Le vieillissement de population fait converger vers les institutions gériatriques un nombre croissant de personnes âgées présentant des fragilités, des polypathologies et une dépendance qui affectent leur santé et leur qualité de vie. L’ environnement physique du résident âgé a été décrit comme un facteur susceptible d’ influencer de façon significative sur son bien-être. Il manque cependant de recommandations précises et scientifiquement validées pour y contribuer. L’ environnement enrichi (EE), a mis en évidence sur le modèle murin, son effet positif sur la plasticité cérébrale ainsi que sur de nombreux marqueurs fonctionnels de santé avec un intérêt majeur pour la gériatrie. La transposition de l’ EE aux institutions gériatriques initiée depuis 10 ans, présente déjà des résultats prometteurs en favorisant la santé et le bien-être des résidents.
The population’ s ageing is leading to a growing number of people entering geriatric care, with frailties, polypathologies and dependency affecting their health and quality of life. The physical environment of residents has been described as a factor likely to influence their well-being significantly. However, there is a lack of accurate, scientifically-validated recommendations to help achieve this. The enriched environment (EE) has proven its positive effect on cerebral plasticity and numerous functional health markers in murine models, and is of major interest to geriatric medicine. The transposition of EE to geriatric institutions, initiated 10 years ago, is already showing promising results in promoting the health and well-being of residents.
Key words: gerontology, geriatric care, frailty, polypathologies, quality of life
Contexte
L’ accroissement continu de la part de la population âgée lié à la transition démographique, s’ accompagne de besoins importants pour la prise en charge par les institutions gériatriques de fragilités, de polypathologies chroniques et de niveaux de dépendance élevés (1). De fortes attentes sociétales s’ expriment pour offrir à ces personnes âgées des réponses adaptées susceptibles d’ assurer leur santé et leur qualité de vie. Il existe depuis les origines, une conviction que l’ environnement et la santé sont étroitement liés (2). L’ homme considère aujourd’ hui comme une évidence que sa relation à l’ environnement est essentielle pour préserver sa santé. Cette relation avec l’ environnement s’ est construite principalement dans l’ objectif de le préserver de nuisances potentielles. Le rapport de l’ Organisation mondiale de la Santé (OMS) sur le vieillissement (3) déclare qu ’ «un milieu physique adapté peut faire toute la différence entre indépendance et dépendance pour tous les individus, mais il revêt une importance particulière pour les personnes âgées». Il convient de citer les nombreux travaux de recherches conduits par des équipes engagées sur la question, tels que Lawton (4), Marquardt (5), Zeisel (6), Fleming (7) ou Sloane (8) pour n’ en mentionner que quelques-uns. Si ces recherches ont permis de faire émerger des lignes directrices utiles, elles s’ inscrivent dans la démarche traditionnelle à savoir «comment aménager l’ environnement afin qu’ il n’ amplifie pas les troubles et fragilités observées chez la personne âgée en particulier lorsqu’ elle est atteinte de démence.
Une relation avec l’ environnement susceptible d’ améliorer la santé humaine est une idée développée par Antonovsky (9) en 1979 à travers sa théorie intitulée Salutogenèse. Ce changement de paradigme où l’ environnement ne serait plus seulement un facteur dont il faut se préserver mais deviendrait promoteur de la santé constitue en soi une approche innovante dont la mise en œuvre peine à se développer dans les institutions gériatriques.
L’ environnement enrichi et le modèle murin
Nos travaux ont pris pour inspiration la richesse extraordinaire de connaissances développées par les recherches en neurosciences sur l’ environnement enrichi. D. Hebb (10) dans ses travaux pionniers en 1949, a développé le concept d’ environnement enrichi décrivant la relation entre l’ environnement et la plasticité cérébrale. Ces recherches se sont poursuivies pendant des décennies en produisant des connaissances d’ un intérêt majeur en particulier en gériatrie. De nombreuses publications ont mis en évidence les avantages de l’ enrichissement environnemental (EE) en tant que stimulation significative de l’ anatomie et de la physiologie du cortex cérébral, aux niveaux biochimiques et moléculaires, prévenant ou inversant les déficits liés au vieillissement (11) (12) (13) (14). Des protocoles robustes et normalisés ont permis d’ étudier diversement les effets de l’ EE sur diverses maladies et troubles liés à l’ âge sur le modèle murin, ce qui suggère que celui-ci pourrait former une réponse valable à de nombreux problèmes de santé chez l’ homme.
Transposition de l’ EE en institution gériatrique
Depuis plus de 10 ans, des études cliniques menées à l’ hôpital universitaire Charles Foix ont confirmé la validité de la transposition à l’ humain de ces travaux jusque-là réservés aux neurosciences.
