoziiert (21), sodass der duplexsonographische Nachweis einer solchen bereits die Behandlung einer LE rechtfertigt. Bei negativem Resultat kann ein Thoraxröntgen zum Ausschluss anderer Ursachen für die Symptomatik dienen. Sollte dennoch der Verdacht auf eine LE bestehen und weitere Diagnostik indiziert sein, ist im Rahmen der Nutzen-Risiko-Abwägung die Durchführung eines LE-CT empfohlen. Alternativ kann insbesondere bei normalem Thoraxröntgenbefund ein weniger strahlungsintensiver Ventilations/Perfusions (VQ)-Scan durchgeführt werden, sofern verfügbar. Bei Nachweis einer LE ist die Fortführung des LMWH empfohlen. Bei Hochrisiko-LE Patientinnen und entsprechender hämodynamischer Instabilität muss die Notwendigkeit einer umgehenden thrombolytischen Therapie oder auch Embolektomie evaluiert werden. Nach überstandenem Primärereignis sollte die weitere Betreuung der Schwangerschaft wie auch die Entbindungsplanung an einem interdisziplinären Zentrum mit entsprechender Expertise in der Behandlung der Lungenembolie und möglicher Komplikationen erfolgen (9).
Abkürzungen CT Computertomographie ESC European Society of Cardiology LE Lungenembolie LMWH Niedermolekulares Heparin LV Linker Ventrikel NOAK Neue orale Antikoagulanzien RV Rechter Ventrikel, rechtsventrikulär (s)PESI (simplified) Pulmonary Embolism Severity Index TTE Transthorakale Echokardiographie TVT Tiefe Venenthrombose UFH Unfraktioniertes Heparin VQ Scan Lung Ventilation Perfusion Scan
Copyright
Aerzteverlag medinfo AG
PD Dr. med. et phil. Dominik F. Draxler MBA
Zentrum für Lungenembolie
Klinik und Poliklinik für Kardiologie
Universitätsspital, Inselspital Bern
Freiburgstrasse 20
3010 Bern
Prof. Dr. med. Stefan Stortecky
Abteilung für Kardiologie, Bürgerspital Solothurn
Schöngrünstrasse 42
4500 Solothurn
stefan.stortecky@insel.ch
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
Etwa 45 % aller Patienten mit akuter LE weisen Zeichen einer RV Belastung auf, mit potenziell resultierender Rechtsherzinsuffizienz, welche bis hin zum kardiogenen Schock und Kreislaufversagen führen kann.
Eine rasche Diagnostik mittels bildgebender Massnahmen, sowie Risikostratifizierung auf Basis von hämodynamischen Merkmalen, RV-Belastung, (s)PESI-Score und Troponin-Bestimmmung ist essenziell und bestimmt das therapeutische Vorgehen.
Eine sofortige Antikoagulation ist die zentrale Therapiekomponente der akuten LE. Bei selektionierten LE Patienten mit intermediär-hohem oder hohem Risiko muss zudem eine rasche Reperfusionsstrategie evaluiert werden, wobei hier neben der systemischen Lyse heutzutage kathetergesteuerte Interventionen, und die chirurgische Embolektomie zur Verfügung stehen.
Schwangerschaft und die peripartale Phase stellen einen besonderen Risikofaktor für die Entwicklung einer akuten LE dar. Für die Diagnostik sollten primär strahlungsarme Massnahmen gewählt werden und Therapiestrategien durch ein interdisziplinäres Lungenembolieteam festgelegt werden, um die beste Option zum Wohlergehen von Mutter und Fötus zu gewährleisten.
1. Wendelboe, A.M. and G.E. Raskob, Global Burden of Thrombosis: Epidemiologic Aspects. Circ Res, 2016. 118(9): p. 1340-7.
2. Keller, K., et al., Trends in thrombolytic treatment and outcomes of acute pulmonary embolism in Germany. Eur Heart J, 2020. 41(4): p. 522-529.
