Statine bei koronarer Herzkrankheit: Mehr ist nicht immer auch besser
In Zeiten, in denen die Norm- oder Zielwerte gemäss Guidelines immer in schwieriger zu erreichende (somit auch teurere und nebenwirkungsreichere) Bereiche definiert werden, ist diese Arbeit für das Vorgehen in der Praxis interessant: Ein Vergleich* einer moderaten Strategie (langsames Eintitrieren der Statindosis auf einen LDL-Zielwert zwischen 1,3 und 1,8 mmol/L) mit einer aggressiven Initialtherapie (20 mg Rosuvastatin oder 40 mg Atorvastatin) mit dem Ziel den LDL-Wert schnell zu halbieren, zeigte keine signifikanten Unterschiede der beiden Optionen in Bezug auf Mortalität und neue kardiovaskuläre Ereignisse. Wenn Patientinnen und Patienten also regelmässig kontrolliert werden (nach 1,5 und 3 Monaten, dann alle 3 Monate), kann man die optimale Statindosis für sie individuell eintitrieren. Interessant ist auch, dass die LDL-Senkung in beiden Therapiegruppen gleich schnell und quantitativ mehr oder weniger identisch erreicht wurde.
*Hier noch die Eckdaten der Studienpopulation: 4400 südkoreanische Patienten, 30 % davon Frauen, Durchschnittsalter 65 Jahre, bemerkenswert hohe 99 % (!) aller Studienteilnehmerinnen und – teilnehmer beendeten den vorgesehenen 3-jährigen follow-up.
JAMA 2023, doi:10.1001/jama.2023.2487, verfasst am 31.03.2023
Medizin jenseits ihrer Grenzen
Wochenendsarbeit verhindern: Burnoutprophylaxe?
In einem eng getakteten Arbeitsumfeld tendieren viele Kolleginnen und Kollegen gewisse nicht so dringliche oder schlimmer: nicht abgeschlossene, Arbeiten auf das Wochenende oder die späten Abendstunden zu verlegen. Es wird vermutet, dass diese «Technik» zur Burnout-Entwicklung beitragen könnte. Ein Hauptproblem ist, dass wir uns oft nicht ganz im Klaren sind, wieviel Zeit wir für eine definierte Aufgabe brauchen. Wenn diese Zeit unterschätzt wird, wird die Arbeit dann oft ausserhalb normaler Arbeitszeiten abgeschlossen. Eine australische Forscherin berichtet, dass (kostenpflichtige) Apps, die die effektiv aufgebrachte Arbeitszeit messen («Timing» für Mac, «Rescue Time» für Windows), die adäquate Budgetierung des Zeitaufwandes signifikant verbessern. Die Produktivität in der normalen Arbeitszeit soll mit der Erfahrung steigen und Überzeiten sollen reduziert werden. Also: Jede Arbeit hat einen vorher definierten Beginn und ein geplantes Ende. Ebenfalls sollte man, wenn immer möglich, diese Arbeit in einem Zug durchführen. Der Wiedereinstieg in die gleiche Arbeit ist speziell aufwendig, weil man wieder alle Details reaktivieren oder gar verschiedene Dokumente wieder auffinden muss.
Nature 2023, doi.org/10.1038/d41586-023-00866-9, verfasst am 06.04.2023
Debatte
Zu viel oder zu wenig Eisen beim M. Parkinson?
Es ist gut bekannt, dass es eine Assoziation von Eisenablagerungen im Gehirn mit einer Reihe von neurologischen Erkrankungen gibt. Namentlich beim M. Parkinson findet man Eisenablagerungen in der Substantia nigra und Tierexperimente wie auch in vitro Zell-Experimente unterstützen die These, dass bei der Pathogenese und der Progression eine Eisenregulationsstörung/Eisenüberlastung vorliegt. Allerdings war eine Eisenchelatortherapie in Frühstadien des M.Parkinson (bei Patientinnen und Patienten die noch keine Levodopa-Therapie erhalten hatten) sogar eher schlechter als Plazebo in Bezug auf die Progression der Krankheit (Beobachtung 36 Wochen, 1). Zu viel Eisen kann zu Oxidation von Lipiden der Zellmembran und Zelluntergang (sog. Ferroptose) führen, während zu wenig Eisen die mitochondriale Energieprodukten in den Neuronen limitieren und damit zur Krankheitsprogres-sion führen könnte. Eine Hypothese ist auch, dass die Eisenchelatortherapie per se die Dopaminsynthese unabhängig vom Eisen hemmen könnte. Was heisst das für den Moment für die Praxis? Weiterhin Eisenmangel und Eisenüberlastung verhindern und Eisen nicht ohne klare Evidenz eines Eisenmangels verordnen (2)!
1. NEJM 2022, doi: 10.1056/NEJMoa2209254, 2. The Lancet Neurology 2023, doi.org/10.1016/S1474-4422(23)00039-X, verfasst am 02.04.2023
Hintergrundswissen: In weniger als einer halben Minute….
Sind Glukokortikoide in der Behandlung ambulant erworbener Pneumonien wirksam?
