Zeitlich begrenzte versus kontinuierliche Behandlung bei chronischer lymphatischer Leukämie (CLL17 Studie)
Hintergrund
Die Behandlung der chronischen lymphatischen Leukämie (CLL) besteht derzeit aus zwei Hauptansätzen: kontinuierliche Therapie mit Bruton-Tyrosinkinase-Inhibitoren (BTKi) und zeitlich begrenzte Regimen, die Venetoclax mit CD20-Antikörpern oder BTKi kombinieren. Direkte Vergleiche dieser beiden therapeutischen Ansätze fehlten bisher.
Methoden
In dieser von Prüfärzten initiierten, offenen Phase-3-Studie wurden erwachsene Patienten mit vorher unbehandelter CLL im Verhältnis 1 : 1 : 1 randomisiert zu:
– Kontinuierliches Ibrutinib
– Zeitlich begrenzte Venetoclax-Obinutuzumab (15 Monate)
– Zeitlich begrenzte Venetoclax-Ibrutinib (15 Monate)
Der primäre Endpunkt war das Progressionsfreie Überleben (PFS) mit einer Nicht-Unterlegenheits-Grenze für die Hazard Ratio von 1.608 (entsprechend 8 Prozentpunkten nach 3 Jahren). Sekundäre Endpunkte umfassten minimale Resterkrankung (MRD), Ansprechrate, Gesamtüberleben (OS) und Sicherheit.
Ergebnisse
Insgesamt wurden 909 Patienten randomisiert: 303 zu Venetoclax-Obinutuzumab, 305 zu Venetoclax-Ibrutinib und 301 zu Ibrutinib. Nach einem medianen Follow-up von 34.2 Monaten zeigte diese präspezifizierte Interimsanalyse:
– Das geschätzte 3-Jahres-PFS betrug 81.1 % (Venetoclax-Obinutuzumab), 79.4 % (Venetoclax-Ibrutinib) und 81.0 % (Ibrutinib)
– Hazard Ratio für Venetoclax-Obinutuzumab vs. Ibrutinib: 0.87 (98.3 % KI 0.54–1.41); für Venetoclax-Ibrutinib vs. Ibrutinib: 0.84 (98.0 % KI 0.53–1.32) – beide Vergleiche erfüllten das Nicht-Unterlegenheitskriterium
– Nach Therapieende war die MRD im peripheren Blut nicht nachweisbar bei 73.3 % (Venetoclax-Obinutuzumab), 47.2 % (Venetoclax-Ibrutinib) und 0 % (Ibrutinib)
– Das geschätzte 3-Jahres-Gesamtüberleben betrug 91.5 %, 96.0 % bzw. 95.7 %
Die häufigsten unerwünschten Ereignisse waren Infektionen, gastrointestinale Störungen und Zytopenien.
Schlussfolgerung
Bei Patienten mit vorher unbehandelter CLL war die zeitlich begrenzte Behandlung mit Venetoclax-Obinutuzumab oder Venetoclax-Ibrutinib hinsichtlich des PFS nicht unterlegen gegenüber kontinuierlichem Ibrutinib.
Literatur
O. Al-Sawaf et al., N Engl J Med 2025 (online publiziert 6. Dezember 2025). DOI: 10.1056/NEJMoa2515458
Studie
Finanzierung: Universität Köln und weitere; CLL17-Studie. ClinicalTrials.gov: NCT04608318; EudraCT: 2019-003854-99
Projizierte lebenslange Krebsrisiken durch Computertomographie-Bildgebung
Hintergrund
Jährlich werden in den USA etwa 93 Millionen Computertomographie-Untersuchungen (CT) bei 62 Millionen Patienten durchgeführt. Ionisierende Strahlung im Rahmen einer CT-Untersuchung ist ein bekanntes Karzinogen. Frühere Schätzungen aus dem Jahr 2007 projizierten etwa 29 000 zukünftige Krebsfälle; seitdem ist die CT-Nutzung um über 30 % gestiegen, und genauere Dosimetrie-Methoden wurden entwickelt.
Methoden
Dieses Risikomodell verwendete eine multizentrische Stichprobe von CT-Untersuchungen aus dem University of California San Francisco International CT Dose Registry (Januar 2018 bis Dezember 2020) mit Daten von 143 US-Krankenhäusern und ambulanten Einrichtungen.
Die Verteilung von CT-Untersuchungen und die damit verbundenen organspezifischen Strahlendosen wurden nach Alter, Geschlecht und CT-Kategorie geschätzt und anhand der IMV-Umfrage 2023 auf die US-Bevölkerung hochgerechnet. Die lebenslange strahleninduzierte Krebsinzidenz und 90 %-Unsicherheitsgrenzen wurden mit der Software des National Cancer Institute basierend auf den BEIR VII-Modellen (Biological Effects of Ionizing Radiation) berechnet und auf die US-Bevölkerung projiziert.
Ergebnisse
Schätzungsweise 61.51 Millionen Patienten unterzogen sich 2023 93 Millionen CT-Untersuchungen, darunter 2.57 Millionen (4.2 %) Kinder, 58.94 Millionen (95.8 %) Erwachsene, 32.6 Millionen (53.0 %) weibliche und 28.91 Millionen (47.0 %) männliche Patienten.
– Es wurden etwa 103 000 (90 % Unsicherheitsgrenze: 96 400–109 500) strahleninduzierte Krebsfälle projiziert
– Das geschätzte Krebsrisiko pro Untersuchung war bei Kindern und Jugendlichen höher, aber die höhere CT-Nutzung bei Erwachsenen machte die meisten (93 000; 91 %) strahlenbedingten Krebsfälle aus
– Die häufigsten Krebsarten waren insgesamt: Lungenkrebs (22 400 Fälle; 90 % UG 20 200–25 000), Kolonkarzinom (8700 Fälle; 90 % UG 7800–9700), Leukämie (7900 Fälle; 90 % UG 6700–9500) und Blasenkrebs (7100 Fälle; 90 % UG 6000–8500). Bei Frauen war Brustkrebs die zweithäufigste Krebsart (5700 Fälle; 90 % UG 5000–6500)
– Nach CT-Kategorie: Abdomen- und Becken-CT bei Erwachsenen führte zur grössten Anzahl projizierter Krebsfälle (37 500 von 103 000 [37 %]) bei 30 Millionen von 93 Millionen Untersuchungen (32 %), gefolgt von Thorax-CT (21 500 Krebsfälle [21 %]; 20 Millionen Untersuchungen [21 %])
– Sensitivitätsanalysen ergaben einen Bereich von 80 000 bis 127 000 projizierten Krebsfällen
Schlussfolgerung
Bei aktueller Nutzung und Strahlendosis wird projiziert, dass CT-Untersuchungen im Jahr 2023 zu etwa 103 000 zukünftigen Krebsfällen über die Lebenszeit der exponierten Patienten führen werden. Wenn die aktuellen Praktiken bestehen bleiben, könnte CT-assoziierter Krebs schliesslich für 5 % aller jährlichen Krebsdiagnosen verantwortlich sein – vergleichbar mit anderen bedeutenden Risikofaktoren wie Alkoholkonsum (5.4 %) und Übergewicht (7.6 %).
Literatur R. Smith-Bindman et al., JAMA Intern Med 2025;185(6):710-719. DOI: 10.1001/jamainternmed.2025.0505
Studie
Finanzierung: National Cancer Institute, Patient-Centered Outcomes Research Institute, und Gerichtsfonds. Datenzugang: University of California San Francisco International CT Dose Registry.
Epcoritamab, Lenalidomid und Rituximab vs. Lenalidomid und Rituximab bei rezidiviertem oder refraktärem follikulärem Lymphom (EPCORE FL-1)
Hintergrund
Das follikuläre Lymphom ist das zweithäufigste B-Zell-Non-Hodgkin-Lymphom. Während die Erstlinientherapie häufig längere Remissionen ermöglicht, werden die Remissionen bei rezidivierten Patienten mit jeder weiteren Therapie kürzer. Die chemotherapiefreie Kombination aus Lenalidomid und Rituximab (R²) ist ein international akzeptierter Standard bei rezidiviertem oder refraktärem follikulärem Lymphom nach mindestens einer Vortherapie. Jedoch erreichen in klinischen Studien nur etwa die Hälfte der Patienten eine komplette Remission mit R². Epcoritamab ist ein subkutan verabreichter bispezifischer Antikörper, der gleichzeitig an das B-Zell-Antigen CD20 und das T-Zell-Antigen CD3 bindet und zu T-Zell-vermittelter Zytotoxizität von CD20-exprimierenden malignen B-Zellen führt.
Methoden
In dieser globalen, offenen, randomisierten Phase-3-Studie wurden 488 Patienten aus 189 akademischen und nicht-akademischen Zentren in 30 Ländern im Verhältnis 1 : 1 randomisiert, um entweder Epcoritamab plus R² (243 Patienten) oder R² allein (245 Patienten) für bis zu 12 Zyklen zu erhalten.
Epcoritamab wurde subkutan wöchentlich in den Zyklen 1–3 und alle 4 Wochen in den Zyklen 4–12 verabreicht. Lenalidomid wurde einmal täglich während der Zyklen 1–12 (Tage 1–21) gegeben, und Rituximab wurde wöchentlich während Zyklus 1 und monatlich in den Zyklen 2–5 verabreicht. Die dualen primären Endpunkte waren die Gesamtansprechrate (ORR) und das Progressionsfreie Überleben (PFS).
Ergebnisse
Von 668 gescreenten Patienten wurden 488 randomisiert. Die Studie erreichte ihre dualen primären Endpunkte und zeigte die Überlegenheit von Epcoritamab plus R² gegenüber R² sowohl in der Gesamtansprechrate als auch im Progressionsfreien Überleben.
– Bei einer medianen Nachbeobachtung von 14.8 Monaten (IQR 11.4–19.0) betrug die ORR 95 % (95 % KI 92–97) mit Epcoritamab plus R² vs. 79 % (74–84) mit R² (p<0.0001)
– Die Rate kompletter Remissionen (CR) betrug 83 % (77–87) mit Epcoritamab plus R² vs. 50 % (43–56) mit R² (p<0.0001)
– Das PFS war mit Epcoritamab plus R² signifikant länger (HR 0.21 [95 % KI 0.14–0.31], p<0.0001). Das mediane PFS wurde mit Epcoritamab plus R² nicht erreicht vs. 11.7 Monate mit R²
– Die geschätzte 16-Monats-PFS-Rate betrug 85.5 % mit Epcoritamab plus R² vs. 40.2 % mit R²
– Das mediane OS wurde in beiden Gruppen nicht erreicht (HR 0.38 [95 % KI 0.18–0.80], p=0.0039). Die 16-Monats-OS-Raten betrugen 95.8 % für Epcoritamab plus R² und 88.8 % für R²
– Nebenwirkungen von Grad 3 oder höher traten häufiger bei Epcoritamab plus R² auf (219 [90 %] von 243 Patienten) vs. R² (161 [68 %] von 238 Patienten)
– Die häufigsten Grad- ≥ 3-Nebenwirkungen waren Neutropenie (69 % vs. 42 %) und Infektionen (33 % vs. 15 %)
– Das Zytokin-Freisetzungssyndrom (CRS) war bei Epcoritamab plus R² niedriggradig (Grad 1 bei 28 [21 %] Patienten und Grad 2 bei 7 [5 %] Patienten), gut handhabbar, und alle Ereignisse waren reversibel
Schlussfolgerung
Epcoritamab plus R² führte bei Patienten mit follikulärem Lymphom, die mindestens eine Therapielinie erhalten hatten, zu einer signifikant höheren Ansprechrate und einem längeren PFS im Vergleich zu R². Obwohl Nebenwirkungen von Grad 3 oder höher mit Epcoritamab plus R² häufiger auftraten als mit R², waren diese handhabbar und konsistent mit den etablierten Sicherheitsprofilen der einzelnen Komponenten, ohne neue Sicherheitsbefunde. Diese Ergebnisse positionieren Epcoritamab plus R² als einen neuen Behandlungsstandard für die Zweit- oder Folgelinientherapie des follikulären Lymphoms.
Literatur
R. Smith-Bindman et al., JAMA Intern Med 2025;185(6):710-719. DOI: 10.1001/jamainternmed.2025.0505
Studie
Finanzierung: AbbVie und Genmab. Die Studie wurde von einem unabhängigen Datenüberwachungskomitee begleitet.
Prof. Dr. med. Christoph Renner
Onkozentrum Hirslanden Zürich und Onkozentrum Zürich
Witellikerstrasse 40
8032 Zürich
ChatGPT als patientenorientiertes Triage-Tool in der Onkologie
In einer kürzlich erschienenen Studie wurde die diagnostische Genauigkeit und medizinische Angemessenheit eines frei zugänglichen grossen Sprachmodells (LLM) bei der Triage häufiger klinischer Szenarien in der Brustkrebsbehandlung bewertet.
Methoden
Von Januar bis Februar 2025 interagierten sieben Ärzte mit OpenAIs ChatGPT-4o und ChatGPT-3.5-turbo und simulierten 66 Patientenszenarien in den Stadien Früh-, Metastasen- und Überlebensstadium. Jede Interaktion begann mit einer standardisierten Phrase, um ChatGPT dazu zu veranlassen, die Rolle des Anbieters einzunehmen. Mithilfe iterativer Befragungen lieferte das Tool eine Diagnose, einen Behandlungsplan, Triage-Empfehlungen und Ratschläge zur unterstützenden Pflege für häufige Triage-Probleme in der Onkologie. Diese wurden anschliessend von den Ärzten auf ihre Angemessenheit überprüft.
Die primären Ergebnisse waren der Anteil der Szenarien, in denen LLM zu einer korrekten oder akzeptablen Diagnose gelangte und klinisch angemessene oder vernünftige Triage-Empfehlungen gab. Der sekundäre Endpunkt war die Angemessenheit der Fragen von LLM während der Anamnese.
Ergebnisse
Von 849 durch LLM generierten Fragen in 132 simulierten Interviews waren 97 % medizinisch hochgradig angemessen und 3 % angemessen, aber repetitiv. Die richtige Diagnose wurde in 89 % der Szenarien als erste Wahl aufgeführt und in 98 % der Fälle in die Differentialdiagnose aufgenommen. Die Triage-Empfehlungen wurden in 92 % der Szenarien als sehr angemessen bewertet, und keine der Empfehlungen wurde als gefährlich eingestuft.
Die klinischen Entscheidungen bezüglich der unterstützenden Behandlung waren in 86 % der Fälle vollständig angemessen und in 14 % der Fälle angemessen, aber nicht optimal. Die Gesamtübereinstimmung zwischen den ärztlichen Bewertern hinsichtlich der Triage-Leistungskennzahlen von ChatGPT lag bei 77 % bis 86 %.
Schlussfolgerung
Die von ChatGPT-4o und ChatGPT-o1pro gesteuerte iterative Befragung zur Triage häufiger ambulant behandelter Brustkrebsfälle war diagnostisch genau und klinisch angemessen. Diese Ergebnisse sprechen für eine weitere Bewertung in der Praxis, um die Sicherheit von LLM-generierten Triage-Empfehlungen bei der Interaktion mit Personen mit unterschiedlichem medizinischem Vokabular und unterschiedlicher Gesundheitskompetenz zu bestätigen.
Quelle:Chan L et al. ChatGPT Performance as Patient-Facing Triaging Tool in Oncology
Trastuzumab Deruxtecan plus Pertuzumab bei HER2-positivem metastasiertem Brustkrebs
Trastuzumab Deruxtecan hat sich bei Patientinnen mit zuvor behandeltem HER2-positivem fortgeschrittenem oder metastasiertem Brustkrebs als wirksam erwiesen. Die Wirksamkeit und Sicherheit von Trastuzumab Deruxtecan bei Patientinnen ohne vorherige Therapie für HER2-positiven fortgeschrittenen oder metastasierten Brustkrebs ist jedoch unklar.
Methoden
Studienleiterin Sara Tolaney und ihre Mitarbeiter führten eine Phase-3-Studie mit Patientinnen durch, die an HER2-positivem, fortgeschrittenem oder metastasiertem Brustkrebs litten und zuvor noch keine Chemotherapie oder HER2-gerichtete Therapie gegen metastasierte Erkrankungen erhalten hatten. Die Patientinnen wurden im Verhältnis 1:1:1 randomisiert und erhielten entweder Trastuzumab Deruxtecan plus Pertuzumab, Trastuzumab Deruxtecan plus Placebo oder die Kombination aus Taxan, Trastuzumab und Pertuzumab (THP). Der primäre Endpunkt war das progressionsfreie Überleben, das durch eine verblindete, unabhängige, zentrale Überprüfung bewertet wurde. Sekundäre Endpunkte waren das objektive Ansprechen, die Ansprechdauer und die Sicherheit.