3. Lehnert, P., et al., Acute Pulmonary Embolism in a National Danish Cohort: Increasing Incidence and Decreasing Mortality. Thromb Haemost, 2018. 118(3): p. 539-546.
4. Payne, J.G., et al., Current estimates of the incidence of acute venous thromboembolic disease in Canada: A meta-analysis. Thromb Res, 2021. 197: p. 8-12.
5. Becattini, C., et al., Acute pulmonary embolism: mortality prediction by the 2014 European Society of Cardiology risk stratification model. Eur Respir J, 2016. 48(3): p. 780-6.
6. Bryce, Y.C., et al., Pathophysiology of right ventricular failure in acute pulmonary embolism and chronic thromboembolic pulmonary hypertension: a pictorial essay for the interventional radiologist. Insights Imaging, 2019. 10(1): p. 18.
7. Bristow, M.R., et al., The pressure-overloaded right ventricle in pulmonary hypertension. Chest, 1998. 114(1 Suppl): p. 101s-106s.
8. van Wolferen, S.A., et al., Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart J, 2008. 29(1): p. 120-7.
9. Konstantinides, S.V., et al., 2019 ESC Guidelines for the diagnosis and management of acute pulmonary embolism developed in collaboration with the European Respiratory Society (ERS). Eur Heart J, 2020. 41(4): p. 543-603.
10. Carrier, M., et al., Subsegmental pulmonary embolism diagnosed by computed tomography: incidence and clinical implications. A systematic review and meta-analysis of the management outcome studies. J Thromb Haemost, 2010. 8(8): p. 1716-22.
11. Kurzyna, M., et al., Disturbed right ventricular ejection pattern as a new Doppler echocardiographic sign of acute pulmonary embolism. Am J Cardiol, 2002. 90(5): p. 507-11.
12. Messika, J., et al., Severe pulmonary embolism managed with high-flow nasal cannula oxygen therapy. Eur J Emerg Med, 2017. 24(3): p. 230-232.
13. Lacroix, G., et al., High-flow oxygen, a therapeutic bridge while awaiting thrombolysis in pulmonary embolism? Am J Emerg Med, 2013. 31(2): p. 463.e1-2.
14. Page, R.L., 2nd, et al., Drugs That May Cause or Exacerbate Heart Failure: A Scientific Statement From the American Heart Association. Circulation, 2016. 134(6): p. e32-69.
15. van Es, N., et al., Direct oral anticoagulants compared with vitamin K antagonists for acute venous thromboembolism: evidence from phase 3 trials. Blood, 2014. 124(12): p. 1968-75.
16. Marti, C., et al., Systemic thrombolytic therapy for acute pulmonary embolism: a systematic review and meta-analysis. Eur Heart J, 2015. 36(10): p. 605-14.
17. Zhang, R.S., et al., Efficacy and Safety of Anticoagulation, Catheter-Directed Thrombolysis, or Systemic Thrombolysis in Acute Pulmonary Embolism. JACC Cardiovasc Interv, 2023. 16(21): p. 2644-2651.
18. Draxler, D.F. and S. Stortecky, Interventional Reperfusion Strategies for Acute Pulmonary Embolism. Praxis (Bern 1994), 2021. 110(13): p. 743-751.
19. Pruszczyk, P., et al., Percutaneous treatment options for acute pulmonary embolism: a clinical consensus statement by the ESC Working Group on Pulmonary Circulation and Right Ventricular Function and the European Association of Percutaneous Cardiovascular Interventions. EuroIntervention, 2022. 18(8): p. e623-e638.
20. Finocchiaro, S., et al., Percutaneous interventions for pulmonary embolism. EuroIntervention, 2024. 20(7): p. e408-e424.
21. Devis, P. and M.G. Knuttinen, Deep venous thrombosis in pregnancy: incidence, pathogenesis and endovascular management. Cardiovasc Diagn Ther, 2017. 7(Suppl 3): p. S309-s319.