Ambulant erworbene Pneumonien sind nach wie vor ein sehr grosses medizinisches Problem und angeblich die Tabellenführer in der Mortalität von Infektionskrankheiten. Müssen diese Patientinnen und Patienten hospitalisiert werden, liegt die Mortalität bei 10 bis 12 %! Seit mehr als 40 Jahren wird über den Stellenwert der Glukorkortikoid (neben adäquat selektionierten Antibiotika) bei Pneumonien diskutiert, die pro und contra Lager gewannen intermittierend in schöner Abfolge die Oberhand. Warum überhaupt Glukokortikoide? Bei Pneumonien – wie bei Infekten anderer Organe – kann die Entzündungsreaktion im befallenen Organ, hier also der Lunge, aber auch systemisch im Sinne eines septischen Syndroms mit Mehrorganerkrankungen Überhand nehmen. Laut einer Metaanalyse von 7 randomisierten, kontrollierten Studien bei ambulant erworbenen Pneumonien führten Glukokortikoide zu einer schnelleren Erholung, kürzeren Hospitalisationszeiten, aber keinem Effekt auf die hohe Mortalität (1). Bei schweren ambulant erworbenen Pneumonien (Intensivstation, Sauerstoffbedürftigkeit, invasive oder nicht invasive Beatmung oder nach Massgabe eines Risikomodells, 2) findet eine gut durchgeführte französische Studie nun fast eine Halbierung der Mortalität (!) innerhalb der ersten 28 Tage nach Diagnose der Pneumonie (Reduktion von 11,9 auf 6,2 %). Die Diskrepanz zu früheren Studien (bzgl. Mortalität) könnte in den unterschiedlich dosierten und gewählten Glukokortikoiden liegen. Hier wurden 200 mg Hydrocortison per infusionem über 24h über 4 oder 8 Tage gebraucht. Die entzündliche Wirtsantwort kann bei der Pneumonie also dominieren, aber durch Glukokortikoide effektiv und relevant für den Verlauf supprimiert werden. Kurz dauernde Glukokortikoidtherapien werden häufig wegen der vermuteten Gefahr von Superinfekten gemieden, was aber nicht der Fall ist. Auch in dieser Studie traten nicht vermehrt Sekundärinfekte (wie auch nicht vermehrt gastronintestinale Blutungen) auf.
Zusätzlicher Pneumokokken-Impfstoff in der Schweiz
Zusätzlich zum zugelassenen und von den Krankenkassen übernommenen Prevenar13 wurde ein neuer Impfstoff (Vaxneuvance) von Swissmedic zugelassen und wird etwa um den 20. April auf dem Markt verfügbar und via OKP abrechenbar sein. Die Zulassung beschränkt sich auf Individuen über 65 Jahre, die die höchste Inzidenz an invasiven Infekten (etwa 20 auf 100’000 pro Jahr) aufweisen. Die Schutzwirkung gegen invasive Pneumokokkeninfekte scheint bei dieser Population wegen den 2 zusätzlichen im Impfstoff enthaltenen Serotypen (22F und 33F) signifikant besser (siehe auch «Hintergrundswissen»).
InfoVac-Bulletin N3, 2023, www.infovac.ch. Verfasst am 06.04.2023
Behandlung der Orthostase im Jahr 2030?
Neural bedingte Orthostasen können invalidisierend sein. Sie treten alterungsbedingt oder anderweitig erworben (z.B. bei der Mulitsystematrophie) aber auch post-traumatisch (namentlich bei Querschnittssyndromen) auf. Die Sturzfolgen führen dann zu weiteren Einschränkungen der Gesundheit. Zwar gibt es eine Reihe von Medikamenten mit unterschiedlichen Angriffspunkten, die aber oft nicht genügend wirksam sind. Neuroprothesen werden auch für diese Indikationen evaluiert: Eine Lausanner Forschergruppe der EPFL entwickelte ein nun industriell lizenziertes System (1, 2), bei dem eine Reihe von Elektroden in das Rückenmark implantiert und via einen ebenfalls implantierbaren Impulsgenerator aktiviert wird. Somit kann der sog. Baroreflex aktiviert und die Orthostase limitiert oder gar verhindert werden. Ein vielversprechender, wenn auch aufwändiger Ansatz!
Diese beiden Konsultationsformen haben vieles für sich, vor allem wenn man die Patientinnen und Patienten vorher physisch gesehen hat und gut kennt. Welche Art der Konsultation aber wird von den Patientinnen oder Patienten bevorzugt? Eine – allerdings im US-Gesundheitssystem durchgeführte – Studie, kommt zum Schluss, dass viele Patientinnen und Patienten eine Telefonkonsultation vorziehen. Nicht überraschend, dass es sich hier vor allem um ältere und ökonomisch weniger privilegierte Individuen handelte. Allerdings offerierten die Arztpraxen, selbst wenn sie Videokonsultationen anbieten, primär die im Alltag wahrscheinlich schnellere Telefonkonsultation an!
JAMA network open 2023, doi: 10.1001/jamanetworkopen.2023.5242, verfasst am 06.04.2023
Kaffee und Vorhof-Extrasystolen: Nicht gehäuft
Der Konsum koffeinhaltigen Kaffees hat in der Medizin bezüglich eines vermuteten Nutzens resp. andererseits der Assoziation mit Gesundheitsrisiken eine sehr wechselvolle Evidenzgeschichte. In der Referenz 1 finden die Leserinnen und Leser eine gute, kritische und aktuelle Zusammenfassung der biologischen Effekte von Koffein beim Menschen (1).