Ergebnisse
Für diese vorab festgelegte Zwischenanalyse werden Daten zu Trastuzumab Deruxtecan plus Pertuzumab und zu THP berichtet. Die Daten zu Trastuzumab Deruxtecan plus Placebo bleiben bis zur endgültigen Analyse des progressionsfreien Überlebens verblindet. Zum Stichtag (26. Februar 2025) betrug das mediane progressionsfreie Überleben 40,7 Monate unter der Kombination aus Trastuzumab Deruxtecan und Pertuzumab (383 Patienten) sowie 26,9 Monate unter THP (387 Patienten) (Hazard Ratio für Progression oder Tod: 0,56; 95 %-Konfidenzintervall [KI]: 0,44 bis 0,71; p < 0,00001 [p-Wert-Grenze für Überlegenheit: 0,00043]). Die Inzidenz eines bestätigten Ansprechens betrug 85,1 % unter Trastuzumab Deruxtecan plus Pertuzumab und 78,6 % unter THP (vollständiges Ansprechen bei 15,1 % bzw. 8,5 %), mit einer medianen Ansprechdauer von 39,2 Monaten bzw. 26,4 Monaten. Die Sicherheit entsprach den bekannten Profilen der einzelnen Behandlungen. Die Inzidenz von unerwünschten Ereignissen des Grades 3 oder höher betrug 63,5 % unter Trastuzumab Deruxtecan plus Pertuzumab und 62,3 % unter THP. Am häufigsten traten unter Trastuzumab Deruxtecan plus Pertuzumab Neutropenie, Hypokaliämie und Anämie sowie unter THP Neutropenie, Leukopenie und Diarrhoe auf. Eine bestätigte, arzneimittelbedingte interstitielle Lungenerkrankung oder Pneumonitis trat bei 12,1 % der Patienten unter Trastuzumab Deruxtecan plus Pertuzumab (Grad 1 oder 2 bei 44 Patienten und Grad 5 [Tod] bei zwei Patienten) auf, während es unter THP nur 1,0 % waren (alle Grad 1 oder 2).
Schlussfolgerungen
Bei der Erstlinienbehandlung von HER2-positivem, fortgeschrittenem oder metastasiertem Brustkrebs führte Trastuzumab Deruxtecan plus Pertuzumab zu einem signifikant geringeren Risiko für Progression oder Tod als THP, ohne dass neue Sicherheitssignale auftraten. (Finanziert von AstraZeneca und Daiichi Sankyo, DESTINY-Breast09, ClinicalTrials.gov-Nummer NCT04784715).
Quelle:Tolaney et al.Trastuzumab Deruxtecan plus Pertuzumab for HER2-Positive Metastatic Breast Cancer. New Engl J Med. Published October 29, 2025. DOI: 10.1056/NEJMoa2508668
Das 6. «ESMO in the Alps» wurde im KKL Luzern mit einer inspirierenden Eröffnungsrede von Prof. Dr. med. Miklos Pless, (Winterthur) dem Präsidenten des Swiss Cancer Institute eröffnet. Schweizer Experten hatten die neuesten Erkenntnisse aus der Onkologie ausgewählt, die auf dem ESMO-Kongress 2025 in Berlin präsentiert wurden, und kommentierten sie. Der zweite Teil dieses Berichts bietet eine Übersicht zu den Brust-, gynäkologischen und urogenitalen Krebserkrankungen.
Urogenitale Karzinome
Die Experten Prof. Silke Gillessen Sommer, EOC und Prof. Arnoud Templeton, St. Claraspital Basel stellten vier Studien zu diesem Thema vor.
Teil 1: Urothelkarzinom
Disitamab vedotin (DV) + Toripalimab (T) vs. 1L-Chemotherapie bei metastasiertem Urothelkarzinom mit HER2-Expression: DV + Tor + CAPOX zeigte eine überlegene ORR und ein längeres PFS bei akzeptablem Sicherheitsprofil; eine reduzierte CAPOX-Dosis verbesserte die Verträglichkeit bei gleichbleibender Wirksamkeit.
KN-905/EV303: Perioperative EV-P bei CIS unfitten Patienten mit cMO MIBC: EFS und OS- Benefit. In der Studie KEYNOTE-905/EV-303 zeigte die Kombination aus einem Antikörper-Wirkstoff-Konjugat und einer Immuntherapie statistisch signifikante und klinisch bedeutsame Verbesserungen hinsichtlich des ereignisfreien Überlebens und des Gesamtüberlebens.
Die Kombination von Enfortumab Vedotin (EV) – einem Antikörper-Wirkstoff-Konjugat – und Pembrolizumab vor und nach einer radikalen Zystektomie und einer pelvinen Lymphknotenentfernung könnte laut den auf dem Präsidialsymposium des ESMO-Kongresses 2025 vorgestellten Ergebnissen ein potenzieller neuer Behandlungsstandard für Patienten mit muskelinvasivem Blasenkrebs (MIBC) sein, die nicht für eine Behandlung mit Cisplatin in Frage kommen. «KEYNOTE-905/EV-303 wurde als positive Studie erwartet, da die Studienbehandlung mit einer alleinigen Operation verglichen wird und beide systemischen Therapien als hochwirksam bekannt sind, aber die von den Forschern berichteten Hazard Ratios (HRs) für das ereignisfreie Überleben und das Gesamtüberleben sind bei dieser gefährdeten Patientengruppe besonders beeindruckend», sagte Dr. Jonathan Rosenberg vom Memorial Sloan Kettering Cancer Center, New York, USA, zur Diskussion der Studienergebnisse (siehe Kasten).
IMvigor011:Dr. Powles (London) und Kollegen berichteten über die primäre Analyse der globalen, randomisierten, doppelblinden Phase-3-Studie IMvigor011, in der ctDNA-gesteuertes Atezolizumab im Vergleich zu Placebo untersucht wurde. Atezolizumab als adjuvante Therapie zeigte bei Patientinnen und Patienten mit muskelinvasivem Blasenkrebs, die durch serielle molekulare Tests auf Restkrankheit als ctDNA+ identifiziert wurden, eine statistisch signifikante Verbesserung des krankheitsfreien Überlebens und des Gesamtüberlebens im Vergleich zu Placebo. Der klinische Nutzen war in wichtigen Untergruppen im Allgemeinen konsistent. Patientinnen und Patienten, die bei den Tests durchgehend ctDNA– waren, wiesen ein geringes Risiko für ein Rezidiv und den Tod auf. Das Sicherheitsprofil von Atezolizumab war verträglich, es gab keine neuen Erkenntnisse.
Diese Ergebnisse deuten darauf hin, dass durch serielle ctDNA-Überwachung Patienten mit muskelinvasivem Blasenkrebs identifiziert werden können, die von einer adjuvanten Behandlung mit Atezolizumab profitieren, während Patienten, die bei den Tests durchgehend ctDNA– sind, vor einer unnötigen Behandlung verschont bleiben.
Enfortumab Vedotin + Pembrolizumab wurde als Standard Erstlinientherapie für lokal fortgeschrittenes oder metastasierendes Urothelkarzinom mit anhaltendem Überlebens-Benefit und konsistenten Resultaten bei verschiedenen Patientensubgruppen, inklusive ältere Patienten bestätigt.
Teil 2: Prostatakarzinom
Beim metastasierenden Hormon-sensitiven Prostatakarzinom mHSPC werden 4 klinische Gruppen unterschieden: Synchrones, hohes Volumen, synchrones geringes Volumen metachronisch (rezidiviert) hohes Volumen, metachronisch (rezidiviert) geringes Volumen. In der EMBARK- Studie, die von Dr. Neil Shore (Myrtle Beach, South Carolina) vorgestellt wurde, wurde die Wirksamkeit und Sicherheit von Enzalutamid + ADT mit Enzalutamid allein bei biochemisch rezidiviertem Prostatakarzinom verglichen. Die Resultate legen nahe, dass Enzalutamid eine wichtige Ergänzung der Behandlung von Patienten mit hochriskantem biochemisch rezidivierendem Prostatakarzinom sein könnte und möglicherweise den Behandlungsstandard für diese Patientengruppe verändern wird. Die Ergebnisse der Studie sprechen für einen früheren Einsatz von Androgenrezeptor-Inhibitoren im Behandlungsverlauf, was die Behandlungsergebnisse für die Patienten deutlich verbessern könnte.
CAPitello-281
In CAPitello wurde Capivasertib + Abiraterone mit Placebo + Abiraterone bei Patienten mit PTEN defizientem de novo metastasierendem Hormon-sensitivem Prostatakarzinom in Phase III verglichen. Die Schlussfolgerungen von Prof. Karim Fizazi (Institut Gustave Roussy and Centre Oscar Lambret), zu CAPitello-281 waren: Patienten mit PTEN-defizientem mHSPC haben eine schlechte Prognose und profitieren nur in geringem Masse von der derzeitigen Standardtherapie. CAPItello-281 erreichte sein primäres Ziel und zeigte einen statistisch signifikanten rPFS-Vorteil mit Capi + Abi gegenüber Placebo + ABI. – Medianes rPFS im Capi + Abi-Arm 33,2 Monate gegenüber 25,7 Monaten im PBO + Abi-Arm; p = 0,034. Konsistente Vorteile wurden auch bei sekundären Endpunkten und in klinisch relevanten vordefinierten Untergruppen beobachtet. Das OS war noch nicht ausgereift, und eine weitere Nachbeobachtung ist geplant. Post-hoc-Analysen bei erhöhten PTEN-Cut-offs zeigen eine grössere Behandlungswirkung mit Capi+Abi. Die häufigsten Nebenwirkungen <3 Grad wie Hautausschlag und Hyperglykämie sind bei AKT-Hemmung zu erwarten. Abi + CAPi stellt eine potenzielle First-in-Class-Therapie bei Patienten mit PTEN-defizientem mHSPC dar.
PSMAddition 177Lu-PSMA-617
Die von Prof. Scott Tagawa (New York) präsentierte Phase-III-Studie zeigte, dass die Zugabe von 177Lu-PSMA-617 zu ADT + ARPI das radiografische progressionsfreie Überleben (rPFS) bei PSMA-positivem metastasiertem hormonsensitivem Prostatakrebs (mHSPC) im Vergleich zur Standardtherapie mit ADT + ARPI allein signifikant verbesserte, mit einem günstigen Trend beim Gesamtüberleben, einer überschaubaren Sicherheit und ohne wesentliche Auswirkungen auf die Lebensqualität. Die Zugabe der Radioligandentherapie führte zu einer 28-prozentigen Verringerung des Risikos einer radiologischen Progression oder des Todes. PSMAddition ist die erste Phase 3 Studie mit einer zielgerichteten Radioligandentherapie bei Patienten mit mHSPC.
Prostatakarzinom: mHSPC Take Home Messages
Eine ADT- und ARPI-basierte systemische Therapie ist für die meisten Patienten immer noch der Standard of Care. Die Zugabe von Capivasertib führt zu einem geringen Nutzen im rPFS (HR 0,81), jedoch noch nicht im OS, und fügt zusätzliche Toxizität hinzu. Das ändert die Praxis nicht. Die Zugabe von sechs Zyklen 177Lu-PSMA-617 führt zu einem Benefit beim rPFS, aber nicht beim OS, und fügt erhebliche Toxizität hinzu. (Inklusive Langzeitoxizität, ändert die Praxis nicht.)
Metastasierendes kastrationsresistentes Prostatakarzinom: mCRPC und 177Lu-PSMA-617
Das «VISION ARPI + Taxan-Protokoll» bezieht sich auf eine Sekundäranalyse der VISION-Studienergebnisse, die auf dem ESMO-Kongress 2025 vorgestellt wurde oder damit in Zusammenhang steht und den Nutzen einer Kombination aus 177Lu-PSMA-617-Radioligandentherapie mit Androgenrezeptor-Pathway-Inhibitoren (ARPIs) und Taxan-basierter Chemotherapie bei Patienten mit metastasiertem kastrationsresistentem Prostatakrebs (mCRPC) untersucht. Die Analyse legt nahe, dass die Zugabe von ARPIs zu 177Lu-PSMA-617 die Gesamtüberlebensrate verbessern kann, selbst nach einer vorherigen taxanbasierten Behandlung.
PSMAfore
Die PSMAfore-Studie, war eine Phase-III-Studie die 177Lu-PSMA-617 (eine gezielte Radioligandentherapie) mit einer Umstellung auf ARPI (Androgenrezeptor-Pathway-Inhibitor) bei PSMA-positivem metastasiertem kastrationsresistentem Prostatakrebs (mCRPC) verglich. Sie zeigte eine verlängerte radiografische progressionsfreie Überlebenszeit, jedoch aufgrund hoher Crossover-Raten keine statistisch signifikante Gesamtüberlebenszeit, unterstützte jedoch die Anwendung in dieser Taxan-naiven Situation. Die im August 2025 vorgestellte und auf der ESMO diskutierte endgültige Analyse der Studie hob die überschaubare Sicherheit und die Vorteile bei anderen sekundären Endpunkten hervor.
CCTG PR21: Phase-II-Studie 177Lu-PSMA-617 vs. Docetaxel bei PSMA+ mCRPC
Dr. Kim Chi (Vancouver) präsentierte die Studie PR.21 der Canadian Cancer Trials Group (CCTG), eine randomisierte Phase-II-Studie zum Vergleich von 177Lu-PSMA-617 mit Docetaxel bei mCRPC-Patienten mit PSMA-positiver Erkrankung.
Dr. Chi stellte fest, dass 177Lu-PSMA-617 das radiografische progressionsfreie Überleben (rPFS) bei Taxan-naiven Patienten mit mCRPC im Vergleich zu einem Wechsel zu einem Androgenrezeptor-Pathway-Inhibitor (ARPI) verlängert. Mit 177Lu-PSMA-617 betrug das mediane rPFS 11,6 Monate und das mediane Gesamtüberleben (OS) 24,5 Monate. Primärer Endpunkt: rPFS (ITT) HR 1.01; p = 0.51. Das mediane rPFS betrug 8,6 Monate unter 177Lu-PSMA-617 und 10,7 Monate unter Docetaxel.
Prostatakrebs: mCRPC, Take Home Messages
OS mit Docetaxel signifikant besser gegenüber 177LuPSMA-617 bei Patienten mit mCRPC nach ARPI. Wir müssen die Chemophobie überwinden; einige (fitte) Patienten profitieren von Taxanen.
Brust und Gynäkologische Krebserkrankungen
Die Experten Dr. med. Elena Kralidis, Brust-Zentrum Zürich, und Prof. Dr. med. Stefan Aebi, Luzerner Kantonsspital, präsentierten Ergebnisse zu Brust- und gynäkologischen Krebserkrankungen.
DESTINY-Breast05
Dr. Sara Tolaney (Dana-Farber Cancer Institute, Boston) stellte eine Zwischenanalyse von DESTINY-Breast05 vor: Trastuzumab Deruxtecan (T-DXd) reduzierte bei Patientinnen mit hochriskantem, HER2-positivem frühem Brustkrebs und residueller invasiver Erkrankung nach neoadjuvanter Therapie das Rezidivrisiko im Vergleich zu Trastuzumab Emtansine (T-DM1) um rund 50 %. Eine längere Nachbeobachtung ist nötig, um einzelne Signale (u. a. ZNS-Ereignisse) zu bestätigen. Diskutiert wurde zudem der prä- vs. postoperative Einsatz von T-DXd.
Prä-operativ:
1. pCR-Rate verbessert, weniger axilläre Operationen
2. Kürzerer Verlauf. T-DXd 4 Zyklen vs. 14 Zyklen
3. Weniger Toxizität da nur während 4 Zyklen gegeben
4. Könnte in besserer Lebensqualität für Patienten resultieren
Post-operativ:
1. Könnte eine de-eskalierte präoperative Therapie ermöglichen, die diejenigen Patienten mit pCR T-DXd verschont, aber diejenigen mit RD erhalten schlussendlich 14 Zyklen
monarchE: Primäre OS-Daten zur adjuvanten Therapie mit Abemaciclib + ET bei HR+/HER2- Brustkrebs
Wie Prof. Stephen Johnston (London) darlegte, hat die monarchE-Studie einen wichtigen Meilenstein in der Behandlung von Hormonrezeptor-positivem (HR+), HER2-negativem, lymphknotenpositivem, hochriskantem Brustkrebs im Frühstadium erreicht. Eine zweijährige adjuvante Behandlung mit Abemaciclib in Kombination mit einer endokrinen Therapie (Verzenio) führte zu einer statistisch signifikanten und klinisch bedeutsamen Verbesserung der Gesamtüberlebensrate (OS) im Vergleich zu einer alleinigen endokrinen Therapie (ET) und bestätigte somit den langfristigen Nutzen einer CDK4/6-Therapie.