Premières études sur le jardin enrichi
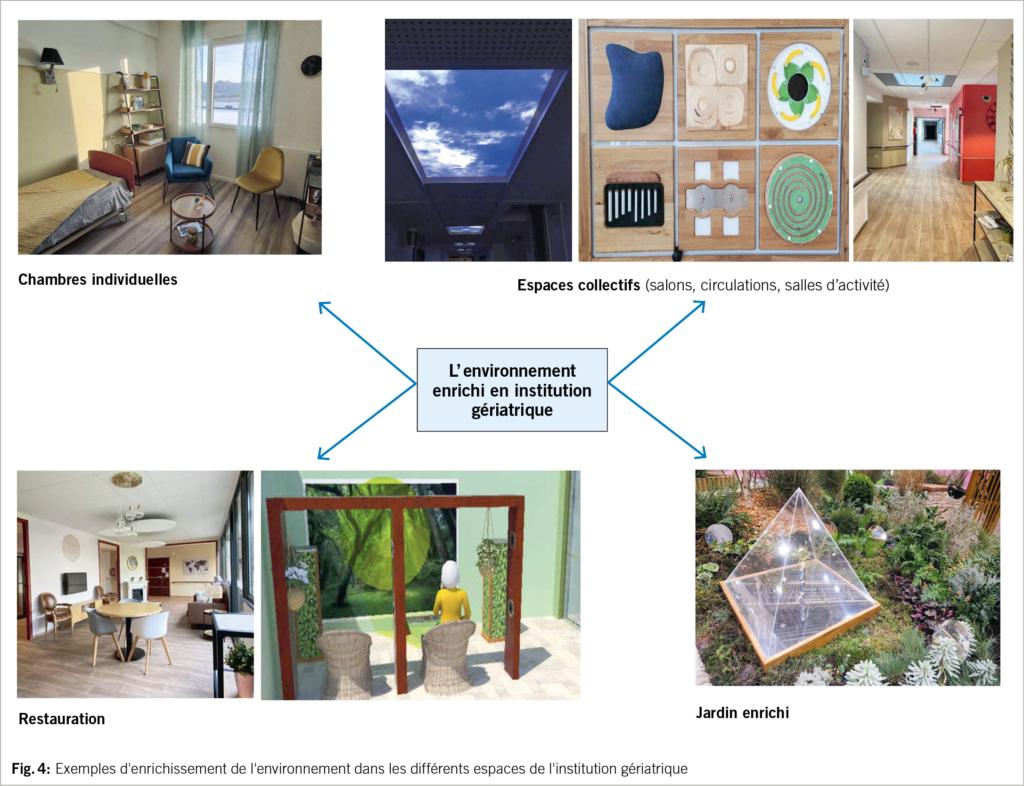
Les premières transpositions du concept d’ EE ont été effectuées sur les espaces extérieurs d’ institution gériatrique – du fait de la moindre pression des normes architecturales et des coûts plus réduits de l’ aménagement d’ un dispositif expérimental. C’ est ainsi qu’ a été décrit le concept de «jardin enrichi»(15). Ce concept de jardin enrichi intégrait un changement de paradigme majeur en plaçant le patient au centre de la conception et en confiant à l’ environnement des missions spécifiques de promotion de la santé et de bien-être des patients accueillis.