In Bezug auf Vorhofs-Extrasystolen (als Vorstufen eines Vorhhofflimmerns) kann Entwarnung gegeben werden. Bei 100 knapp 40-jährigen Freiwilligen, Allgemeinpopulation mit gleich vielen Männern wie Frauen, konnte prospektiv randomisiert kein Effekt von Konsum koffeinhaltigen Kaffees auf die Anzahl von Vorhofs-Extraystolen gefunden werden (2). Die Studie wies als Pluspunkt ein sog. cross-over Design auf, das erlaubte, dass jedes Individuum sowohl in einer Periode mit koffeinhaltigem Kaffee als auch einer ohne, untersucht werden konnte. Weniger gute Note erhalten die Autorinnen und Autoren wie auch die Herausgeber des New England Journal of Medicine für die Titelwahl der Arbeit: Es wird angekündigt, dass die Studie die akuten Auswirkungen von Kaffee auf die Gesundheit untersuchte. Der primäre Endpunkt war dann aber sehr bescheiden, nämlich «nur» die Zahl von Vorhofs-Extrasystolen.
Die klinische Diagnose eines Hirnschlags in der Notfallsituation stellt die Ärzte vor eine grosse Herausforderung, zumal unter Zeitdruck die richtige Therapie getroffen werden sollte und eine Fehldiagnose mit einer ungünstigen Prognose einhergehen kann. Dieser Artikel handelt über «stroke mimics» und «stroke chameleons», welche häufige Fallstricke in der klinischen Hirnschlagdiagnostik darstellen.
The clinical diagnosis of stroke in an emergency situation is a major challenge for physicians, especially since the correct therapy should be chosen under time pressure and a misdiagnosis can be associated with an unfavorable prognosis. This article is about “stroke mimics” and “stroke chameleons”, which are common pitfalls in clinical stroke diagnosis. Key Words: Stroke Mimics, Stroke Chameleons, Stroke.
Der Hirnschlag weist global eine rasant zunehmende Inzidenz auf und stellt einen absoluten medizinischen Notfall dar. Gerade in der präklinischen Situation ohne Bildgebung (z.B. Praxis oder Arztbesuch) ist eine rasche und korrekte klinische Diagnosestellung von grosser Bedeutung, um dem Patienten eine rasche Lysetherapie zu ermöglichen. Das typische klinische Bild eines Hirnschlags besteht aus dem plötzlichen Auftreten eines fokal-neurologischen Defizits mit maximaler Intensität bei Beginn. Es bestehen jedoch atypische Krankheitsbilder, die die Diagnose erschweren oder zu einer Fehldiagnose führen können.
Dabei sind folgende 2 Gruppen voneinander zu unterscheiden:
I) stroke mimics: darunter verstehen wir ein klinisches Syndrom, das einem akuten Hirnschlag gleicht, dessen Ursache jedoch nicht auf eine zerebrale Ischämie zurückzuführen ist («falsch-positive Diagnose»). II) stroke chameleons: damit sind klinische Syndrome gemeint, welche atypisch sind für einen Hirnschlag und somit den Kliniker nicht an die Differentialdiagnose eines Hirnschlags denken lassen, deren Ursache aber doch auf eine zerebrale Ischämie zurückzuführen ist («falsch-negative Diagnose»).
Eine korrekte Diagnose ist entscheidend für die adäquate Therapie und die Prognose. Denn Fehldiagnosen als stroke mimic führen zur unnötigen Diagnostik (CT/MRI) und Therapie (Lyse, Antithrombotika), welche einerseits unnötige Kosten verursachen und andererseits die Patienten zusätzlichen Therapierisiken (v.a. Blutungsrisiko) aussetzen. Fehldiagnosen als stroke chameleon hingegen sind mit verpasster bzw. unterlassener Akuttherapie (Lyse) verbunden, was zu einer ungünstigen Prognose führt. Das Unterlassen der Sekundäprävention ist zudem mit erhöhtem Rezidivrisiko verbunden.
Im Folgenden werden wir diese Differentialdiagnosen des Hirnschlags näher erläutern.
Stroke mimics
Die häufigsten Ursachen für stroke mimics sind Migräne-Attacken, epileptische Anfälle oder funktionelle Störungen, gefolgt von selteneren Ursachen wie Hypoglykämie, Infektionen oder Elektrolytentgleisungen.
Migräne mit Aura
Die Migräne mit Aura stellt eine der häufigsten Ursachen von Stroke mimics dar. Typischerweise können Migräne-Attacken mit motorischer oder dysphasischer Aura als Hirnschlag fehldiagnostiziert werden. Erschwerend kommt hinzu, dass gerade die häufigsten Hirnschlagursachen bei jüngeren Patienten (Persistierendes Foramen ovale und Dissektionen) häufig mit Migräne assoziiert sind. Zudem können Kopfschmerzen nicht selten auch als Begleitsymptom bei akutem Hirnschlag auftreten, insbesondere bei Infarkten im posterioren Stromgebiet oder häufiger bei Hirnblutungen (1).