POSITIVE trial
Die POSITIVE-Studie untersuchte die Sicherheit des Abbruchs der endokrinen Therapie bei hormonrezeptorpositivem Brustkrebs, um eine Schwangerschaft zu versuchen. An dieser Studie nahmen 518 Frauen im Alter von bis zu 42 Jahren (Durchschnittsalter 37 Jahre) teil, die eine 18- bis 24-monatige endokrine Therapie abgeschlossen und diese dann abgebrochen hatten, um eine Schwangerschaft zu versuchen. Die Patientinnen wurden zwischen 2014 und 2019 in die Studie aufgenommen (bevor die CDK 4/6-Inhibitoren Abemaciclib und Ribociclib zugelassen und bei Brustkrebs im Frühstadium eingesetzt wurden). Es war eine dreimonatige Auswaschphase (Zeit ohne Medikamenteneinnahme vor dem Versuch einer Schwangerschaft) erforderlich, und die Studienteilnehmerinnen hatten bis zu zwei Jahre Zeit, um für eine Schwangerschaft auf die endokrine Therapie zu verzichten. Auf der Grundlage dieser Ergebnisse kommen die Autoren zu dem Schluss, dass eine vorübergehende Unterbrechung der ET für eine Schwangerschaft das Risiko für Brustkrebsereignisse nach einer medianen Nachbeobachtungszeit von 71 Monaten nicht erhöht. Mehr als zwei Drittel der Frauen hatten eine Lebendgeburt und die meisten nahmen die endokrine Therapie gemäss Protokoll wieder auf. Wir müssen die Nachbeobachtung fortsetzen, aber diese Daten sind sicherlich beruhigend. Eine weitere Nachbeobachtung ist bis 2029 geplant.
Fünf-Jahres-Follow-up der Wirksamkeitsergebnisse der NATALEE-Studie und aktualisierte Gesamtüberlebensrate
Adjuvante Therapie mit Ribociclib plus nichtsteroidalen Aromatasehemmern bei Patientinnen mit HR-positivem/HER2-negativem Brustkrebs im Frühstadium. Diese vorab festgelegte 5-Jahres-Nachbeobachtung der Wirksamkeitsergebnisse aus der NATALEE-Studie, die von Prof. John Crown (Dublin) präsentiert wurde, zeigte, dass Ribociclib in Kombination mit nichtsteroidalen Aromatasehemmern das Risiko eines Rezidivs über den 3-Jahres-Behandlungszeitraum hinaus weiter senkte, was den Einsatz als adjuvante Therapie bei Patientinnen mit HR-positivem/HER2-negativem Brustkrebs im Frühstadium unterstützt. Es wurde ein anhaltender positiver Trend hinsichtlich der Verbesserung der Gesamtüberlebenszeit zugunsten von Ribociclib + NSAI beobachtet.
TROPION BREAST02
In der Studie TROIAN BREAST02 wurde der Wirkstoff Datopotamab Deruxtecan (auch Dato-DXd oder Datroway genannt) bei Patientinnen mit zuvor unbehandeltem, lokal rezidivierendem, inoperablem oder metastasiertem, dreifach negativem Brustkrebs (TNBC) untersucht. Teilnahmeberechtigt waren Patientinnen, die nicht für eine Therapie mit PD-1/PD-L1-Inhibitoren infrage kamen. Datopotamab Deruxtecan wurde mit einer vom Prüfer ausgewählten Chemotherapie (Paclitaxel, Nab-Paclitaxel, Capecitabin, Carboplatin oder Eribulin) verglichen.
Die primären Endpunkte waren progressionsfreies Überleben (PFS) und Gesamtüberleben (OS). Datopotamab Deruxtecan zeigte im Vergleich zur Chemotherapie eine statistisch signifikante und klinisch bedeutsame Verbesserung sowohl des progressionsfreien Überlebens (PFS) als auch des Gesamtüberlebens (OS), einschliesslich einer Verbesserung des medianen Gesamtüberlebens um fünf Monate. Dies sind die ersten positiven Ergebnisse einer First-Line-Studie zu TNBC, die einen signifikanten Überlebensvorteil für ein ADC zeigen.
TROP2-gerichtete ADCs Sacituzumab Govitecan und Datopotamab Deruxtecan erreichen ihre primären Endpunkte in Phase-III-Studien ASCENT-03
Antikörper-Wirkstoff-Konjugate (ADCs) gegen TROP2 rücken weiter in frühere Behandlungslinien vor, darunter auch die Erstlinienbehandlung des dreifach negativen Brustkrebses. In ASCENT-03 wurden ADCs gegen TROP2 mit einer Chemotherapie bei Patientinnen mit zuvor unbehandeltem, fortgeschrittenem TNBC verglichen, die nicht für eine Immuntherapie infrage kommen. In der ASCENT-03-Studie mit 558 Patientinnen und Patienten war das mediane progressionsfreie Überleben (PFS) unter Sacituzumab Govitecan im Vergleich zur Chemotherapie signifikant länger (9,7 vs. 6,9 Monate; HR 0,62; 95-%-KI 0,50–0,77; p<0,0001). Nach einer medianen Nachbeobachtungszeit von 13,2 Monaten sind die Daten zum Gesamtüberleben (OS) noch nicht ausgereift. Die objektiven Ansprechraten (ORR) waren ähnlich (48 % vs. 46 %), die mediane Ansprechdauer (DOR) war unter Sacituzumab Govitecan länger (12,2 vs. 7,2 Monate). Die Rate behandlungsbedingter unerwünschter Ereignisse Grad ≥3 betrug 66 % unter Sacituzumab Govitecan und 62 % unter Chemotherapie; Therapieabbrüche waren unter Sacituzumab Govitecan seltener (4 % vs. 12 %).
Die Therapieoptionen zur Behandlung des fortgeschrittenen kleinzelligen Lungenkarzinoms (SCLC, small cell lung cancer) haben sich in den letzten Jahren stark gewandelt [1, 2]. Anlässlich der vielen Neuerungen kamen am Swiss Oncology and Haematology Congress (SOHC) am 19. November 2025 in Basel mehrere Expert/-innen aus der ganzen Schweiz zusammen, um anhand eines Patientenfalls über die Anwendung der neuen Therapieoptionen zu diskutieren.
Lungenkarzinome sind noch immer die häufigste krebsbedingte Todesursache [1]. In 12–15% der Fälle liegt ein SCLC vor, welches bei den meisten Patient/-innen (60–70%) bereits bei Diagnose in einem fortgeschrittenen Stadium (ES, extensive stage) ist. Noch bis 2019 standen für die Behandlung des ES-SCLC nur die Chemo- und Strahlentherapie zur Verfügung [2]. Mit den Ergebnissen der IMpower-133- und CASPIAN-Studien wurde die Chemoimmuntherapie (CIT) plus anschliessende Erhaltungstherapie mit Atezolizumab oder Durvalumab zum neuen Standard [1, 3, 4]. Zudem zeigte die aktuelle IMforte-Studie, dass eine Hinzugabe von Lurbinectedin zur Erhaltungstherapie mit Atezolizumab das Outcome der Patient/-innen weiter verbessern kann [5]. Unter der Moderation von Prof. Dr. Alessandra Curioni Fontecedro (Kantonsspital Fribourg) tauschten sich die Expert/-innen Prof. Dr. Martin Früh (Kantonsspital St. Gallen), Prof. Dr. Alfredo Addeo (Universitätsspital Genf), Dr. Nuria Neisy Mederos Alfonso (Universitätsspital Vaud), Dr. Martina Imbimbo (Kantonsspital Ticino) und Dr. Ulrich Richter (Universitätsspital Zürich) anhand eines Patientenfalls von Dr. Alexander Meisel (Kantonsspital Glarus) zu den neuen Behandlungsoptionen aus.
ES-SCLC in der klinischen Praxis: ein Patientenfall
Dr. Meisel startete die Gesprächsrunde mit einem Patientenfall: der 79-jährige Patient stellte sich im Dezember 2024 mit Thoraxschmerzen und Belastungsdyspnoe vor. Er war ehemaliger Raucher mit zusätzlicher Asbest-Belastung in der Vergangenheit und zeigte stark erhöhtes LDH sowie erhöhte Leberwerte. Eine peritoneale Biopsie führte zur Diagnose eines Slfn11-positiven SCLC mit TP53 und CDKN2A-Mutation. Dr. Meisel entschied sich für das IMpower-133-Regimen (Atezolizumab + Carboplatin + Etoposide) [3], mit welchem die detektierte pulmonale Masse sowie die Lebermetastasen nach 6 Zyklen zurückgingen (Teilremission). Er bekam die Genehmigung für den Einsatz des IMforte-Regimens und nach 4 Zyklen dieser Behandlung kam es zu einer weiteren Verkleinerung der Läsionen (nahezu vollständiges Ansprechen). Über 11 Monate wurde dann keine Progression festgestellt. Der Tumormarker NSE war bereits nach dem ersten Zyklus der 1L-Therapie unter den Schwellenwert von 20 µg/l abgesunken und blieb auch unter der Erhaltungstherapie auf tiefem Niveau. Insgesamt vertrug der Patient die Therapie gut und zeigte hauptsächlich Nebenwirkungen geringen Grades (≤2). Unter der Erhaltungstherapie wurde eine Neutropenie 3. Grades festgestellt, welche jedoch mit zwei GCS-F-Gaben behoben werden konnte.
Bewertung der neuen Therapieoptionen durch Expert/-innen
Prof. Früh stellte fest, dass sich durch das Add-On Lurbinectedin zur Erhaltungstherapie neue Möglichkeiten für SCLC-Patient/-innen ergeben. In seiner Erfahrung ist die Verträglichkeit des erweiterten Regimes gut, aber es sollte zwingend GCS-F gegeben werden, um der Neutropenie entgegenzuwirken. Patient/-innen sollten dabei immer in den Therapieentscheid miteinbezogen werden. Prof. Früh erwartet sich durch die Zugabe von Lurbinectedin zur Erhaltungstherapie eine Zunahme an Patient/-innen, welche zur Zweitlinientherapie (2L) fortschreiten können. Dr. Imbimbo stimmt ihm zu, dass eine Intensivierung der Erhaltungstherapie vor allem bei jüngeren Patient/-innen sinnvoll ist, um eine schnelle Progression zu verhindern. Prof. Addeo wirft ein, dass die Datenlage sehr vielversprechend sei, er aber bei unfitten Patient/-innen oder bei Hirnmetastasen die Erhaltungstherapie nicht intensivieren würde. Das Alter selbst sei für ihn aber keine Limitation. Auch Dr. Richter fügt hinzu, dass er anfangs aufgrund der assoziierten Hämatotoxizität Bedenken hatte und weist darauf hin, dass in der 2L aktuell nur Chemotherapeutika zur Verfügung stehen. In Zukunft sind jedoch etwa mit dem bispezifischen T-Cell-Engager Tarlatamab [6] wirksamere 2L-Therapien in Aussicht, welche ein anderes Spektrum an Nebenwirkungen zeigen. Im Hinblick darauf würde auch Dr. Richter die Erhaltungstherapie intensivieren, sodass Patient/-innen eine bessere Chance auf eine 2L-Therapie erhalten. Dr. Mederos fügt hinzu, dass sie für ihre Patient/-innen in der 2L vor allem klinische Studien mit Antikörper-Wirkstoff-Konjugaten bevorzugt. Sie stimmt zu, dass die Zugabe von Lurbinectedin zur Erhaltungstherapie eine gute Option darstellt, und die Nebenwirkungen normalerweise handhabbar sind. Die einzige Limitierung für den Einsatz der intensivierten Erhaltungstherapie sieht auch sie bei Hirnmetastasen und Patient/-innen mit einem ECOG PS von >2.
Fazit
Prof. Curioni fasst zusammen, dass die Zusammenkunft der Expert/-innen aus so vielen Kantonen der Schweiz eine wahre Bereicherung für den Austausch war und eine Möglichkeit bot, die derzeit stark im Wandel stehende Behandlungslandschaft des ES-SCLC besser zu verstehen. ES-SCLC-Patient/-innen erhalten dank der neuen Behandlungsoptionen immer öfter die Chance auf eine 2L-Therapie und mit den erwarteten Zulassungen sollten sich diese Chance in Zukunft noch verbessern.
Quelle: «Small Cell Lung Cancer: How the treatment paradigm is evolving? A Patient Case-based Discussion.» Präsentiert am SOHC, 19. November 2025, Basel, Schweiz.
1. Onkopedia Leitlinie. Lungenkarzinom, kleinzellig (SCLC). Stand: September 2025. Abrufbar unter: https://www.onkopedia.com/de/onkopedia/guidelines/lungenkarzinom-kleinzellig-sclc/@@guideline/html/index.html. Letzter Zugriff: November 2025.
2. Deutsches Krebsforschungszentrum, Krebsinformationsdienst. Neue Therapieoptionen beim kleinzelligen Lungenkrebs – erste Immuntherapie für das SCLC zugelassen. Stand: November 2019. Abrufbar unter: https://www.krebsinformationsdienst.de/fachkreise/nachrichten/detail/neue-therapieoptionen-beim-kleinzelligen-lungenkrebs, letzter Zugriff: November 2025.
3. Horn, L., et al., First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med, 2018. 379(23): p. 2220-2229.
4. Paz-Ares, L., et al., Durvalumab, with or without tremelimumab, plus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer: 3-year overall survival update from CASPIAN. ESMO Open, 2022. 7(2): p. 100408.
5. Paz-Ares, L., et al., Efficacy and safety of first-line maintenance therapy with lurbinectedin plus atezolizumab in extensive-stage small-cell lung cancer (IMforte): a randomised, multicentre, open-label, phase 3 trial. Lancet, 2025. 405(10495): p. 2129-2143.
6. Mountzios, G., et al., Tarlatamab in Small-Cell Lung Cancer after Platinum-Based Chemotherapy. N Engl J Med, 2025. 393(4): p. 349-361.
Im Rahmen des Projekts «Ermittlung der Bedürfnisse junger Onkologiepflegefachpersonen in der Schweiz und verbandsspezifische Unterstützungsmöglichkeiten» führte die Onkologiepflege Schweiz in Zusammenarbeit mit dem Institut für Pflege der ZHAW eine schweizweite Befragung durch. Dieser Artikel fasst die wesentlichsten Ergebnisse zusammen – erkannte Bedürfnisse sowie bereits vorhandene Unterstützungsangebote – und stellt verbandsspezifische Unterstützungsmöglichkeiten vor.
Einleitung
Bereits heute fehlt es an ausreichenden Pflegefachpersonen in der Onkologie, und mit dem prognostizierten deutlichen Anstieg der Krebsfälle in den kommenden Jahren wird sich dieser Mangel weiter verschärfen. Umso wichtiger ist es, die Bedürfnisse von Berufseinsteiger/-innen in der Onkologiepflege genauer zu verstehen. Ein gelingender Berufseinstieg hat entscheidenden Einfluss auf Arbeitszufriedenheit und langfristigen Verbleib im Beruf. Gezielte Unterstützungsmassnahmen können dazu beitragen, Motivation, fachliche Entwicklung und Versorgungsqualität nachhaltig zu sichern.
Vor diesem Hintergrund hat die Onkologiepflege Schweiz (OPS) im Rahmen einer grösseren Initiative für Berufseinsteiger/-innen in der Onkologiepflege eine Studie in Auftrag gegeben.
Das Ziel des Projekts war es zu untersuchen: (1a) welche fachspezifischen Bedürfnisse junge Pflegefachpersonen haben, die onkologische Patientinnen und Patienten betreuen (1b) welche Unterstützungsangebote sie bereits nutzen und zusätzlich wünschen, (2) welche Strategie und Unterstützungsangebote Arbeitgebende ihnen bieten, sowie (3) welche Unterstützungsangebote der Verband Onkologiepflege Schweiz dieser Gruppe Pflegefachpersonen und ihren Arbeitgebern bieten könnte.
Methode
Im Rahmen dieses Projekts führten wir zwischen dem 28. Februar und 4. Mai 2025 eine Onlinebefragung in der Schweiz durch (deskriptive Querschnittsstudie).
Die Befragung richtete sich an zwei Zielgruppen: 1) Pflegefachpersonen aus den Versorgungssettings Onkologie – Akut- & Langzeitpflege (ambulant und stationär) – mit unterschiedlichen Ausbildungsniveaus, die entweder nicht älter als 35 Jahre waren (Young Care Nurses) oder über eine Berufserfahrung von maximal drei Jahren in der Onkologie verfügten (New Care Nurses). 2.) Zusätzlich wurden auch Führungspersonen im Bereich der Onkologie zur Teilnahme eingeladen.
Die Rekrutierung der Teilnehmenden erfolgte in erster Linie über den Verband Onkologiepflege Schweiz, welcher über den Newsletter, die Webseite sowie auf dem Jahreskongress des Verbands als auch über Kontaktpersonen von Partnerorganisationen (z. B. VFP) über das Projekt informierte und den Zugangslink zur online Befragung zur Verfügung stellte. Zusätzlich erhielten ehemalige Studierende, die kürzlich den Bachelor in Pflege an der ZHAW abgeschlossen haben oder sich in einer Weiterbildung in Onkologie befanden, die Einladung an der Befragung teilzunehmen.
Die Fragebögen wurden spezifisch für dieses Projekt entwickelt. Der Fragebogen für die Pflegefachpersonen umfasst 48 Fragen und jener für die Führungspersonen 47 Fragen, jeweils zu folgenden Themenbereichen: (1) demografischen Merkmalen, (2) Kompetenzen, Kenntnisse oder Fähigkeiten in der Onkologiepflege, (3) Schulungen in den Kompetenzen bei der Einarbeitung, (4) Relevante Unterstützungsangebote hinsichtlich Dokumente & Schulungen, Einarbeitung & Mentoring und zur allgemeinen Unterstützung. Zusätzlich konnten weitere relevante und fehlende Unterstützungsangebote sowie allgemeine Hinweise zum Thema ergänzt werden. Der Fragebogen stand in deutscher, französischer und italienischer Sprache zur Verfügung. Die Erhebung der Daten erfolgte elektronisch mithilfe des Datenerhebungstools REDCap .