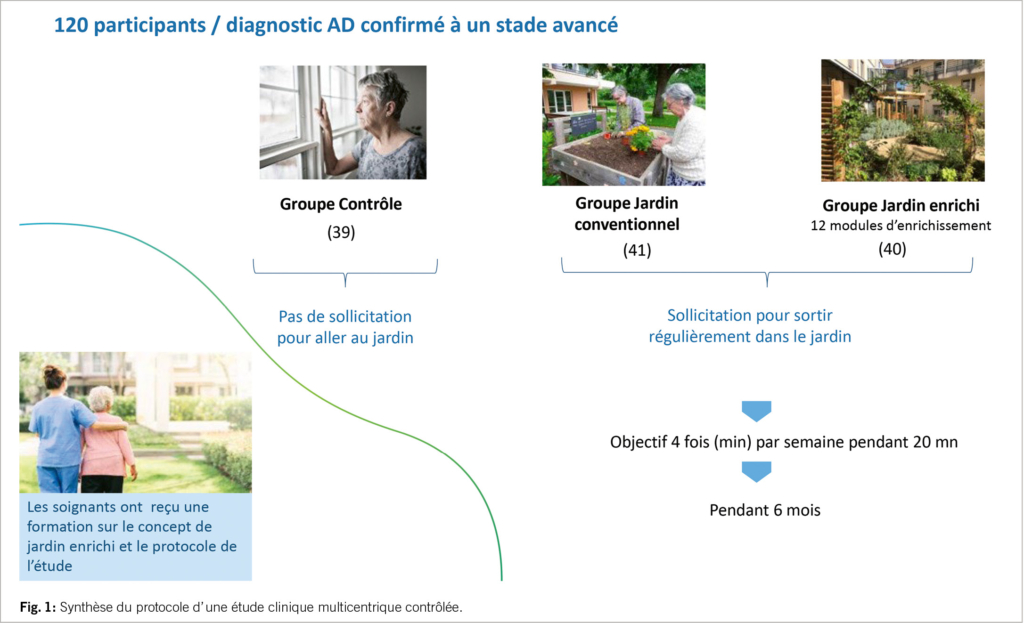
L’ enrichissement du jardin se forme par l’ aménagement de «modules» spécifiques constituant la matière active du jardin. Ils ont été conçus en fonction d’ objectifs thérapeutiques précis correspondants aux troubles et fragilités observés parmi les résidents (troubles cognitifs, du comportement, de l’ humeur, perte d’ indépendance …) Les interactions que le résident établit spontanément avec ces modules participent de stimulations cognitivo- comportementales et sensorielles destinées à produire des effets bénéfiques sur sa santé. Reprenant les principes décrits dans les études sur le modèle murin, l’ EE est associé à des interactions régulières du visiteur (env. 4 fois / semaine) et un renouvellement régulier des modules d’ enrichissement.
Le jardin enrichi est un dispositif expérimental visant à évaluer par la transposition à la personne âgée du concept d’ EE, les liens entre l’ environnement physique avec la santé et le bien-être des résidents.
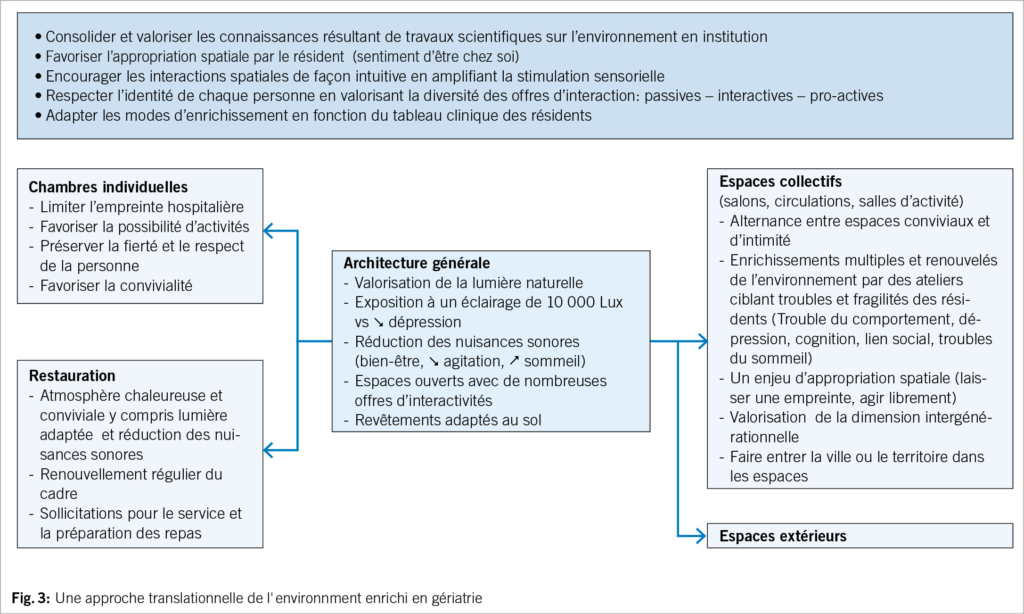
Les études sur le «jardin enrichi» ont également exploré la notion d’ appropriation spatiale par le résident (16) – une dimension essentielle dans le contexte d’ un événement majeur dans un parcours de vie, reliée au sentiment d’ être chez soi et fondateur de la construction de l’ identité de chaque individu. Nos observations, davantage que de valider la logique domiciliaire actuellement très défendue dans les projets architecturaux, ont souligné plusieurs médiateurs déterminants de l’ appropriation spatiale. Il convient de mentionner l’ esthétique de l’ espace suggérant la fierté d’ y résider, la convivialité et le sentiment de liberté s’ écartant de la pression de la vie en collectivité et enfin la possibilité d’ y laisser librement une trace ou son empreinte.