Bei Migräne sind die meisten Auren sensibel oder visuell, die als fokales Defizit fehlinterpretiert werden können. Eine rasch fortschreitende Ausdehnung der Symptome über einige Minuten hinweg deutet auf eine Aura hin, gerade wenn sie zuvor oder anschliessend von Kopfschmerzen begleitet werden. Bei visueller Aura sind typischerweise beide Augen betroffen (binokulär). Zudem berichten die Patienten über positive Reizphänomene wie Blendungsgefühl oder Blitzphänomene, die auch bei Lidschluss persistieren. Bei Sehstörungen infolge der Ischämie ist der Beginn jedoch perakut, bei Amaurosis fugax nur ein Auge betroffen (monokulär) und die Patienten berichten über Negativphänomene («alles schwarz/dunkel»). Zu berücksichtigen ist, dass bei Migräne mit zunehmendem Alter die Kopfschmerzen immer seltener werden und die Auren isoliert auftreten können. Andererseits ist Vorsicht geboten bei erstmaliger Präsentation einer Aura-Symptomatik, bei der man im Zweifelsfall eher grosszügig eine Bildgebung (MRI) empfehlen würde. Last not least ist zu bedenken, dass auch eine Migräne sich sehr unterschiedlich manifestieren kann (mit grossen Variationen in der Dauer und Dynamik der Kopfschmerzen und Aura-Phänomene sowie Intensität der Beschwerden), sodass auch die Migräne selbst als mimic und chameleon von neurologischen Erkrankungen diskutiert wird (2,3).
Besonders schwierig ist die Einschätzung bei Patienten, welche die Kriterien für eine Migräne nicht erfüllen und die sich mit der ersten Episode von transienten sensorischen oder aphasischen Symptomen präsentieren. Die hemiplegische Migräne ist sehr selten: die ersten Episoden treten in der Regel vor dem 20. Lebensjahr auf und neigen dazu, mit zunehmendem Alter weniger häufig aufzutreten, dafür aber länger zu dauern (4).
Epileptischer Anfall
Epileptische Anfälle sind ebenfalls eine sehr häufige Ursache von stroke mimics. Ein postiktales motorisches Defizit kann auf einen nicht diagnostizierten fokalen Anfall von kurzer Dauer folgen. Die Entwicklung einer fokalen Schwäche nach einem Anfall wurde 1849 von Robert Bentley Todd beschrieben und ist vermutlich auf eine Überaktivität und anschliessende Erschöpfung des primären motorischen Kortex zurückzuführen. Die Diagnose kann schwierig sein, wenn ein Anfall die erste Manifestation eines Schlaganfalls darstellt oder wenn die Ursache des epileptischen Anfalles ein früherer Schlaganfall ist, im Sinne einer strukturellen Epilepsie. Die MRT mit DWI- und ADC-Sequenzen (scheinbarer Diffusionskoeffizient) sind für die Unterscheidung zwischen alten und neuen ischämischen Schlaganfällen entscheidend (5). Abhängig von betroffenen Hirnarealen können andere Defizite auftreten, wie z.B. Sprachstörung, sensible Symptome oder Gesichtsfeldausfälle.
Funktionelle Störung
Funktionelle Störungen äussern sich oft als akute Schwäche oder Empfindungsstörungen, welche neuro-anatomisch schwierig zu lokalisieren sind. Häufig gibt es einen Auslöser wie z.B. eine Panikattacke oder eine akute Belastungssituation. Bei der Diagnose funktioneller Störungen können positive Befunde im neurologischen Status, die Inkonsistenz (z.B. kann während der Untersuchung das Bein nicht bewegt werden, Patient kann aber normal zur Toilette gehen) und Inkongruenzen (z.B. Hemiparese bei vollständiger Schonung des Gesichts) sowie auch die Indifferenz gegenüber dem Schweregrad der Beeinträchtigung für die Diagnose hilfreich sein. Das Hoover-Zeichen und Drift ohne Pronation bei Armlähmung sind weitere Beispiele, die für eine funktionelle Schwäche sprechen.
Andere Ursachen
Eine Hypoglykämie präsentiert sich normalerweise mit autonomen Symptomen, kann aber auch allein mit fokalen neurologischen Symptomen auftreten. Deshalb empfiehlt sich bei akuten Ausfällen immer den Blutzucker zu bestimmen, insbesondere bei Risikopatienten (z.B. Therapie mit Insulin oder Sulfonylharnstoffen). Auch eine Sepsis oder Elektrolytentgleisungen wie Hyponatriämien können ein stroke mimic vortäuschen, gerade bei älteren multimorbiden Patienten. Die Sepsis kann aber über Hyperkoagulabilität auch einen Hirnschlag begünstigen.
Bei akutem Schwindel kann der HINTS-Test (Head impulse, Nystagmus, Test of Skew) (6, 7) hilfreich sein zur Unterscheidung eines zentralen vs. peripheren Schwindels. Ein negativer Kopfimpulstest mit richtungswechselndem Nystagmus und skew deviation hat eine sehr hohe Sensitivität und Spezifität für einen Hirnschlag. Zu bedenken ist zudem, dass auch chronische Erkrankungen gelegentlich akut beginnen können (z.B. akute myasthene Krise, Multiple Sklerose mit apoplektiformer Präsentation, Einblutung von Tumoren oder epidurale Abszesse).
Stroke mimics und Lysetherapie
Die Prävalenz von stroke mimics ist in Zentren, welche in der Akutsituation ein CT durchführen, hoch (bis zu 25%) (5). Dies bedeutet, dass auch Patienten ohne Hirnschlag mit intravenöser Lyse behandelt werden können. Ein führendes Symptom bei diesen Patienten ist z.B. eine schwere Aphasie ohne Hemiparese (3). Glücklicherweise ist die Komplikationsrate äusserst niedrig bei diesen Patienten (Hirnblutung 0.5%, orolinguale Ödeme 0.3%) (8). In Zentren mit MRI in der Akutdiagnostik kann die Rate von falsch-pos. Diagnosen deutlich reduziert werden.