Die Datenanalyse erfolgte deskriptiv unter Verwendung der Datenanalysetools R und RStudio Version 4.4.3 (2025). Entsprechend der jeweiligen Skalenniveaus wurden absolute und prozentuale Häufigkeiten, der Mittelwert (M), der Median (Mdn), die Standardabweichung (SD), der Interquartilsabstand (IQR) sowie der Wertebereich (Range) berechnet. Die Auswertungen wurden getrennt für Pflegefachpersonen und Führungspersonen durchgeführt. Anschliessend erfolgte ein deskriptiver Vergleich der beiden Gruppen.
Resultate
Teilnehmende
Von insgesamt 129 Teilnehmenden, welche die Befragung öffneten, füllten schlussendlich 71 Teilnehmende, 49 Pflegefachpersonen und 22 Führungspersonen, den Hauptfragebogen aus und konnten damit in die Analyse eingeschlossen werden. Dabei nutzen 41 Teilnehmende den Fragebogen in deutscher Sprache, 21 Teilnehmende in italienischer Sprache und neun Teilnehmende in französischer Sprache.
Die Pflegefachpersonen waren mehrheitlich weiblich (n = 46), grösstenteils dipl. Pflegefachfrau/-mann HF/FH (n = 43), meist junge Onkologiepflegefachpersonen (n = 33) und überwiegend im ambulanten Spitalsetting (n = 33) sowie im Bereich Hämato-Onkologie (n = 29) tätig. Die Führungspersonen waren mehrheitlich Stationsleitungen (n = 13), mehrheitlich im ambulanten Spitalsetting (n = 33) tätig und verfügten über 1–21 Jahre an Führungserfahrung.
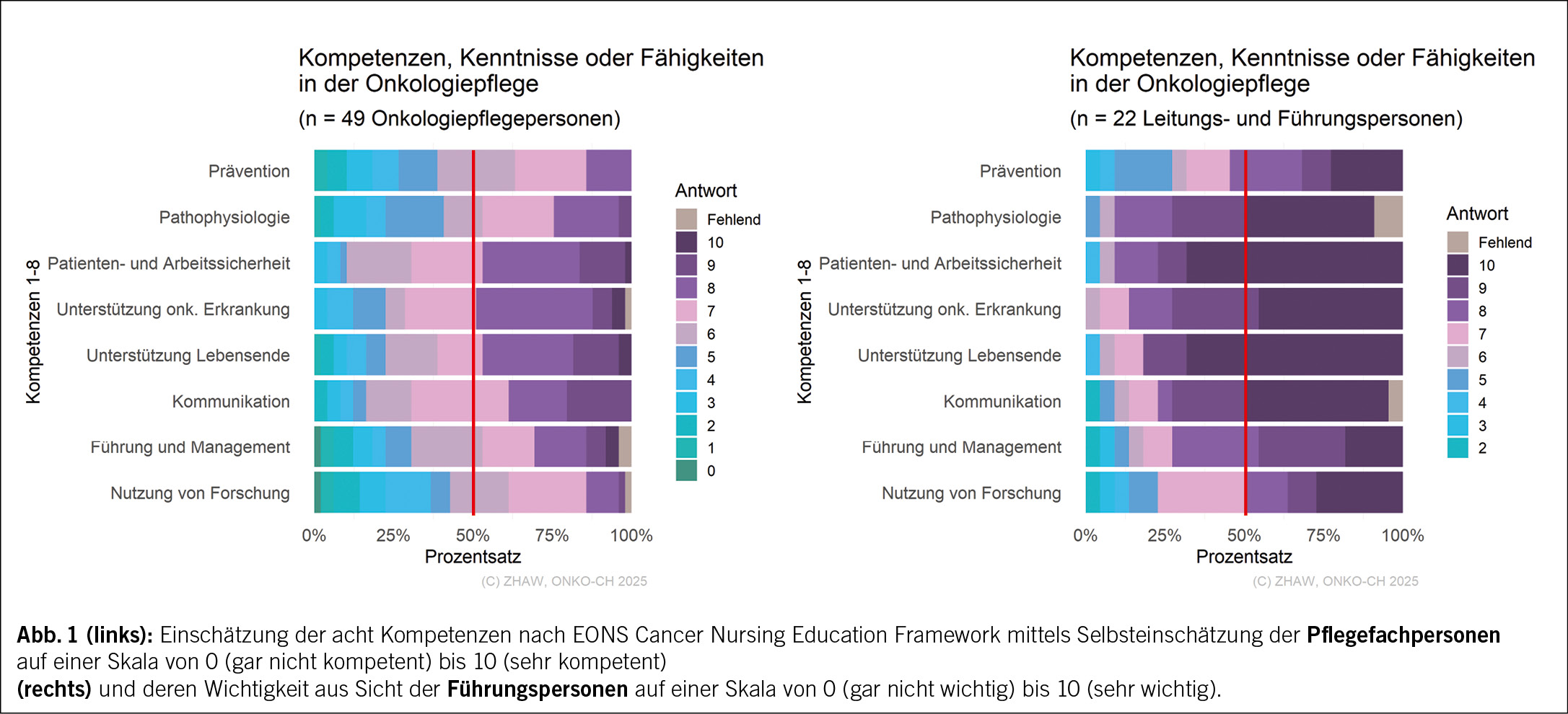
Fachspezifische Bedürfnisse: Einschätzung der Kompetenzen
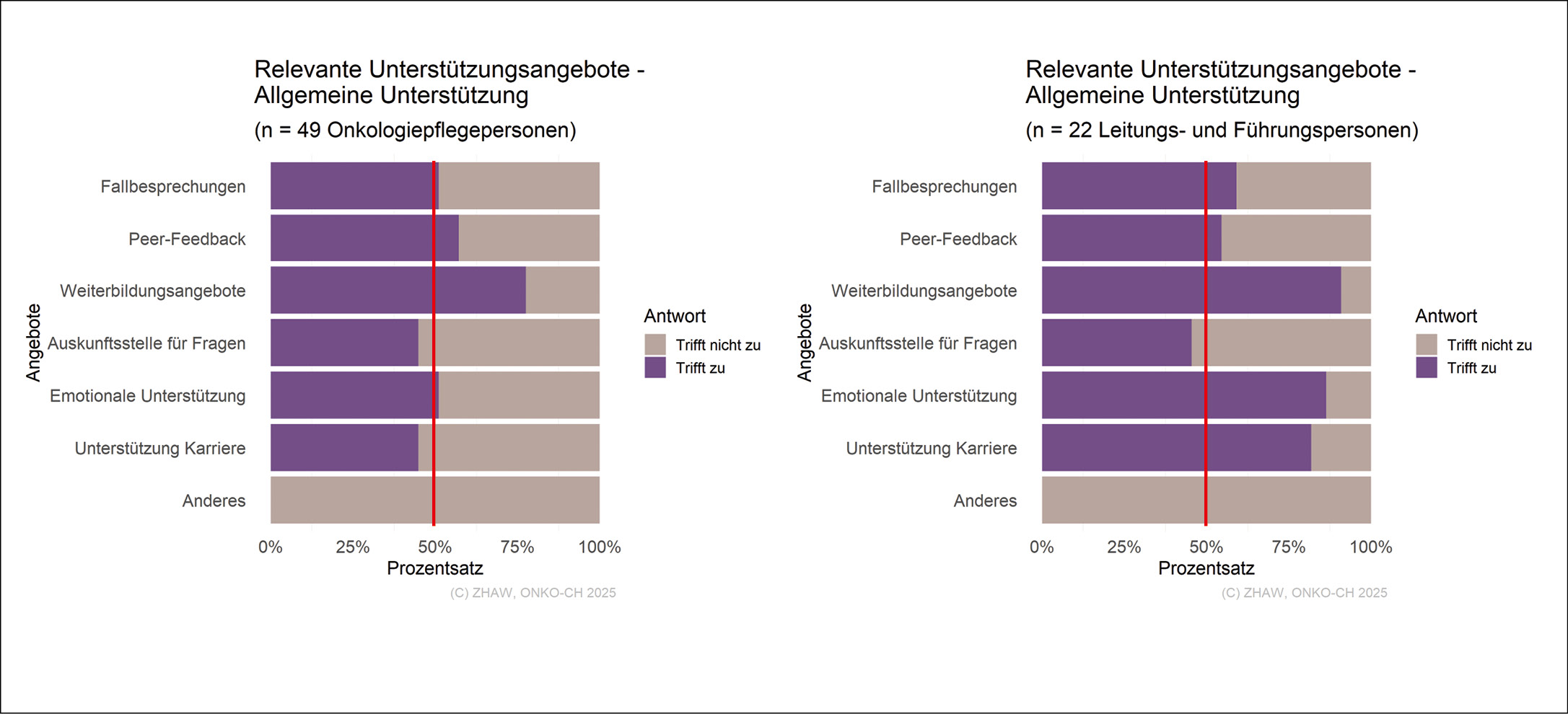
Bei der Einschätzung der acht Kompetenzen nach EONS Cancer Nursing Education Framework (2022) weicht die von den Pflegefachpersonen vorgenommene Selbsteinschätzung der eigenen Kompetenzen und die von den Führungspersonen eingeschätzte Wichtigkeit dieser Kompetenzen zum Teil stark voneinander ab (Abb. 1).
So schätzten bei vier dieser Kompetenzen 50 % der Pflegefachpersonen die eigene Kompetenz auf der gegebenen Skala von 0 (gar nicht kompetent) bis 10 (sehr kompetent) mit einem Wert von sieben oder weniger ein. Bei den anderen vier Kompetenzen schätzten 50 % der Pflegefachpersonen die eigene Kompetenz mit einem Wert von sechs oder weniger ein. Demgegenüber schätzen 50 % der Führungspersonen alle Kompetenzen auf der gegebenen Skala von 0 (gar nicht wichtig) bis 10 (sehr wichtig) mit einem Wert von acht oder höher ein.
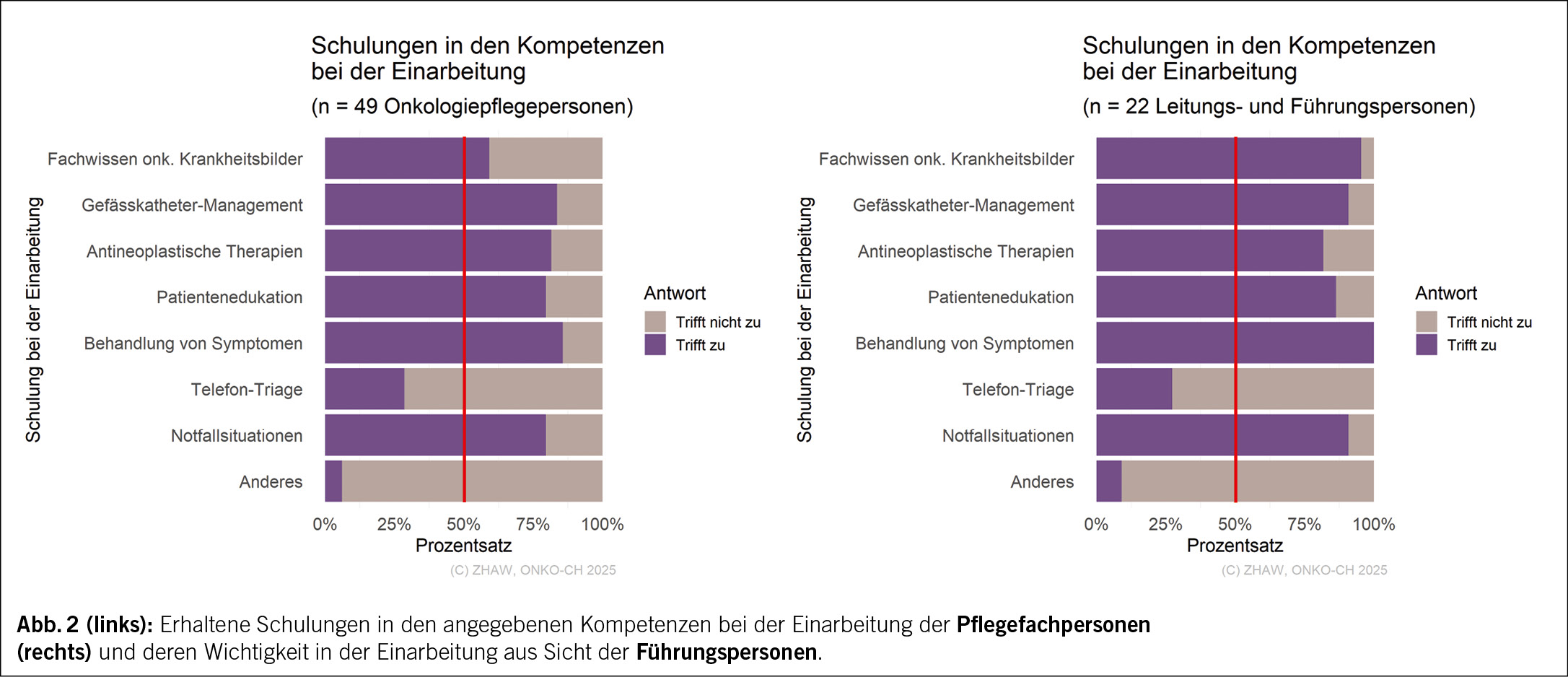
Bei den Einschätzungen der fachspezifischen Bedürfnisse während der Einarbeitungszeit und den damit verbundenen Schulungen, zeigt sich zwischen den Schulungen, die die Pflegefachpersonen erhalten haben und deren Wichtigkeit aus Sicht der Führungspersonen eine gute Übereinstimmung (Abb. 2).
Hiervon ausgenommen sind Schulungen bezüglich «Fachwissens zu den häufigsten onkologischen Krankheitsbildern», welche nur 59 % der Pflegefachpersonen erhielten. Demgegenüber gaben 95 % der Führungspersonen an, auf diese Schulungen innerhalb der Einarbeitung Wert zu legen.
Unterstützungsangebote: Angebot und Relevanz
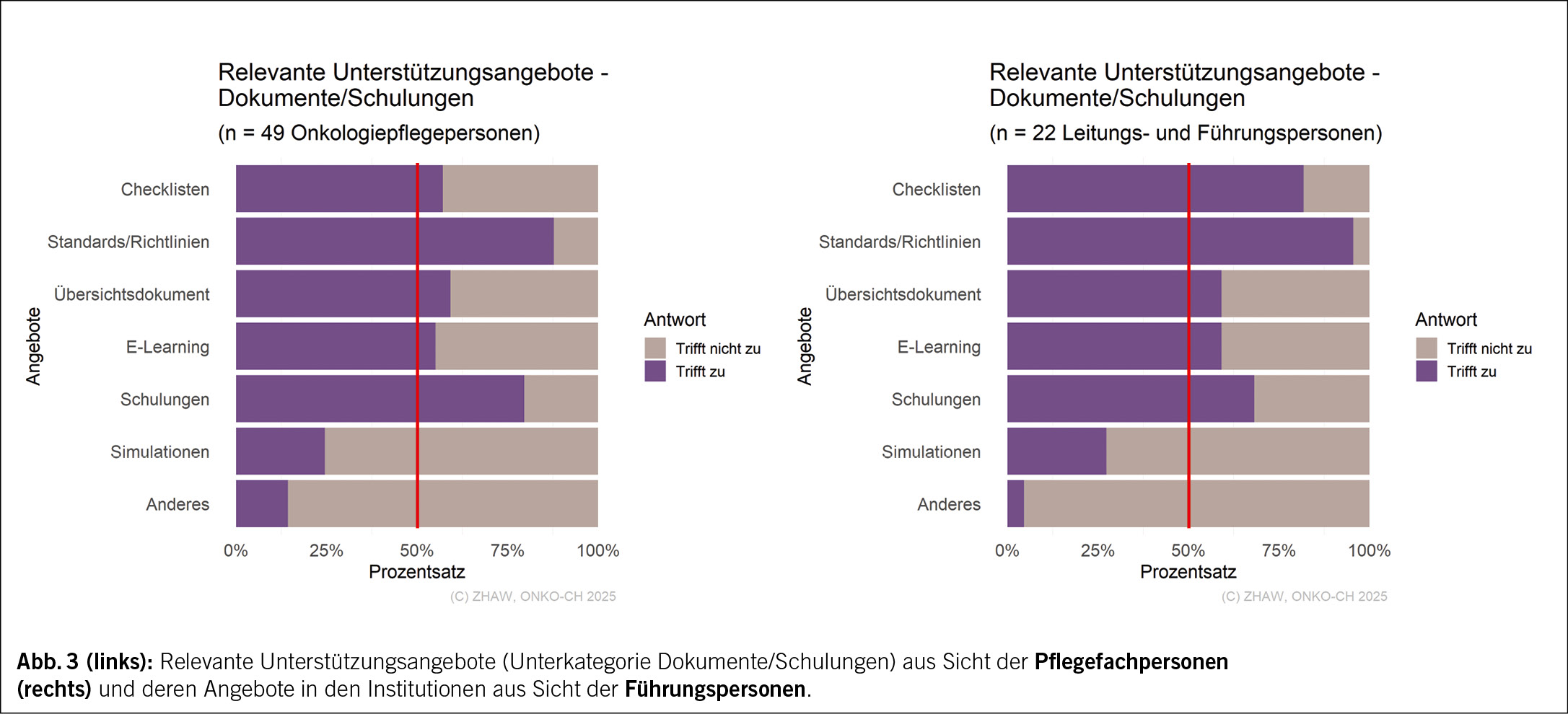
Zwischen den aus Sicht der Pflegefachpersonen relevanten Unterstützungsangeboten und der Unterstützung, welche die Institutionen gemäss der Führungspersonen anbieten, sind grössere Abweichungen feststellbar.