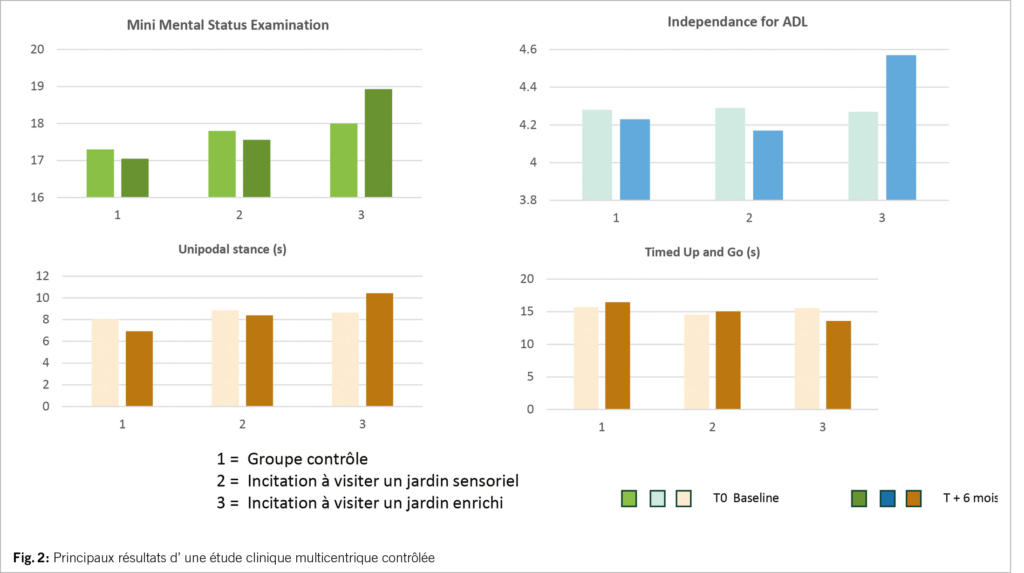
Les études cliniques multicentriques, randomisées et contrôlées conduites par notre équipe sur le concept de jardin enrichi sur des populations atteintes de la maladie d’ Alzheimer à un stade avancé ont mis en évidence une relation significative entre sa fréquentation régulière et la récupération de capacité cognitive, l’ amélioration de l’ indépendance fonctionnelle, les troubles perturbateurs du comportement et la prévention des chutes. Ces études ont consisté à recruter des résidents en institution gériatrique présentant un diagnostic confirmé de la maladie d’ Alzheimer à un stade avancé (10<MMSE< 20) disposant d’ une autonomie suffisante à la marche pour fréquenter un jardin sans aide humaine et ne souffrant pas de troubles majeurs perturbateurs du comportement (17).
Les participants étaient répartis en plusieurs groupes: contrôle, fréquentant un jardin sensoriel ou un jardin enrichi. Cette fréquentation était motivée par des incitations régulières par les professionnels de santé.
Les résultats ont mis en évidence que les groupes «contrôle» et «jardin sensoriel» présentaient à l’ issue de l’ intervention des profils identiques sur les marqueurs fonctionnels suivants: MMSE, ADL, CMAI, station unipodale, test UpnGo. Le groupe «jardin enrichi» se caractérisait systématiquement par une amélioration significative de ces scores par rapport aux autres groupes d’ une part et par rapport aux mesures faites au démarrage de l’ étude.
Travaux en cours et perspectives
De nombreux travaux complémentaires restent à réaliser afin de réduire les risques de biais observés dans ces études pilotes. Ces résultats ouvrent la voie à une transposition plus large des connaissances acquises par les neurosciences sur l’ EE. Daffner et al (18) soulignent que les études sur l’ EE sur le modèle murin peuvent aider à identifier les facteurs favorisant un vieillissement réussi. En effet, ces études sur l’ influence de l’ EE sur les marqueurs fonctionnels liés à l’ âge ont présenté des effets significatifs et bénéfiques sur la dépression, l’ anxiété, les troubles du comportement, la désorientation spatiale, la mémoire spatiale et la mémoire de travail, les troubles du sommeil et de la nutrition, l’ indépendance fonctionnelle et la préservation des relations sociales.
Les études complémentaires en cours visent à étendre le concept d’ environnement enrichi à l’ ensemble des espaces des institutions gériatriques – notamment l’ architecture générale de l’ établissement, les espaces collectifs et individuels, les espaces extérieurs. L’ enrichissement de l’ environnement permet ainsi d’ ajuster une action de promotion de la santé et du bien-être par l’ intégration et le renouvellement des «modules d’ enrichissement» en fonction du tableau clinique des résidents. Il en est ainsi du projet de rénovation de l’ hôpital gériatrique du Centre Hospitalier de la Métropole de Savoie (CHMS) à Aix les Bains (France).