Stroke Chameleons
Ein Hirnschlag kann fälschlicherweise mit einer peripheren Nervenläsion, Delirium oder Synkope verwechselt werden. Das Risiko einer Fehldiagnose ist gerade bei jüngeren Patienten und solchen mit leichten Symptomen oder Koma höher. Dabei werden häufiger Schlaganfälle im hinteren Kreislauf übersehen, gerade wenn sich diese Patienten mit eher atypischen Symptomen wie Verwirrtheit ohne Lateralisierungszeichen präsentieren (9, 10).
Vigilanzminderung
Das «Top-of-the-Basilar»-Syndrom wird durch einen Verschluss des distalen Abschnitts der Arteria basilaris verursacht; die Pa-
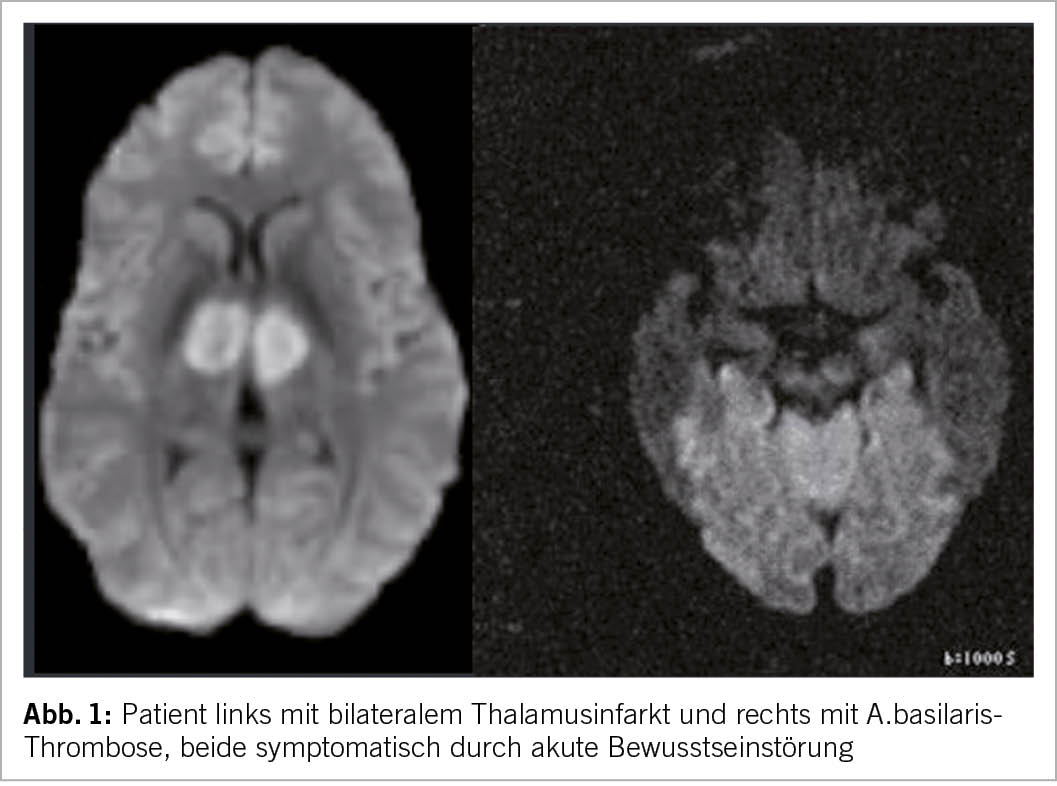
tienten können sich mit Bewusstlosigkeit und Tetraplegie vorstellen (11). Pupillenveränderungen und okulomotorische Zeichen liefern Hinweise, doch in der Regel ist zur Bestätigung der Diagnose eine CT- oder Magnetresonanzangiographie erforderlich (11). Bewusstseinsstörungen können auch bei beidseitigen Thalamusinfarkten auftreten, in der Regel in Verbindung mit vertikalen Blickparesen (12). Heimtückisch sind Situationen, bei denen der Patient intraoperativ einen Hirnschlag im hinteren Stromgebiet erleidet und postoperativ eine persistierende Bewusstseinstrübung aufweist, welche auf die Narkose zurückgeführt wird (Abb. 1).