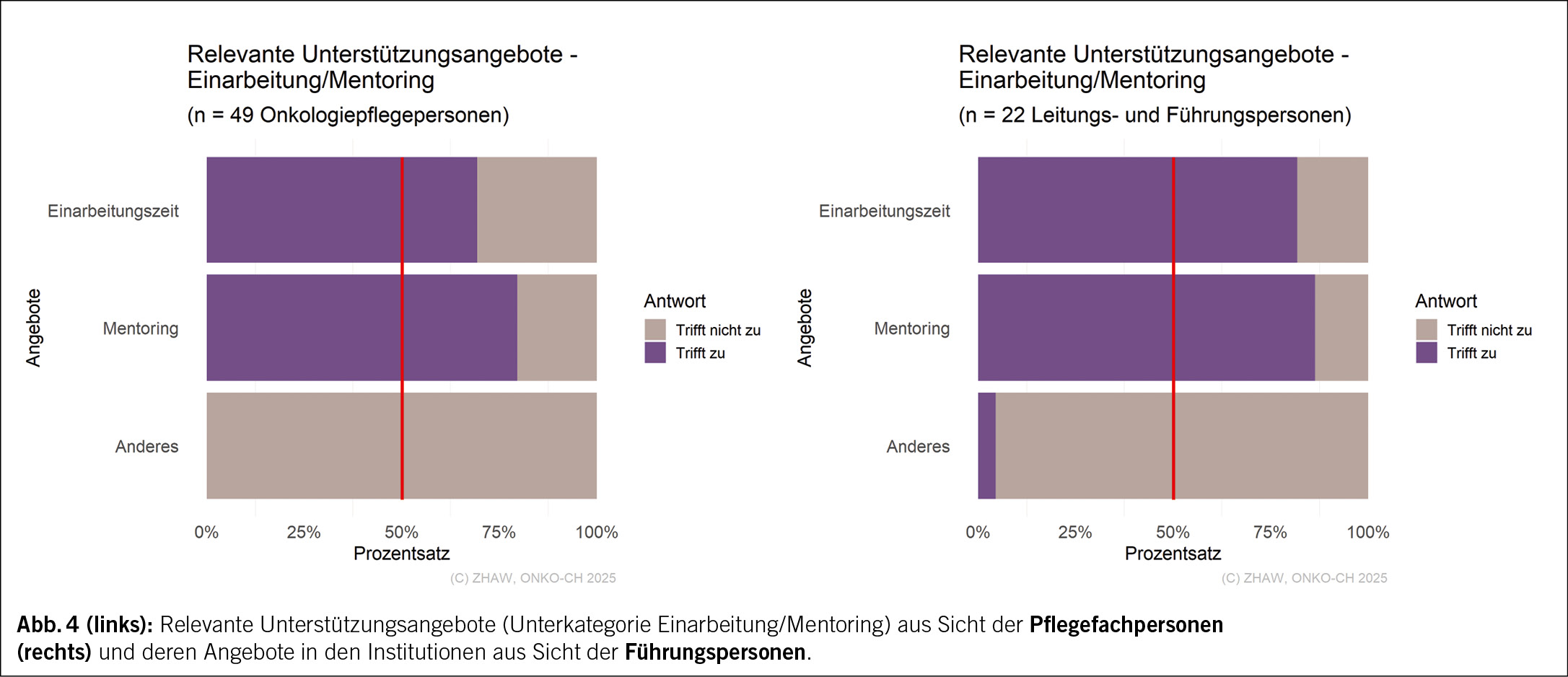
Bezogen auf die Unterkategorie Dokumente/Schulungen (Abb. 3), nannten nur 57 % der Pflegefachpersonen «Checklisten» als ein relevantes Unterstützungsangebot, hingegen nannten 82 % der Führungspersonen diese als ein relevantes Angebot. Während in der Unterkategorie Einarbeitung/Mentoring keine wesentlichen Unterschiede zwischen den Pflegefachpersonen und den Führungspersonen erkennbar sind (Abb. 4), gaben in der Unterkategorie Allgemeine Unterstützung (Abb. 5) nur circa die Hälfte der Pflegefachpersonen die Angebote «Emotionale Unterstützung» mit 51 % und «Unterstützung in der Karriere» mit 45 % als relevant an, während 86 % beziehungsweise 82 % der Führungspersonen die beiden Angebote als vorhandene Unterstützung nannten.
Bezogen auf diese drei Angebotskategorien zur Unterstützung gaben die Pflegefachpersonen eine Vielzahl an Freitextergänzungen zu den Bedarfen an. Die Führungspersonen nannten diesbezüglich fehlende Werkzeuge. Die Freitextergänzungen der Pflegepersonen und der Führungspersonen konnten wie folgt zusammengefasst und geordnet werden:
Zusätzliche Bedarfe der Pflegefachpersonen:
• Dokumente/Schulungen: Regelmässige interne Fortbildungen und (Mikro-)Schulungen (onkologische Erkrankungen, Pflege, Kommunikation), mehr strukturierte Informationen, Zugriff auf Fachinformationen, z. B. Onkologika.ch
• Einarbeitung/Mentoring: Einführung in der Hämatologie, mehr Begleitung durch geschultes Pflegepersonal
• Allgemeine Unterstützung: Fallbesprechungen, Super-/Intervision, Rituale im Team, Beratungen mit Fachpersonal, weniger wertende Einstellung, mehr emotionale Unterstützung, Zugang zu externen Fort-/Weiterbildungen (theoretische Grundlagen, neue Therapien) und Teilnahme an Kongressen, Unterstützung der beruflichen Karriere, mehr Personal und Zeit im Arbeitsalltag.
Fehlende Werkzeuge aus Sicht der Führungspersonen:
• Dokumente/Schulungen: Werkzeuge zur Aufrechterhaltung der Kompetenzen
• Einarbeitung/Mentoring: Mehr Fachkräfte finden, um 1 : 1 Coaching anbieten zu können, evidenzbasierte Werkzeuge zur Integration für die neuen, unerfahrenen Pflegefachpersonen in der Onkologie.
• Allgemeine Unterstützung: Aus-/Fortbildungsangebot in Hämatologie.
Wie kann der Verband Onkologiepflege Schweiz unterstützen
Die Befragung von Pflegefachpersonen und Führungskräften zeigt, dass bereits verschiedene Angebote bestehen, um Berufseinsteiger/-innen in der Onkologie zu unterstützen. Gleichzeitig wird aber deutlich, dass es unterschiedliche Einschätzungen zu den besonders wirksamen Massnahmen gibt. Übereinstimmend lässt sich festhalten, dass in den Bereichen Fortbildung (insb. Hämatologie), Mentoring/Coaching sowie Tools zur Aufrechterhaltung der Kompetenzen ein erhöhter Unterstützungsbedarf besteht.
Die OPS wird sich im Rahmen ihres Fortbildungsprogramms und durch gezielte Projekte dafür einsetzen, dass Praxis und Management künftig auf zusätzliche Ressourcen und Angebote zählen können. Ziel ist es, Berufseinsteiger/-innen in ihrer Einführungsphase bestmöglich zu begleiten, ihre Motivation zu fördern und sie langfristig für die Onkologiepflege zu gewinnen.
Nicole Zigan MSc Pflege, dipl. Pflegefachfrau 1 Milena Marta Bruschini MSc Pflege, dipl. Pflegefachfrau 2 Prof. Dr. Maria Schubert dipl. Pflegefachfrau, PhD 3 Prof. Manuela Eicher 4
1 Wissenschaftliche Mitarbeiterin,
Forschung & Entwicklung und MSc Pflege
Institut für Pflege, ZHAW Gesundheit,
Katharina-Sulzer-Platz 9, 8400 Winterthur
nicole.zigan@zhaw.ch
2 Wissenschaftliche Mitarbeiterin,
Forschung & Entwicklung und MSc Pflege
Institut für Pflege, ZHAW Gesundheit
Katharina-Sulzer-Platz 9, 8400 Winterthur
milena.bruschini@zhaw.ch
3 Professur Pflege in der Akutversorgung und Dozentin
Co-Leiterin Forschung & Entwicklung und MSc Pflege
Institut für Pflege, ZHAW Gesundheit
Katharina-Sulzer-Platz 9, 8400 Winterthur
maria.schubert@zhaw.ch
Projektteam: Natalie Battaglia, Nicole Corballis, Sara Kohler, Giovanni Presta, Petra Stolz Baskett, Natacha Szüts
Dies ist eine Zweitpublikation. Die Originalversion wurde in der Fachzeitschrift Onkologiepflege Ausgabe 03/2025.
EONS Cancer Nursing Education Framework, European Oncology Nursing Society (EONS) 2022, https://www.cancernurse.eu/cancer-nursing-education-framework R und R Studio Version 4.4.3, 2025, (https://www.r-project.org/).
Kardiales Troponin ist ein strukturelles Protein des kontraktilen Apparats der Herzmuskelzelle und der spezifischste Biomarker zur Detektion eines Myokardschadens. Mit der Einführung hochsensitiver kardialer Troponin-Assays hat sich die Diagnostik des akuten Myokardinfarkts (AMI) grundlegend verändert. Diese ermöglichen den Nachweis minimaler Troponinfreisetzungen bereits kurze Zeit nach Symptombeginn und haben die bisherige Diagnostik revolutioniert. Insbesondere die in den aktuellen Leitlinien der europäischen Gesellschaft für Kardiologie verankerten 0/1-h- und 0/2-h-Algorithmen erlauben einen raschen Ein- und Ausschluss des AMI bei Patientinnen und Patienten, die sich mit akuten Brustschmerzen auf der Notfallstation vorstellen. Hierdurch wird der klinische Workflow in der Notfallstation erheblich beschleunigt. Dennoch erfordert die Interpretation von kardialen Troponin-Erhöhungen stets die Berücksichtigung klinischer und kontextueller Faktoren, da zahlreiche andere Krankheitsbilder ebenfalls mit erhöhten Troponinwerten einhergehen können. Neben den laborbasierten Verfahren gewinnen zunehmend auch Point-of-Care Tests an Bedeutung.
Cardiac troponin (cTn) is a structural protein of the contractile apparatus of cardiomyocytes and the most specific biomarkers for the detection of myocardial injury. The introduction of high-sensitivity cardiac troponin assays has fundamentally transformed the diagnosis of acute myocardial infarction (AMI). These assays enable the detection of minimal troponin release within a short time after symptom onset and have revolutionized the diagnostic approach. In particular, the 0/1-h and 0/2-h algorithms endorsed by the European Society of Cardiology guidelines allow for rapid and reliable rule-in and rule-out decisions in patients presenting with suspected AMI, thereby substantially accelerating emergency department workflows. Nevertheless, interpretation of hs-cTn elevations always requires careful consideration of clinical and contextual factors, as numerous other conditions may also lead to elevated troponin levels. In addition to laboratory-based measurements, point-of-care tests are gaining increasing importance. Keywords: high-sensitivity cardiac troponin; acute myocardial infarction; 0/1-hour algorithm; biomarker; point-of-care testing
Warum hochsensitives Troponin der kardiale Biomarker Nr. 1 ist
Das kardiale Troponin (cTn) ist ein strukturelles Protein des kontraktilen Apparats der Herzmuskelzelle und spielt eine zentrale Rolle in der Regulation der Myofilamentkontraktion (1, 2). Im Gegensatz zu Troponin C, das auch in der Skelettmuskulatur vorkommt, sind cTn T und I nahezu ausschliesslich im Myokard exprimiert und gelten daher als herzmuskelspezifische Marker (1–3). Die Freisetzung von Troponin in den Blutkreislauf ist ein direktes Zeichen für eine strukturelle Schädigung von Kardiomyozyten, unabhängig von deren Ursache (1–3). Damit ist Troponin der kardiale Biomarker zur Erfassung eines akuten oder chronischen Myokardschadens und essenzieller Bestandteil der Definition des akuten Myokardinfarkts (AMI) (4, 5). Vor der Einführung der hochsensitiven Assays konnten konventionelle Troponin-Tests nur deutlich erhöhte Konzentrationen nachweisen, sodass zur Diagnosestellung häufig serielle Messungen über 6–12 Stunden notwendig waren. Die Einführung der hochsensitiven Troponin-Assays (hs-cTn T und I) ab etwa 2010 markierte einen Paradigmenwechsel: Diese Methoden detektieren Troponinkonzentrationen, die bei bis zu 50–90 % gesunder Individuen messbar sind, und unterscheiden sich in zwei wesentlichen Punkten von konventionellen Troponin-Assays (1, 6–8). Erstens ermöglichen sie den Nachweis von Troponin bei einem erheblichen Anteil gesunder Personen und zweitens erlauben sie eine präzisere Definition des Normalbereichs (entsprechend der 99. Perzentile), wobei die analytische Präzision der Assays durch den Variationskoeffizienten beschrieben wird, der idealerweise <10 % betragen sollte (1). So wird eine präzise Quantifizierung minimaler Freisetzungen bereits kurz nach Symptombeginn ermöglicht. In der aktuellen ESC-Leitlinie 2023 werden hs-cTn T und I als Biomarker der Wahl für die Diagnose des AMI empfohlen (5). Zusammen mit Anamnese, klinischer Untersuchung und 12-Kanal-EKG bilden sie die diagnostische Trias (5). Dank der hohen analytischen Sensitivität und Reproduzierbarkeit lassen sich dynamische Veränderungen (Anstieg/Abfall) bereits innerhalb von 1–2 Stunden erfassen, was die Grundlage der modernen 0/1-h- und 0/2-h-Algorithmen bildet (6, 7, 9–12). Durch diese Innovation konnten die Infarktdiagnostik entscheidend beschleunigt und die Patientensicherheit verbessert werden (10). Frühe Rule-out-Entlassungen, rasche Identifikation von Hochrisikopatienten und eine zeitkritische Therapieeinleitung sind möglich.
Analytik
Hochsensitive Troponin-Assays zeichnen sich durch eine Messpräzision mit einem Variationskoeffizienten ≤ 10 % im Bereich der 99. Perzentile des oberen Referenzwerts sowie durch eine Nachweisrate ≥ 50 % bei gesunden Personen aus (1). Die Grosszahl der kommerziell erhältlichen hs-cTn T- und auch hs-cTn I-Assays erfüllt diese Kriterien. Wichtig ist zu beachten, dass die 99. Perzentile für jeden hs-cTn Assay in unterschiedlichen gesunden Populationen deriviert wird. Somit sind die Referenzwerte eines hs-cTn Assays nicht auf einen anderen übertragbar. Für eine valide Interpretation sind zudem präanalytische Faktoren wie korrekte Probenentnahme, zeitgerechte Zentrifugation und Vermeidung von Hämolyse entscheidend, welche einen Einfluss haben können (3, 13, 14). Auch wenn die analytischen Details komplex sind, ist die klinisch entscheidende Botschaft klar: Hochsensitive Troponin-Assays vereinen Genauigkeit mit hervorragender Reproduzierbarkeit und bilden damit die Grundlage für eine verlässliche und differenzierte Diagnostik des AMI.
Myokardschaden und Freisetzungskinetik
Die kardialen Troponine sind intrazelluläre Proteine, die im Zytosol und an den kontraktilen Myofibrillen der Herzmuskelzellen gebunden vorliegen (15). Kommt es zum Beispiel infolge einer ischämischen Schädigung zu einer gestörten Zellmembranintegrität, werden zytosolische wie auch gebundene Troponinanteile freigesetzt (15). Die Halbwertszeit von cTn T im Blut beträgt etwa 120 Minuten, wobei das verlängerte Nachweisfenster darauf zurückzuführen ist, dass während der zellulären Nekrose fortlaufend Troponin T aus dem myofibrillären Pool freigesetzt wird, während sich der kontraktile Apparat der Kardiomyozyten schrittweise abbaut (15). Die Freisetzung erklärt die Kinetik des hs-cTn-Anstiegs beim AMI: Der Troponinwert beginnt meist innerhalb von 1–3 Stunden nach Symptombeginn zu steigen, erreicht nach etwa 12–24 Stunden seinen Gipfel und fällt anschliessend über mehrere Tage wieder ab. Neben der Ischämie-induzierten Nekrose können auch andere Mechanismen, etwa Apoptose, Dehnung, Entzündung oder toxische Schädigung, zur Freisetzung von Troponin führen, wobei die Dynamik des Anstiegs meist geringer ausfällt. Die hochsensitiven Assays ermöglichen es, diese feinen Konzentrationsänderungen zu erkennen und damit zwischen akuter und chronischer Myokardschädigung zu unterscheiden, ein entscheidender Fortschritt für die differenzierte Interpretation erhöhter Troponinwerte.