Il convient aussi de renforcer les connaissances sur la notion d’ appropriation spatiale. L’ ensemble de ces efforts s’ inscrivent dans une vision salutogénique plus large visant produire des recommandations basées sur des connaissances scientifiques solides, pour l’ environnement d’ institution gériatrique dans lequel le résident se sente chez lui et favorisant la préservation et la promotion de la santé et de la qualité de vie de la personne âgée.
Copyright Aerzteverlag medinfo AG
PhD Santé publique –
MSc Gérontologie – Psychogériatrie
Hôpital gériatrique Charles Foix-
Assistance Publique Hôpitaux de Paris (APHP)
Laboratoire Éducation et Promotion de la Santé (LEPS UR 3412)
Université Sorbonne Paris Nord
etienne.bourdon-ext@aphp.fr
etiennepbourdon@gmail.com
L’ auteur ne présente aucun conflit d’ intérêt avec le sujet présenté dans cet article
1. Belmin J, Chassagne P, Friocourt P. Gériatrie (collection Pour le praticien 4ème édition). Elsevier Masson. Issy les Moulineaux; 2023. 939 p.
2. Berridge V. Environment, health and history. Basingstoke: Palgrave Macmillan; 2012.
3. World Health Organization. World report on ageing and Health.pdf [Internet]. Genève: WHO; 2015 p. 260. Disponible sur: http://apps.who.int/iris/bitstream/handle/10665/186463/9789240694811_eng.pdf
4. Lawton MP. The elderly in context: Perspectives from environmental psychology and gerontology. Environ Behav. 1985;17(4):501‑19.
5. Marquardt G, Schmieg P. Dementia-friendly architecture. Environments that facilitate wayfinding in nursing homes. Z Gerontol Geriatr. 2009;42(5):402‑7.
6. Zeisel J. Environmental design effects on Alzheimer symptoms in long-term care residences. World Hosp Health Serv Off J Int Hosp Fed. 2000;36(3):27‑31, 36, 38.
7. Fleming R, Goodenough B, Low LF, Chenoweth L, Brodaty H. The relationship between the quality of the built environment and the quality of life of people with dementia in residential care. Dement Lond Engl. 2016;15(4):663‑80.
8. Sloane PD, Williams CS, Mitchell CM, Preisser JS, Wood W, Barrick AL, et al. High-intensity environmental light in dementia: effect on sleep and activity. J Am Geriatr Soc. oct 2007;55(10):1524‑33.
9. Mittelmark MB, Bauer GF, Vaandrager L, Pelikan JM, Sagy S, Eriksson M, et al., éditeurs. The handbook of Salutogenesis [Internet]. Cham: Springer International Publishing; 2022. Disponible sur: https://link.springer.com/10.1007/978-3-030-79515-3
10. Hebb D. The organization of behavior; a neuropsychological theory. New-York: Wiley; 1949.
11. Diamond MC, Krech D, Rosenzweig MR. The effects of an enriched environment on the histology of the rat cerebral cortex. J Comp Neurol. 1964;123(1):111‑9.
12. Berardi N, Braschi C, Capsoni S, Cattaneo A, Maffei L. Environmental enrichment delays the onset of memory deficits and reduces neuropathological hallmarks in a mouse model of Alzheimer-like neurodegeneration. J Alzheimers Dis. 2007;11(3):359‑70.
13. Faherty CJ, Raviie Shepherd K, Herasimtschuk A, Smeyne RJ. Environmental enrichment in adulthood eliminates neuronal death in experimental Parkinsonism. Mol Brain Res. 2005;134(1):170‑9.
14. van Gool WA, Mirmiran M. Effects of aging and housing in an enriched environment on sleep-wake patterns in rats. Sleep. 1986;9(2):335‑47.
15. Bourdon E, Belmin J. Le concept de jardin enrichi, une innovation en gériatrie. Soins Gerontol. 2022;27(157).
16. Bourdon E, Belmin J. L’ appropriation de l’ espace par le résident en institution gériatrique : une étude qualitative sur le jardin enrichi en Ehpad. NPG Neurol – Psychiatr – Gériatrie. 2022;(24).
17. Bourdon E, Belmin J. Enriched gardens improve cognition and independence of nursing home residents with dementia: a pilot controlled trial. Alzheimers Res Ther. 2021;13:116.
18. Daffner KR. Promoting successful cognitive aging: A comprehensive review. J Alzheimers Dis. 11 mars 2010;19(4):1101‑22.