Akute Verwirrtheit
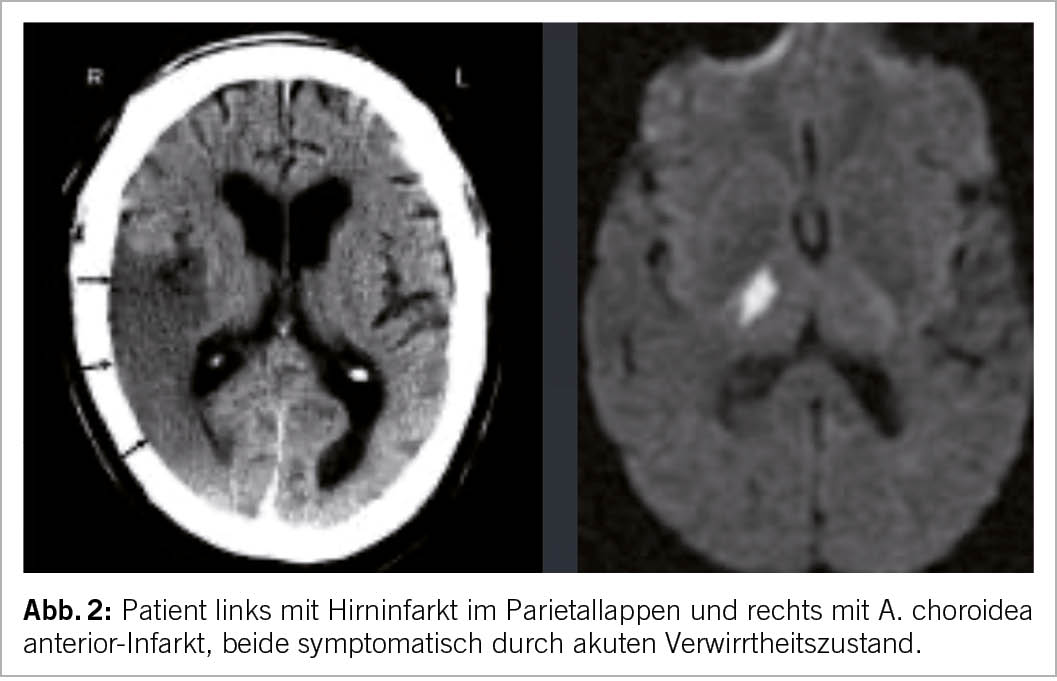
Schlaganfälle können mit Verwirrtheit, Erregung oder Unruhe einhergehen und fälschlicherweise für ein Delirium gehalten werden. Typisch sind z.B. Infarkte im Parietallappen oder auch Infarkte im Stromgebiet der A. choroidea anterior, welche eine akute Verwirrtheit auslösen können. Ein hilfreicher klinischer Hinweis ist das plötzliche Auftreten der Verwirrtheit (perakut) bei zuvor asymptomatischem Patienten (Abb. 2).
Akute Bewegungsstörungen
Hemichorea, Hemidystonie oder Hemibalismus treten bei Läsionen auf, die die basalen Ganglien betreffen. Rhythmische tonische Bewegungen können gelegentlich auf eine Hirnstamm-Ischämie hinweisen. Bei diesen Bewegungen kann es sich um anfallsartige rhythmische Zuckungen handeln, manchmal mit verlängerten tonischen Muskelkontraktionen.
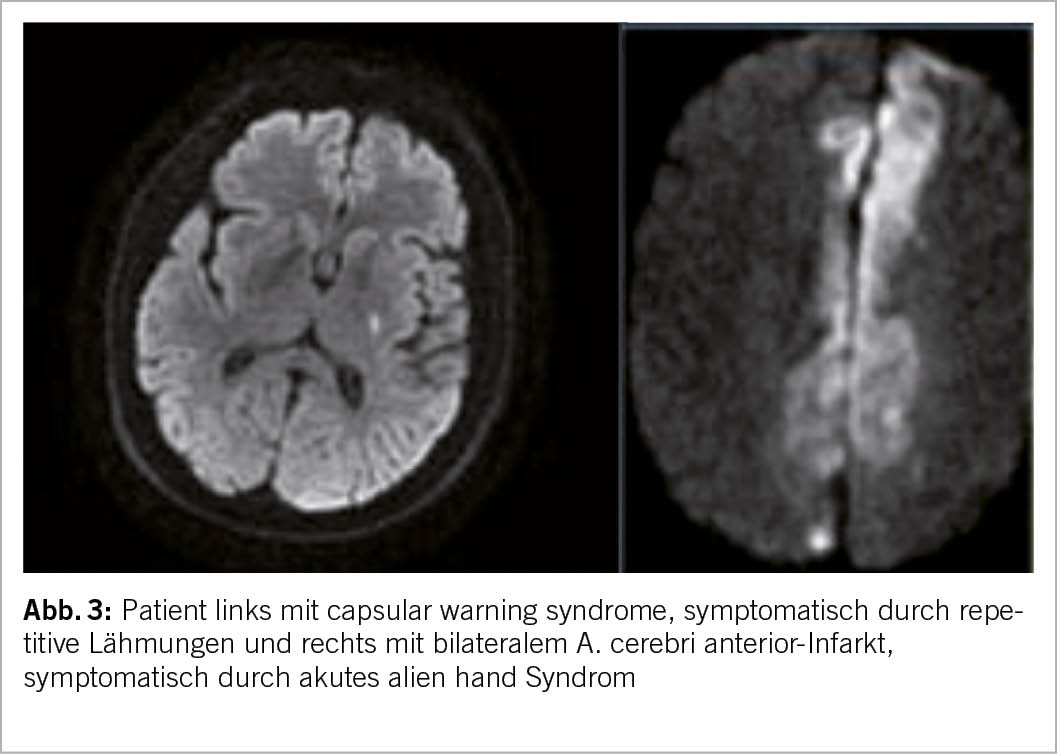
Limb shaking TIA sind rhythmische, unwillkürliche, ruckartige Bewegungen der Gliedmassen aufgrund einer schweren Stenose der A. carotis interna. Diese hämodynamischen TIA können durch eine verminderte zerebrale Durchblutung ausgelöst werden z.B. durch Haltungswechsel oder körperliche Anstrengung, und mit fokalen epileptischen Anfällen verwechselt werden. Das Alien-Hand-Syndrom ist definiert als unwillkürliche, unkontrollierbare, aber scheinbar zielgerichtete Bewegung einer oberen Extremität. Bei einem Infarkt im Bereich des Corpus callosum kann es zu verschiedenen abnormen unwillkürlichen motorischen Verhaltensweisen kommen (12) (Abb. 3).