Revolutionierte Infarktdiagnostik – Rolle des Troponins und der ESC-Algorithmen
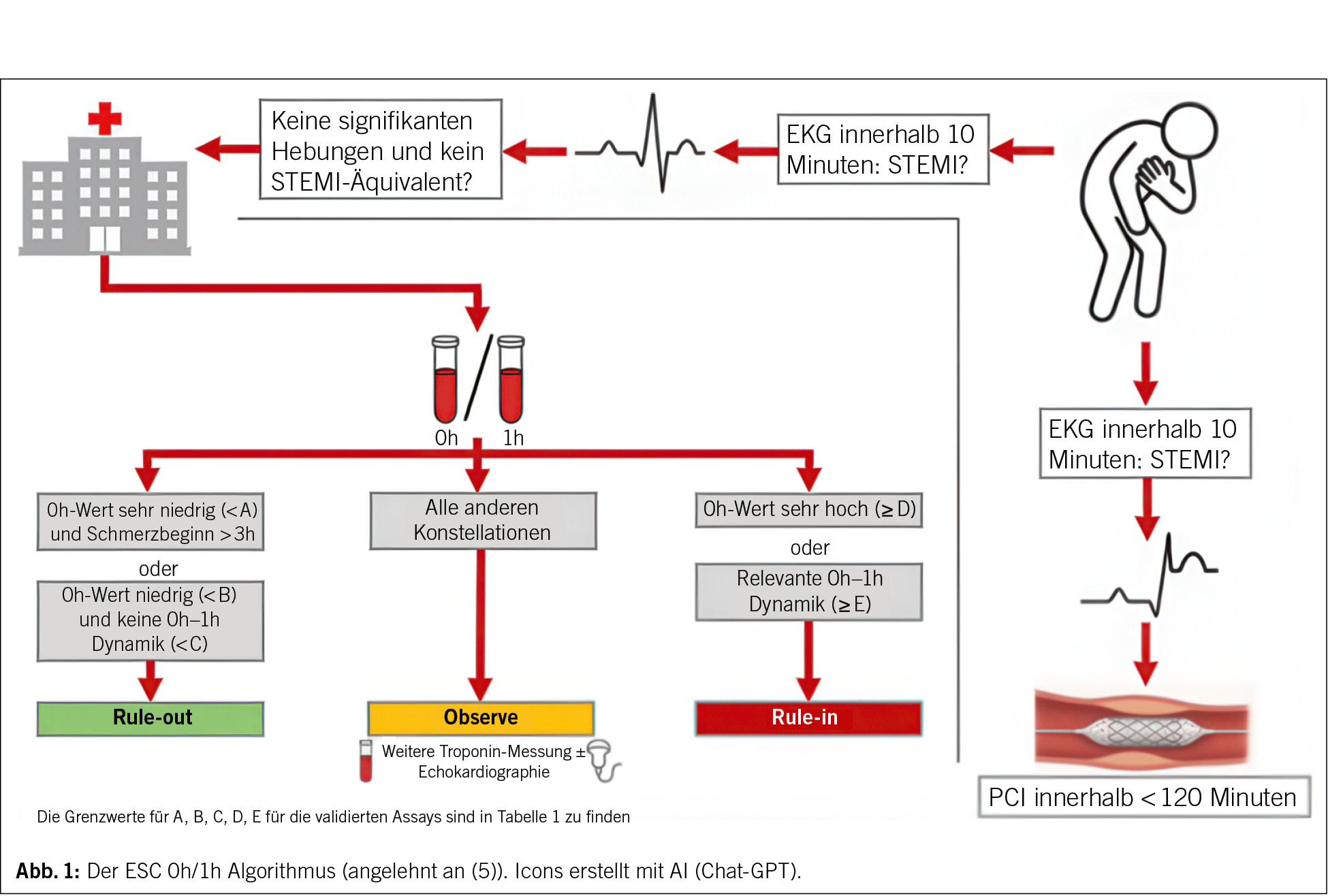
Mit Einführung der hochsensitiven Troponin-Assays wurde die Diagnostik des AMI grundlegend verändert. Während frühere Testverfahren erst nach mehreren Stunden pathologische Werte zeigten, erlauben die hochsensitiven Assays den Nachweis minimaler Myokardschädigungen bereits kurz nach Symptombeginn (16–22). Damit wurde die Zeit bis zur gesicherten Diagnose deutlich verkürzt, was einen entscheidenden Vorteil mit sich bringt, denn weiterhin gilt: «Time is muscle».
Die aktuellen ESC-Leitlinien von 2023 empfehlen den 0/1-h-Algorithmus als bevorzugte Strategie zum schnellen und sicheren Ein- oder Ausschluss des AMI (Abb. 1) (5). Dieser Algorithmus basiert auf zwei Troponinmessungen: einer initialen Bestimmung zum Zeitpunkt 0 (Stunden) und einer zweiten Messung 1 Stunde später (5). Alternativ kann, etwa bei organisatorischen Einschränkungen oder verzögerter Blutabnahme, der 0/2-h-Algorithmus angewendet werden (5). Beide Verfahren sind für verschiedene Assays validiert und liefern eine ausgezeichnete diagnostische Sicherheit, wobei der 0/1-h-Algorithmus aufgrund seiner frühzeitigeren Entscheidungsmöglichkeit bevorzugt wird. Das Konzept beruht auf zwei zentralen Prinzipien: Erstens ist das Troponin eine kontinuierliche Variable, deren absolute Konzentration eng mit der Wahrscheinlichkeit eines Myokardinfarkts korreliert (23). Zweitens liefern frühe absolute Änderungen («Deltas») der Troponinwerte zwischen der Erst- und der Folgemessung entscheidende Informationen zur Dynamik des Myokardschadens (24, 25). Während relative Veränderungen (z. B. + 20 %) in der Vergangenheit häufig verwendet wurden, empfehlen die Leitlinien heute die Verwendung Assay-spezifischer absoluter Deltas, da diese weniger durch Messvariabilität beeinflusst sind und eine höhere diagnostische Genauigkeit bieten (5).
Ein Rule-out ist möglich, wenn der Troponinwert entweder bereits bei der ersten Messung sehr niedrig ist oder wenn der Ausgangswert niedrig bleibt und sich innerhalb von 1 Stunde keine relevante Dynamik zeigt (5, 26). In mehreren grossen Validierungsstudien lag der negative Vorhersagewert (NPV) dieser Strategien bei über 99 %, was eine sichere Entlassung vieler Patienten bereits nach kurzer Zeit ermöglicht (22, 27, 28). Ein Rule-in erfolgt, wenn der Troponinwert bereits bei Aufnahme deutlich erhöht ist, oder wenn innerhalb von 1 bzw. 2 Stunden ein klarer absoluter Anstieg beobachtet wird (5). Der positive Vorhersagewert (PPV) liegt zwischen 70 und 75 %. Patienten, die weder die Rule-out- noch die Rule-in-Kriterien erfüllen, fallen in die sogenannte «Observe-Zone» und benötigen eine dritte Troponin-Messung nach 3 Stunden und weitere Abklärung, wie zum Beispiel eine ergänzende bildgebende kardiale Diagnostik (5, 29).
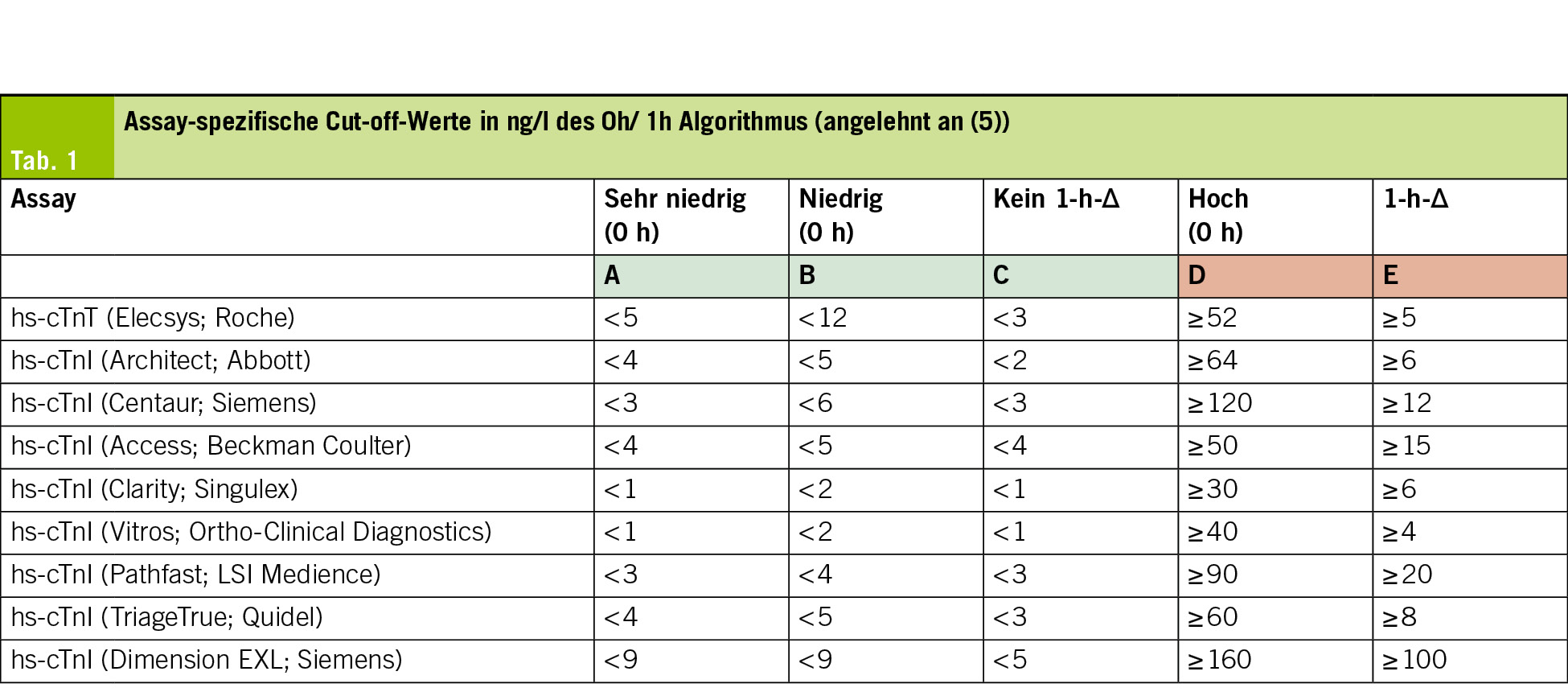
Wie bereits erwähnt sind die Grenzwerte Assay-spezifisch und dürfen nicht zwischen verschiedenen Assays übertragen werden (Tab. 1). Für den weit verbreiteten Roche Elecsys hs-cTnT-Assay gelten nach ESC-Empfehlung folgende Schwellen: Ein Wert < 5 ng/L (bei Symptombeginn > 3 h) oder ein Ausgangswert < 12 ng/L ohne einen Anstieg von ≥ 3 ng/L nach 1 Stunde schliesst einen AMI aus (5). Ein Wert ≥ 52 ng/L oder ein Anstieg von ≥ 5 ng/L innerhalb 1 Stunde gilt als klarer Hinweis auf einen akuten Infarkt (5). Für den Abbott Architect hs-cTnI-Assay werden etwas andere Grenzwerte verwendet: Ein Wert < 4 ng/L (bei Schmerzbeginn vor > 3h) oder < 5 ng/L ohne Δ ≥ 2 ng/L nach 1 Stunde spricht für Rule-out, während Werte ≥ 64 ng/L oder ein 1-h-Δ ≥ 6 ng/L ein Rule-in definieren (5).
Bei Anwendung des 0/2-h-Algorithmus bleiben die Entscheidungsgrenzen ähnlich, die zulässigen Deltas sind jedoch etwas grösser, da die Zeit zwischen den Messungen länger ist und das Troponin kontinuierlich über die Zeit ansteigt (5). Auch hier liegt die diagnostische Sicherheit für den Ausschluss eines AMI bei über 99 %. In der Praxis wird der 0/2-h-Algorithmus insbesondere eingesetzt, wenn die 1-h-Blutabnahme organisatorisch nicht zuverlässig möglich ist.
Mehrere grosse prospektive Studien, darunter randomisierte Implementierungsstudien, haben gezeigt, dass der 0/1-h-Algorithmus im Vergleich zu älteren Strategien sowohl die Zeit bis zur Diagnose als auch die Aufenthaltsdauer in der Notaufnahme signifikant verkürzt, ohne die Sicherheit zu beeinträchtigen (22, 27, 28). Entsprechend wurde der frühere Algorithmus in den ESC-Leitlinien 2023 hinsichtlich des Empfehlungsgrades abgestuft, während der 0/1-h-Algorithmus eine Klasse-I-Empfehlung besitzt (5). Damit haben die hochsensitiven Troponin-Assays die Diagnostik des akuten Myokardinfarkts revolutioniert: Sie ermöglichen eine frühe, hochpräzise Abgrenzung zwischen akuter und chronischer Myokardschädigung, reduzieren unnötige Hospitalisierungen und verbessern die Risikostratifizierung. In Kombination mit klinischer Beurteilung und einem 12-Kanal-EKG bilden sie heute das zentrale Fundament der modernen Infarktdiagnostik.
Altersabhängige Effekte und Einfluss der Nierenfunktion
Die 99. Perzentile der oberen Referenzgrenze (upper reference limit, URL) bildet die zentrale diagnostische Schwelle für die Definition eines Myokardschadens und eines AMI. Troponin-Konzentrationen oberhalb dieses Wertes gelten als erhöht (4). Diese Schwelle ist Assay-spezifisch und beruht auf Messungen in grosse Kollektiven gesunder Personen. Dabei zeigte sich, dass biologische Unterschiede, insbesondere Alter und Nierenfunktion, einen relevanten Einfluss auf die Troponin-Konzentration haben können.
Mit zunehmendem Alter steigt die Prävalenz leicht erhöhter Troponinspiegel auch in Abwesenheit einer akuten Ischämie (31). Dieser Anstieg reflektiert meist subklinische strukturelle Myokardschäden, Fibrose, hypertensive oder diastolische Belastung. Dennoch wird derzeit kein altersadjustierter Grenzwert in den Leitlinien empfohlen, da die Sicherheit weiterhin gegeben ist und die Verwendung altersspezifischer Grenzwerte zu Herausforderungen in der klinischen Praxis führen könnte (32, 33).
Die chronische Niereninsuffizienz stellt einen weiteren häufigen und klinisch relevanten Confounder der Troponindiagnostik dar (34). Bei eingeschränkter glomerulärer Filtrationsrate finden sich häufig persistierend erhöhte hs-cTn-Konzentrationen, vermutlich bedingt durch eine Kombination aus verminderter renaler Clearance und chronischem Myokardschaden infolge Druck- und Volumenbelastung. Somit empfiehlt es sich, bei Patienten mit eingeschränkter Nierenfunktion (ebenso bei Patienten im fortgeschrittenen Alter) nicht den absoluten Ausgangswert, sondern vor allem die Dynamik (Anstieg oder Abfall) zwischen den Messungen zu bewerten (5). Ein signifikanter Anstieg über die Assay-spezifische Δ-Schwelle hinaus gilt auch in dieser Population als starkes Indiz für eine akute Myokardschädigung z. B. im Rahmen eines ischämischen Prozesses. Folglich sollte die Diagnose eines AMI in diesem Kontext nur bei nachweisbarer Dynamik in Verbindung mit Symptomen und/oder EKG-Veränderungen gestellt werden. Ein persistierend stabil erhöhter Troponinwert ohne Klinik hingegen spricht eher für eine chronische kardiale Schädigung und ist prognostisch bedeutsam, ohne zwingend auf ein akutes Koronarsyndrom hinzuweisen.