Pseudoperiphere Monoparesen
Weniger als 5% aller Schlaganfälle äussern sich durch eine isolierte Monoparese (meistens der Arm betroffen), die fälschlicherweise als periphere Nervenlähmung diagnostiziert werden kann (12). Die meisten dieser Schlaganfälle sind auf subkortikale Läsionen zurückzuführen, 30% werden jedoch durch kortikale Läsionen verursacht. Eine isolierte Handparese ist in der Regel aber kortikal bedingt (hand knob infarction) und kann typischerweise eine periphere Nervenschädigung vortäuschen (13, 14).
Luiz Alexandre Dalla Vecchia Dr. med. Adrian Scutelnic PD Dr. med. Kateryna Antonenko Prof. Dr. med. Mirjam R. Heldner Prof. Dr. med. Marcel Arnold Prof. Dr. med. Hakan Sarikaya
Universitätsklinik für Neurologie, Inselspital, Freiburgstrasse, 3010 Bern
Copyright bei Aerzteverlag medinfo AG
Luiz Alexandre Dalla Vecchia
Universitätsklinik für Neurologie
Inselspital
Freiburgstrasse
3010 Bern
Prof. Dr. med. Marcel Arnold
Universitätsklinik für Neurologie
Inselspital
Freiburgstrasse
3010 Bern
Prof. Dr. med. Hakan Sarikaya
Universitätsklinik für Neurologie
Inselspital Bern
Rosenbühlgasse 25
3010 Bern
Die Autoren haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
◆ Die klinische Diagnose eines Hirnschlags kann in der Akutsituation ohne Bildgebung herausfordernd sein. Die Kenntnis über stroke mimics und chameleons ist hilfreich für eine präzise Diagnostik,
um einerseits unnötige Abklärungen und Therapien zu verhindern
(im Falle von stroke mimics), jedoch auch in der Früherkennung
und Behandlung von atypischen Schlaganfallpräsentationen
(im Falle von stroke chameleons).
1. Tentschert, S., Wimmer, R., Greisenegger, S., Lang, W., & Lalouschek, W. (2005).
Headache at stroke onset in 2196 patients with ischemic stroke or transient ischemic attack. Stroke; a Journal of Cerebral Circulation, 36(2). https://doi.org/10.1161/01.str.0000151360.03567.2b
2. Fernandes, P. M., Whiteley, W. N., Hart, S. R., & Al-Shahi Salman, R. (2013, February). Strokes: Mimics and chameleons. Practical Neurology. https://doi.org/10.1136/practneurol-2012-000465
3. Sarikaya, H., Yilmaz, M., Luft, A. R., & Gantenbein, A. R. (2012). Different
pattern of clinical deficits in stroke mimics treated with intravenous thrombolysis. European Neurology, 68(6), 344–349. https://doi.org/10.1159/000337677
4. Thomsen, L. L., Eriksen, M. K., Roemer, S. F., Andersen, I., Olesen, J., & Russell, M. B. (2002). A population-based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain, 125(6), 1379–1391. https://doi.org/10.1093/brain/awf132
5. Moulin, S., & Leys, D. (2019, February 1). Stroke mimics and chameleons. Current Opinion in Neurology. Lippincott Williams and Wilkins. https://doi.org/10.1097/WCO.0000000000000620
6. Kattah, J. C. (2018). Use of HINTS in the acute vestibular syndrome. An Overview. Stroke and Vascular Neurology, 3(4), 190–196. https://doi.org/10.1136/svn-2018-000160
7. Kattah, J. C., Talkad, A. V., Wang, D. Z., Hsieh, Y. H., & Newman-Toker, D. E. (2009). HINTS to diagnose stroke in the acute vestibular syndrome: Three-step bedside oculomotor examination more sensitive than early MRI diffusion-weighted imaging. Stroke, 40(11), 3504–3510. https://doi.org/10.1161/STROKEAHA.109.551234
8. Erbguth, F. (2017). Stroke Mimics und Stroke Chamäleons-Differenzialdiagnose des Schlaganfalls. Stroke Mimics Und … Fortschr Neurol Psychiatr, 85, 747–764.
9. Arch, A. E., Weisman, D. C., Coca, S., Nystrom, K. V., Wira, C. R., & Schindler,
J. L. (2016). Missed Ischemic Stroke Diagnosis in the Emergency Department by Emergency Medicine and Neurology Services. Stroke, 47(3), 668–673. https://doi.org/10.1161/STROKEAHA.115.010613
10. Richoz, B., Hugli, O., Dami, F., Carron, P. N., Faouzi, M., & Michel, P. (2015). Acute stroke chameleons in a university hospital: Risk factors, circumstances, and outcomes. Neurology, 85(6), 505–511.