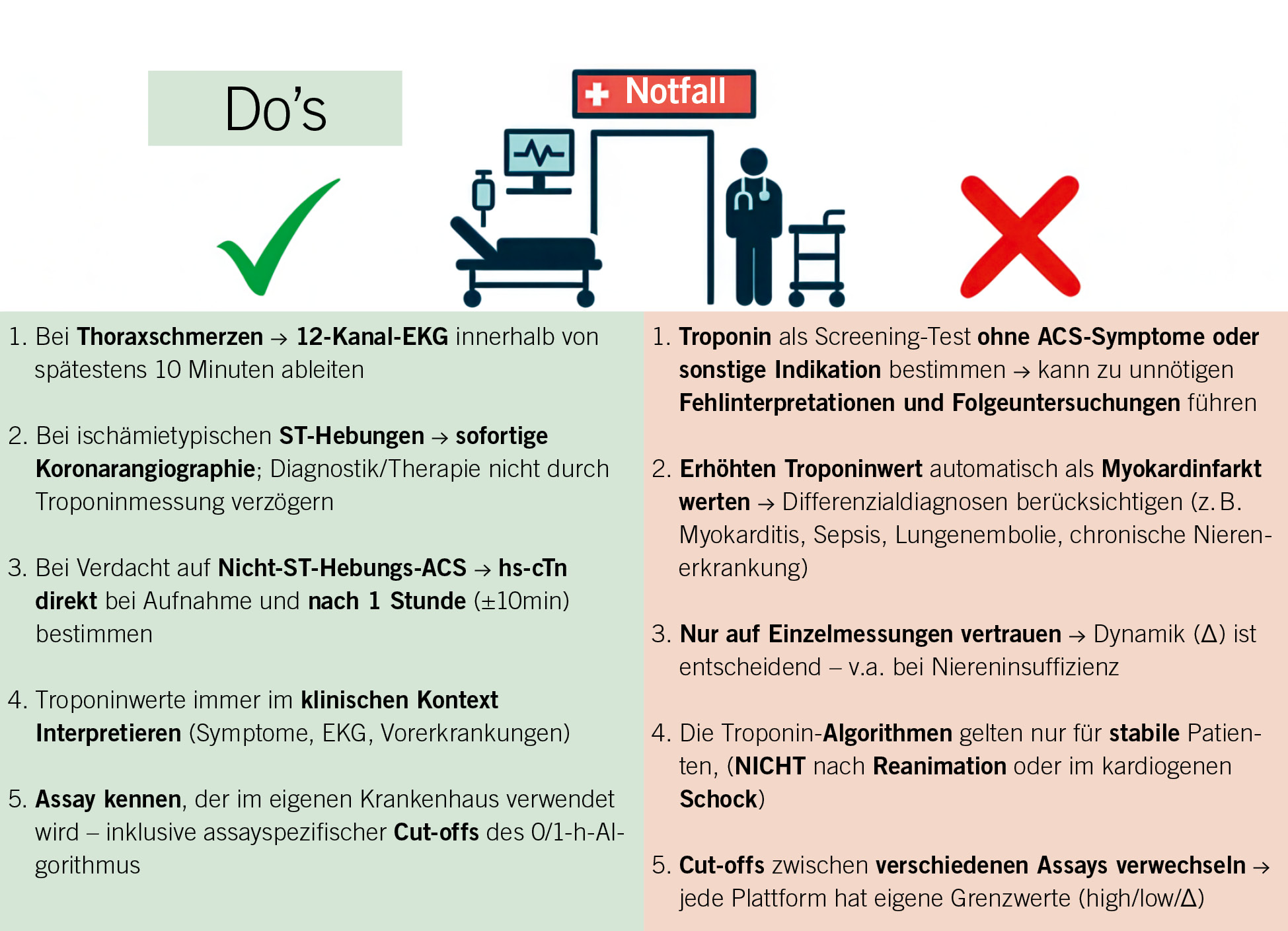
Klinische Szenarien und Pitfalls
Die Interpretation erhöhter hochsensitiver Troponinwerte erfordert stets eine enge Verknüpfung mit Anamnese, klinischem Kontext und EKG-Befunden (Abb. 2). Eine Troponinerhöhung weist grundsätzlich auf einen Myokardschaden, aber nicht zwingend auf einen AMI hin. Erst das Zusammenspiel aus klinischer Präsentation mit Symptomen, ischämietypischer Dynamik und ggf. korrespondierenden EKG-Veränderungen erlaubt die sichere Diagnose eines AMI (4, 5). Der Typ-1-Myokardinfarkt entsteht infolge eines (plaqueassoziierten) koronaren (teilweise)-Verschlusses mit Thrombusbildung und akuter Myokardischämie. Der Typ-2-Myokardinfarkt hingegen beruht auf einer Ungleichgewichts-Ischämie zwischen myokardialem Sauerstoffbedarf und -angebot ohne akute Plaqueruptur (35). Er tritt sekundär im Rahmen anderer Erkrankungen auf, die zu einem verminderten Sauerstoffangebot (z. B. bei Hypoxie) oder einem erhöhten Sauerstoffbedarf (z. B. Tachykardie) führen (35). Aggraviert kann dies werden durch bestehende, unter «normalen Umständen» nicht-signifikanten Stenosen. Typische klinische Konstellationen sind die Anämie (reduziertes Sauerstoffangebot bei stabiler KHK oder auch blanden Koronarien), das tachykarde Vorhofflimmern oder andere tachykarde Herzrhythmusstörungen (erhöhter Sauerstoffverbrauch und diastolische Minderversorgung) und die Lungenembolie (akute Druckbelastung und Dilatation des rechten Ventrikels mit subendokardialer Ischämie) (35). In all diesen Fällen kann das hs-cTn deutlich erhöht sein, ohne dass eine akute Koronarobstruktion vorliegt. Entscheidend ist die Gesamtinterpretation der klinischen Situation: Ein Typ-2-Infarkt ist kein primär koronar-interventionelles Krankheitsbild, sondern Ausdruck eines sekundären myokardialen Schadens, der eine kausale Behandlung der Grunderkrankung erfordert (35).
Neben ischämischen Mechanismen existieren zahlreiche nicht-ischämische Ursachen für erhöhte hs-cTn-Werte, die eine differenzierte Beurteilung erfordern. Ein möglicher Befund in der klinischen Praxis ist die Troponinerhöhung nach Liegetrauma oder bei ausgeprägter Skelettmuskelschädigung, etwa bei einer Rhabdomyolyse oder rheumatischen Erkrankung (36, 37). Dabei kann das Troponin T erhöht sein, das Troponin I bleibt in der Regel normal (36). Auch die Myokarditis präsentiert sich mit Troponinerhöhung und Brustschmerz und kann somit ein akutes Koronarsyndrom imitieren (38). Charakteristisch ist häufig eine moderate, teilweise länger anhaltende Troponinfreisetzung ohne koronares Korrelat (38). Die Differenzierung gelingt insbesondere durch die kardiale MRT, welche ein myokardiales Ödem und Spätkontrastmittelaufnahme (late gadolinium enhancement, vorwiegend subepikardial oder mittmyokardial in einem nicht-ischämischen Verteilungsmuster) nachweist (39). Nach perkutaner Koronarintervention (PCI) oder koronarer Bypass-Operation (CABG) treten Troponinerhöhungen häufig auf (40). Diese spiegeln in der Mehrzahl der Fälle prozedurale Myokardschäden wider, die definitionsgemäss erst ab Überschreiten des fünffachen 99. Perzentils (PCI) bzw. des zehnfachen (CABG) als prozedurbezogener Myokardinfarkt (Typ 4a/5 MI) gewertet werden (in Kombination mit neuen EKG-Veränderungen/ neuen Veränderungen in der Bildgebung, zum Beispiel Wandbewegungsstörungen im Echo) (4). Eine isolierte moderate Troponinerhöhung ohne klinische oder elektrokardiographische Hinweise auf Ischämie sollte hier nicht als Infarkt fehlinterpretiert werden (4). Ein weiterer häufiger Sonderfall betrifft intensiven Sport, bei dem transiente, meist moderate Anstiege des hs-cTn beobachtet werden können (41). Die zugrunde liegende Mechanik ist multifaktoriell und umfasst myozytäre Membranpermeabilität, Dehnung, zellulären Stress und Skelettmuskelschäden, ohne dass strukturelle Nekrose vorliegen muss (41). Zusammenfassend gilt: Nicht jede Troponinerhöhung ist Ausdruck eines AMI. Die differenzierte Betrachtung von Ursache, Dynamik, Begleitsymptomen und EKG-Verlauf ist entscheidend, um Überdiagnosen zu vermeiden und eine gezielte Therapie einzuleiten.
Point-of-Care-Assays
Neben den laborbasierten Hochdurchsatzsystemen gewinnen in den letzten Jahren Point-of-Care-(POC)-Assays zunehmend an Bedeutung (42, 43). Sie ermöglichen eine schnelle Messung von Troponin ohne zeitliche Verzögerung durch den Probentransport und Laborprozess. Ziel ist es, den diagnostischen Ablauf weiter zu beschleunigen und insbesondere in präklinischen oder ressourcenlimitierten Settings, wie im Rettungsdienst, peripheren Spitälern, ambulanten Praxen oder ländliche Regionen eine zeitnahe Entscheidungsfindung zu ermöglichen.
Moderne POC-Systeme verwenden Technologien, die in ihrer analytischen Leistungsfähigkeit den Labor-Assays zunehmend nahekommen (42, 43). Einige der neueren Geräte, etwa der Siemens Atellica VTLi, der Abbott i-STAT Alinity oder der Roche LumiraDx hs-cTnI-Assay, erfüllen bereits viele Kriterien für «high-sensitivity». Damit können sie, im Gegensatz zu älteren POC-Tests, in standardisierte 0/1-h- oder 0/2-h-Algorithmen integriert werden, jedoch sind auch hier die Assay-spezifischen Grenzwerte zu beachten (42, 43). Der Hauptvorteil dieser Systeme liegt in der Zeitersparnis. Es konnte gezeigt werden, dass sich mit POC-Assays die «door-to-troponin result time» um bis zu 54 Minuten verkürzen lässt, bei neueren POC-Systemen ohne relevante Einbussen in diagnostischer Sicherheit (42–44). Dadurch können Rule-out-Entscheidungen bereits innerhalb einer Stunde getroffen werden (bei Schmerzbeginn vor > 3h), ein Vorteil insbesondere in überlasteten Notaufnahmen oder präklinischen Situationen (42, 43). Insgesamt stellen POC-Assays eine vielversprechende Ergänzung der etablierten Laboranalytik dar. Sie können die Versorgung von Patienten mit Verdacht auf akutes Koronarsyndrom deutlich beschleunigen und sind insbesondere für den Einsatz in präklinischen und dezentralen Strukturen geeignet. Mit der fortschreitenden Miniaturisierung und Standardisierung dieser Technologien ist absehbar, dass hochpräzise POC-Systeme künftig einen festen Platz in der AMI-Diagnostik einnehmen und die zeitkritischen ESC-Algorithmen auch ausserhalb des Krankenhauses ermöglichen werden.
Ausblick: KI-gestützte Pfade und transdermale Messung von Troponin I
Mit der Einführung hochsensitiver Troponin-Assays wurde die Diagnostik des akuten Myokardinfarkts bereits erheblich beschleunigt und standardisiert. Dennoch beruhen die aktuell empfohlenen ESC-Algorithmen auf festen Schwellenwerten und Zeitpunkten, die individuelle Unterschiede, etwa Alter, Nierenfunktion, EKG-Veränderungen (ausser ST-Hebungen), Komorbiditäten oder Zeitpunkt des Symptombeginns, nicht berücksichtigen (5). Hier setzt der Einsatz von künstlicher Intelligenz (KI) an: Machine-Learning-Modelle ermöglichen eine individualisierte Interpretation von Labor- und EKG-Daten und könnten die bisher starren Entscheidungsgrenzen ablösen.
Hierzu wurde der CoDE-ACS-Algorithmus (Collaboration for the Diagnosis and Evaluation of Acute Coronary Syndrome) entwickelt und in internationalen Kohorten validiert (45, 46). Dieses System kombiniert Troponinkonzentrationen, sowohl bei Aufnahme als auch in seriellen Messungen, mit klinischen Variablen wie Alter, Geschlecht, Blutdruck, Herzfrequenz, Nierenfunktion, Hämoglobin, Symptombeginn und EKG-Befunden (45, 46). Aus diesen Informationen berechnet das Modell eine individuelle Wahrscheinlichkeit für das Vorliegen eines Myokardinfarkts (0–100 Punkte). In einer prospektiven Analyse zeigte CoDE-ACS eine ausgezeichnete Diskriminationsleistung (AUC ≈ 0.95) und identifizierte deutlich mehr Patienten mit niedriger Infarkt-Wahrscheinlichkeit (61 vs. 27 %) bei gleichem negativem prädiktivem Wert (45). Die Folgestudie bestätigte diese Ergebnisse in über 4000 Patienten und zeigte, dass CoDE-ACS unabhängig vom Zeitpunkt der Messung (0, 1 oder 2 h) stabil performt und mehr Patienten sicher als «low probability» klassifizieren kann als die ESC-0/1- oder 0/2-h-Algorithmen (46). KI-gestützte Entscheidungssysteme wie CoDE-ACS könnten künftig Labor-, EKG- und klinische Daten in Echtzeit integrieren und die Wahrscheinlichkeit eines AMI patientenspezifisch quantifizieren. Damit eröffnen sie den Weg zu dynamischen, adaptiven Diagnosepfaden, die sich an individuellen Risikoprofilen orientieren und Über- wie Unterdiagnosen reduzieren. Ob dieser Ansatz im klinischen Alltag zu einer verbesserten Versorgung und Ressourcennutzung führt, bleibt abzuwarten.
Eine vielversprechende Weiterentwicklung stellt die transdermale Messung von kardialem Troponin mittels optischer IR-Sensoren dar (47). Das neu entwickelte, nichtinvasive Wearable-Device («Infrasensor») ermöglicht eine Troponin I-Bestimmung innerhalb von nur 3 Minuten und zeigte in einer multizentrischen Pilotstudie eine gute Sensitivität (90 %) und eine C-Statistik-AUC von 0.90 zur Identifikation erhöhter cTnI-Spiegel (47). Künftige Studien müssen klären, ob diese Technologie in der Notaufnahme oder sogar präklinisch eingesetzt werden kann, um den diagnostischen Prozess beim akuten Koronarsyndrom weiter zu beschleunigen.
Copyright
Aerzteverlag medinfo AG
Dr. med. Michael Kunz
Klinik für Kardiologie und
Cardiovascular Research Institute Basel (CRIB)
Universitätsspital Basel
Petersgraben 4
4031 Basel
Prof. Dr. med.Christian Müller
Klinik für Kardiologie und
Cardiovascular Research Institute Basel (CRIB)
Universitätsspital Basel
Petersgraben 4
4031 Basel
Dr. med. Jasper Boeddinghaus
Klinik für Kardiologie und
Cardiovascular Research Institute Basel (CRIB)
Universitätsspital Basel
Petersgraben 4
4031 Basel
jasper.boeddinghaus@usb.ch
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
Hochsensitive Troponin-Assays (hs-cTnT und hs-cTnI) sind heute der zentrale Biomarker in der Diagnostik des AMI und ermöglichen den Nachweis minimaler Myokardschädigungen bereits kurz nach Symptombeginn.
Die ESC-0/1-h- und 0/2-h-Algorithmen haben die AMI-Diagnostik revolutioniert, indem sie eine schnelle und sichere Rule-out/Rule-in-Entscheidung mit hoher Sensitivität und Spezifität erlauben.
Die Interpretation der hs-cTn-Werte muss immer im klinischen Kontext erfolgen. Besonders Alter, Nierenfunktion und Begleiterkrankungen beeinflussen die Troponinkonzentration und erfordern eine individuelle Bewertung des Absolutwertes und auch der Dynamik.
Nicht jede Troponinerhöhung bedeutet einen AMI. Differenzialdiagnosen wie Myokarditis, Tachyarrhythmien, Lungenembolie oder Niereninsuffizienz sind häufige Fallstricke in der Interpretation.
KI-basierte Systeme wie CoDE-ACS kombinieren Labor-, EKG- und klinische Daten zu einem patientenspezifischen Risikoscore und könnten künftig die diagnostische Präzision und Effizienz weiter verbessern.
1. Mueller C. Biomarkers and acute coronary syndromes: an update. European Heart Journal [Internet]. 1. März 2014 [zitiert 22. Oktober 2025];35(9):552–6. Verfügbar unter: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/eht530
2. Thygesen K, Mair J, Katus H, Plebani M, Venge P, Collinson P, u. a. Recommendations for the use of cardiac troponin measurement in acute cardiac care. European Heart Journal [Internet]. 2. September 2010 [zitiert 22. Oktober 2025];31(18):2197–204. Verfügbar unter: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehq251
3. Giannitsis E, Kurz K, Hallermayer K, Jarausch J, Jaffe AS, Katus HA. Analytical Validation of a High-Sensitivity Cardiac Troponin T Assay. Clinical Chemistry [Internet]. 1. Februar 2010 [zitiert 22. Oktober 2025];56(2):254–61. Verfügbar unter: https://academic.oup.com/
clinchem/article/56/2/254/5622529
4. Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, u. a. Fourth Universal Definition of Myocardial Infarction (2018). Journal of the American College of Cardiology [Internet]. Oktober 2018 [zitiert 27. Oktober 2025];72(18):2231–64. Verfügbar unter: https://linkinghub.elsevier.com/retrieve/pii/S0735109718369419
5. Byrne RA, Rossello X, Coughlan JJ, Barbato E, Berry C, Chieffo A, u. a. 2023 ESC Guidelines for the management of acute coronary syndromes. European Heart Journal [Internet]. 25. August 2023;1–107. Verfügbar unter: https://academic.oup.com/eurheartj/advance-article/doi/10.1093/eurheartj/ehad191/7243210
6. Reichlin T, Schindler C, Drexler B, Twerenbold R, Reiter M, Zellweger C, u. a. One-Hour Rule-out and Rule-in of Acute Myocardial Infarction Using High-Sensitivity Cardiac Troponin T. Arch Intern Med [Internet]. 10. September 2012 [zitiert 22. Oktober 2025];172(16):1211. Verfügbar unter: http://archinte.jamanetwork.com/article.aspx?doi=10.1001/
archinternmed.2012.3698
7. Thygesen K, Mair J, Giannitsis E, Mueller C, Lindahl B, Blankenberg S, u. a. How to use high-sensitivity cardiac troponins in acute cardiac care. European Heart Journal [Internet]. 2. September 2012 [zitiert 22. Oktober 2025];33(18):2252–7. Verfügbar unter: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehs154
8. Than M, Cullen L, Reid CM, Lim SH, Aldous S, Ardagh MW, u. a. A 2-h diagnostic protocol to assess patients with chest pain symptoms in the Asia-Pacific region (ASPECT): a prospective observational validation study. The Lancet [Internet]. März 2011 [zitiert 22. Oktober 2025];377(9771):1077–84. Verfügbar unter: https://linkinghub.elsevier.com/retrieve/pii/S0140673611603103
9. Reichlin T, Twerenbold R, Reiter M et al. Introduction of High-sensitivity Troponin Assays: Impact on Myocardial Infarction Incidence and Prognosis. The American Journal of Medicine [Internet]. Dezember 2012 [zitiert 22. Oktober 2025];125(12):1205-1213.e1. Verfügbar unter: https://linkinghub.elsevier.com/retrieve/pii/S000293431200647X
10. Reichlin T, Hochholzer W, Bassetti S, Steuer S, Stelzig C, Hartwiger S, u. a. Early Diagnosis of Myocardial Infarction with Sensitive Cardiac Troponin Assays. N Engl J Med [Internet]. 27. August 2009 [zitiert 22. Oktober 2025];361(9):858–67. Verfügbar unter: http://www.nejm.org/doi/abs/10.1056/NEJMoa0900428
11. Keller T, Zeller T, Peetz D, Tzikas S, Roth A, Czyz E, u. a. Sensitive Troponin I Assay in Early Diagnosis of Acute Myocardial Infarction. N Engl J Med [Internet]. 27. August 2009 [zitiert 22. Oktober 2025];361(9):868–77. Verfügbar unter: http://www.nejm.org/doi/abs/10.1056/NEJMoa0903515
12. Giannitsis E, Becker M, Kurz K et al. High-Sensitivity Cardiac Troponin T for Early Prediction of Evolving Non–ST-Segment Elevation Myocardial Infarction in Patients with Suspected Acute Coronary Syndrome and Negative Troponin Results on Admission. Clinical Chemistry [Internet]. 1. April 2010 [zitiert 22. Oktober 2025];56(4):642–50. Verfügbar unter: https://academic.oup.com/clinchem/article/56/4/642/5622453
13. Hawkins RC. Hemolysis Interference in the Ortho-Clinical Diagnostics Vitros ECi cTnI Assay. Clinical Chemistry [Internet]. 1. Juli 2003 [zitiert 22. Oktober 2025];49(7):1226–7. Verfügbar unter: https://academic.oup.com/clinchem/article/49/7/1226/5642043
14. Dasgupta A, Wells A, Biddle DA. Negative interference of bilirubin and hemoglobin in the MEIA troponin I assay but not in the MEIA CK‐MB assay. Clinical Laboratory Analysis [Internet]. März 2001 [zitiert 22. Oktober 2025];15(2):76–80. Verfügbar unter: https://onlinelibrary.wiley.com/doi/10.1002/jcla.5
15. Apple FS, Collinson PO, for the IFCC Task Force on Clinical Applications of Cardiac Biomarkers. Analytical Characteristics of High-Sensitivity Cardiac Troponin Assays. Clinical Chemistry [Internet]. 1. Januar 2012 [zitiert 22. Oktober 2025];58(1):54–61. Verfügbar unter: https://academic.oup.com/clinchem/article/58/1/54/5620628
16. Boeddinghaus J, Nestelberger T, Twerenbold R, Wildi K, Badertscher P, Cupa J, u. a. Direct Comparison of 4 Very Early Rule-Out Strategies for Acute Myocardial Infarction Using High-Sensitivity Cardiac Troponin I. Circulation [Internet]. 25. April 2017 [zitiert 26. Oktober 2025];135(17):1597–611. Verfügbar unter: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.116.025661
17. Wildi K, Boeddinghaus J, Nestelberger T, Twerenbold R, Badertscher P, Wussler D, u. a. Comparison of fourteen rule-out strategies for acute myocardial infarction. International Journal of Cardiology [Internet]. Mai 2019 [zitiert 26. Oktober 2025];283:41–7. Verfügbar unter: https://linkinghub.elsevier.com/retrieve/pii/S0167527318347843
18. Rubini Gimenez M, Twerenbold R, Jaeger C, Schindler C, Puelacher C, Wildi K, u. a. One-hour Rule-in and Rule-out of Acute Myocardial Infarction Using High-sensitivity Cardiac Troponin I. The American Journal of Medicine [Internet]. August 2015 [zitiert 26. Oktober 2025];128(8):861-870.e4. Verfügbar unter: https://linkinghub.elsevier.com/retrieve/pii/S0002934315002569
19. Badertscher P, Boeddinghaus J, Twerenbold R, Nestelberger T, Wildi K, Wussler D, u. a. Direct Comparison of the 0/1h and 0/3h Algorithms for Early Rule-Out of Acute Myocardial Infarction. Circulation [Internet]. 5. Juni 2018 [zitiert 26. Oktober 2025];137(23):2536–8. Verfügbar unter: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.118.034260
20. Chapman AR, Anand A, Boeddinghaus J, Ferry AV, Sandeman D, Adamson PD, u. a. Comparison of the Efficacy and Safety of Early Rule-Out Pathways for Acute Myocardial Infarction. Circulation [Internet]. 25. April 2017 [zitiert 26. Oktober 2025];135(17):1586–96. Verfügbar unter: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.116.025021
21. Chapman AR, Fujisawa T, Lee KK, Andrews JP, Anand A, Sandeman D, u. a. Novel high-sensitivity cardiac troponin I assay in patients with suspected acute coronary syndrome. Heart [Internet]. 15. November 2018 [zitiert 26. Oktober 2025];heartjnl-2018-314093. Verfügbar unter: https://heart.bmj.com/lookup/doi/10.1136/heartjnl-2018-314093
22. Chew DP, Lambrakis K, Blyth A, Seshadri A, Edmonds MJR, Briffa T, u. a. A Randomized Trial of a 1-Hour Troponin T Protocol in Suspected Acute Coronary Syndromes: The Rapid Assessment of Possible Acute Coronary Syndrome in the Emergency Department With High-Sensitivity Troponin T Study (RAPID-TnT). Circulation [Internet]. 5. November 2019 [zitiert 26. Oktober 2025];140(19):1543–56. Verfügbar unter: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.119.042891
23. Neumann JT, Twerenbold R, Ojeda F, Sörensen NA, Chapman AR, Shah ASV, u. a. Application of High-Sensitivity Troponin in Suspected Myocardial Infarction. N Engl J Med [Internet]. 27. Juni 2019 [zitiert 26. Oktober 2025];380(26):2529–40. Verfügbar unter: http://www.nejm.org/doi/10.1056/NEJMoa1803377
24. Reichlin T, Irfan A, Twerenbold R, Reiter M, Hochholzer W, Burkhalter H, u. a. Utility of Absolute and Relative Changes in Cardiac Troponin Concentrations in the Early Diagnosis of Acute Myocardial Infarction. Circulation [Internet]. 12. Juli 2011 [zitiert 26. Oktober 2025];124(2):136–45. Verfügbar unter: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.111.023937
25. Reichlin T, Twerenbold R, Wildi K, Gimenez MR, Bergsma N, Haaf P, u. a. Prospective validation of a 1-hour algorithm to rule-out and rule-in acute myocardial infarction using a high-sensitivity cardiac troponin T assay. CMAJ [Internet]. 19. Mai 2015 [zitiert 26. Oktober 2025];187(8):E243–52. Verfügbar unter: http://www.cmaj.ca/lookup/doi/10.1503/cmaj.141349
26. Rubini Giménez M, Hoeller R, Reichlin T, Zellweger C, Twerenbold R, Reiter M, u. a. Rapid rule out of acute myocardial infarction using undetectable levels of high-sensitivity cardiac troponin. International Journal of Cardiology [Internet]. Oktober 2013 [zitiert 26. Oktober 2025];168(4):3896–901. Verfügbar unter: https://linkinghub.elsevier.com/retrieve/pii/S0167527313011030
27. Stoyanov KM, Hund H, Biener M, Gandowitz J, Riedle C, Löhr J, u. a. RAPID-CPU: a prospective study on implementation of the ESC 0/1-hour algorithm and safety of discharge after rule-out of myocardial infarction. European Heart Journal: Acute Cardiovascular Care [Internet]. Februar 2020 [zitiert 26. Oktober 2025];9(1):39–51. Verfügbar unter: https://academic.oup.com/ehjacc/article/9/1/39-51/5933825
28. Twerenbold R, Costabel JP, Nestelberger T, Campos R, Wussler D, Arbucci R, u. a. Outcome of Applying the ESC 0/1-hour Algorithm in Patients With Suspected Myocardial Infarction. Journal of the American College of Cardiology [Internet]. Juli 2019 [zitiert 26. Oktober 2025];74(4):483–94. Verfügbar unter: https://linkinghub.elsevier.com/retrieve/pii/S0735109719353811
29. Lopez-Ayala P, Nestelberger T, Boeddinghaus J, Koechlin L, Ratmann PD, Strebel I, u. a. Novel Criteria for the Observe-Zone of the ESC 0/1h-hs-cTnT Algorithm. Circulation [Internet]. 7. September 2021 [zitiert 26. Oktober 2025];144(10):773–87. Verfügbar unter: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.120.052982
30. Reiter M, Twerenbold R, Reichlin T, Haaf P, Peter F, Meissner J, u. a. Early diagnosis of acute myocardial infarction in the elderly using more sensitive cardiac troponin assays. European Heart Journal [Internet]. Juni 2011 [zitiert 27. Oktober 2025];32(11):1379–89. Verfügbar unter: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehr033
31. Gore MO, Seliger SL, deFilippi CR, Nambi V, Christenson RH, Hashim IA, u. a. Age- and Sex-Dependent Upper Reference Limits for the High-Sensitivity Cardiac Troponin T Assay. Journal of the American College of Cardiology [Internet]. April 2014 [zitiert 27. Oktober 2025];63(14):1441–8. Verfügbar unter: https://linkinghub.elsevier.com/retrieve/pii/S0735109714003386
32. Boeddinghaus J, Nestelberger T, Twerenbold R, Neumann JT, Lindahl B, Giannitsis E, u. a. Impact of age on the performance of the ESC 0/1h-algorithms for early diagnosis of myocardial infarction. European Heart Journal [Internet]. 7. November 2018 [zitiert 27. Oktober 2025];39(42):3780–94. Verfügbar unter: https://academic.oup.com/eurheartj/article/39/42/3780/5086724
33. Twerenbold R, Badertscher P, Boeddinghaus J, Nestelberger T, Wildi K, Puelacher C, u. a. 0/1-Hour Triage Algorithm for Myocardial Infarction in Patients With Renal Dysfunction. Circulation [Internet]. 30. Januar 2018 [zitiert 26. Oktober 2025];137(5):436–51. Verfügbar unter: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.117.028901
34. Chapman AR, Taggart C, Boeddinghaus J, Mills NL, Fox KAA. Type 2 myocardial infarction: challenges in diagnosis and treatment. European Heart Journal [Internet]. 7. Februar 2025 [zitiert 28. Oktober 2025];46(6):504–17. Verfügbar unter: https://academic.oup.com/eurheartj/article/46/6/504/7920120
35. Du Fay De Lavallaz J, Prepoudis A, Wendebourg MJ, Kesenheimer E, Kyburz D, Daikeler T, u. a. Skeletal Muscle Disorders: A Noncardiac Source of Cardiac Troponin T. Circulation [Internet]. 14. Juni 2022 [zitiert 26. Oktober 2025];145(24):1764–79. Verfügbar unter: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.121.058489
36. Giger RD, Du Fay De Lavallaz J, Prepoudis A, Stoll T, Lopez-Ayala P, Glarner N, u. a. Rhabdomyolysis. Journal of the American College of Cardiology [Internet]. Dezember 2020 [zitiert 26. Oktober 2025];76(22):2685–7. Verfügbar unter: https://linkinghub.elsevier.com/retrieve/pii/S0735109720374167
37. Prepoudis A, Koechlin L, Nestelberger T, Boeddinghaus J, Lopez-Ayala P, Wussler D, u. a. Incidence, clinical presentation, management, and outcome of acute pericarditis and myopericarditis. European Heart Journal Acute Cardiovascular Care [Internet]. 8. Februar 2022 [zitiert 28. Oktober 2025];11(2):137–47. Verfügbar unter: https://academic.oup.com/ehjacc/article/11/2/137/6445206
38. Schulz-Menger J, Collini V, Gröschel J, Adler Y, Brucato A, Christian V, u. a. 2025 ESC Guidelines for the management of myocarditis and pericarditis. European Heart Journal [Internet]. 22. Oktober 2025 [zitiert 28. Oktober 2025];46(40):3952–4041. Verfügbar unter: https://academic.oup.com/eurheartj/article/46/40/3952/8234483
39. Koechlin L, Boeddinghaus J, Nestelberger T, Lopez-Ayala P, Shrestha S, Wussler D, u. a. Lower diagnostic accuracy of hs-cTnI in patients with prior coronary artery bypass grafting. International Journal of Cardiology [Internet]. Mai 2022 [zitiert 26. Oktober 2025];354:1–6. Verfügbar unter: https://linkinghub.elsevier.com/retrieve/pii/S0167527322002601
40. Gresslien T, Agewall S. Troponin and exercise. International Journal of Cardiology [Internet]. Oktober 2016 [zitiert 28. Oktober 2025];221:609–21. Verfügbar unter: https://linkinghub.elsevier.com/retrieve/pii/S0167527316312505
41. Koechlin L, Boeddinghaus J, Lopez-Ayala P, Reber C, Nestelberger T, Wildi K, u. a. Clinical and Analytical Performance of a Novel Point-of-Care High-Sensitivity Cardiac Troponin I Assay. Journal of the American College of Cardiology [Internet]. August 2024 [zitiert 26. Oktober 2025];84(8):726–40. Verfügbar unter: https://linkinghub.elsevier.com/retrieve/pii/S0735109724076307
42. Boeddinghaus J, Nestelberger T, Koechlin L, Wussler D, Lopez-Ayala P, Walter JE, u. a. Early Diagnosis of Myocardial Infarction With Point-of-Care High-Sensitivity Cardiac Troponin I. Journal of the American College of Cardiology [Internet]. März 2020 [zitiert 26. Oktober 2025];75(10):1111–24. Verfügbar unter: https://linkinghub.elsevier.com/retrieve/pii/S0735109720302527
43. Koehler J, Flarity K, Hertner G, Aker J, Stout JP, Gifford M, u. a. Effect of Troponin I Point-of-Care Testing on Emergency Department Throughput Measures and Staff Satisfaction. Advanced Emergency Nursing Journal [Internet]. Juli 2013 [zitiert 28. Oktober 2025];35(3):270–7. Verfügbar unter: https://journals.lww.com/01261775-201307000-00010
44. Doudesis D, Lee KK, Boeddinghaus J, Bularga A, Ferry AV, Tuck C, u. a. Machine learning for diagnosis of myocardial infarction using cardiac troponin concentrations. Nat Med [Internet]. Mai 2023 [zitiert 28. Oktober 2025];29(5):1201–10. Verfügbar unter: https://www.nature.com/articles/s41591-023-02325-4
45. Boeddinghaus J, Doudesis D, Lopez-Ayala P, Lee KK, Koechlin L, Wildi K, u. a. Machine Learning for Myocardial Infarction Compared With Guideline-Recommended Diagnostic Pathways. Circulation [Internet]. 2. April 2024 [zitiert 28. Oktober 2025];149(14):1090–101. Verfügbar unter: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.123.066917
46. Peacock WF, Soto-Ruiz KM, Jaffe AS, Tiffany BR, Mahler SA, Patterson BW, u. a. A rapid noninvasive wearable device for assessing cardiac troponin I level. Clin Exp Emerg Med [Internet]. 6. September 2024 [zitiert 28. Oktober 2025];12(3):251–8. Verfügbar unter: http://ceemjournal.org/journal/view.php?doi=10.15441/ceem.24.294