11. Luengo-Fernandez, R., Paul, N. L. M., Gray, A. M., Pendlebury, S. T., Bull, L. M., Welch, S. J. V., … Rothwell, P. M. (2013). Population-Based Study of Disability and Institutionalization After Transient Ischemic Attack and Stroke. Stroke, 44(10), 2854–2861. https://doi.org/10.1161/strokeaha.113.001584
12. Tucha, O., Naumann, M., Berg, D., Alders, G. L., & Lange, K. W. (2001). Bilateral thalamic infarction: Clinical, etiological and mri correlates. Acta Neurologica Scandinavica, 103(1), 35–42. https://doi.org/10.1034/j.1600-0404.2001.00141.x
13. Edlow, J. A., & Selim, M. H. (2011, June). Atypical presentations of acute cerebrovascular syndromes. The Lancet Neurology. https://doi.org/10.1016/S1474-4422(11)70069-2
14. Peters, N., Müller-Schunk, S., Freilinger, T., Düring, M., Pfefferkorn, T., & Dichgans, M. (2009). Ischemic stroke of the cortical “hand knob” area: Stroke mechanisms and prognosis. Journal of Neurology, 256(7), 1146–1151. https://doi.org/10.1007/s00415-009-5104-8
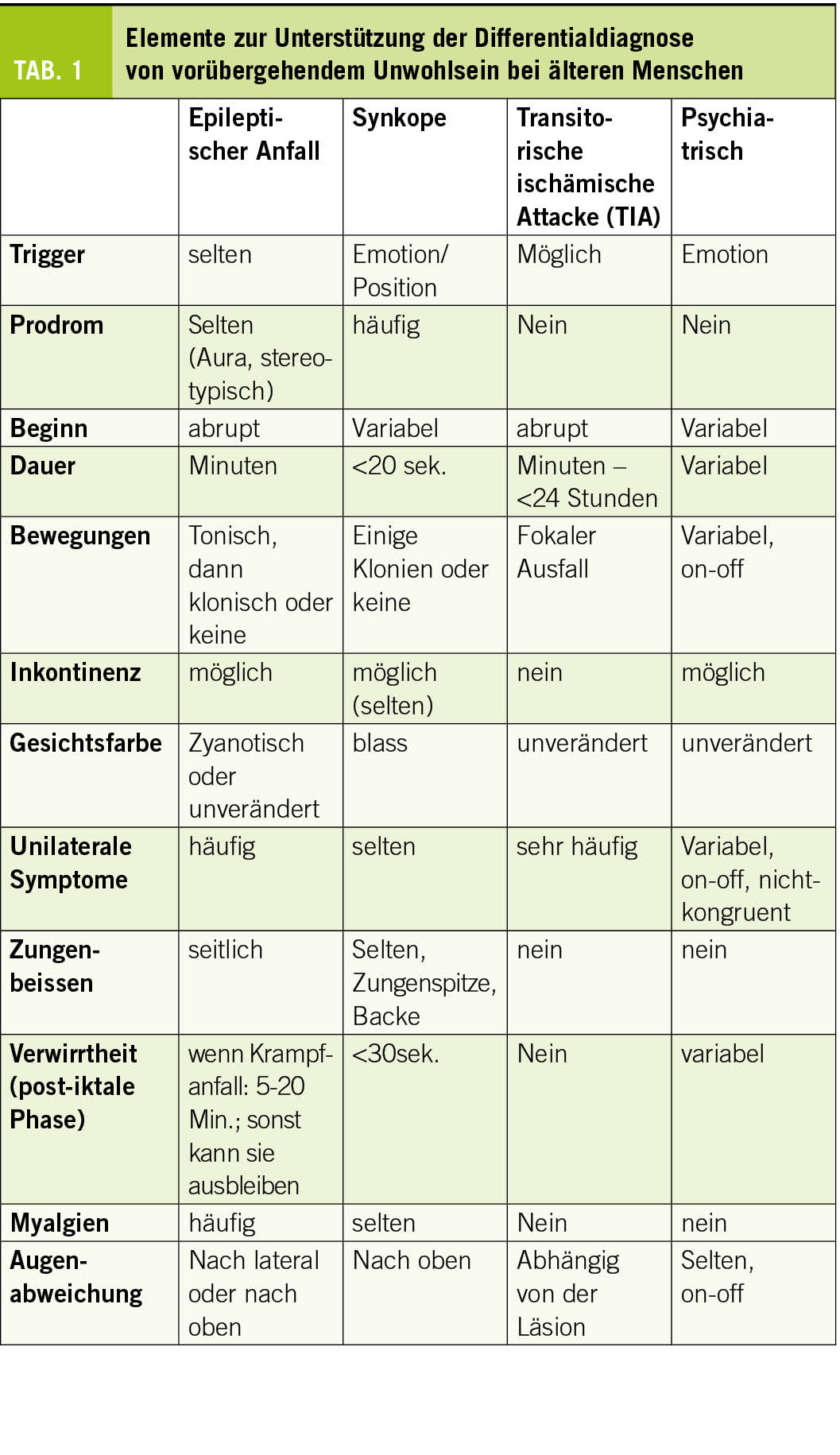
Sowohl die Inzidenz als auch die Prävalenz der Epilepsie steigen nach 65 Jahren an. Angesichts der Alterung der Bevölkerung wird die Behandlung von Epilepsie bei älteren Menschen zu einer Herausforderung für die öffentliche Gesundheit. Bei der Behandlung dieser Patienten müssen die Ätiologie, die Komorbiditäten, die Risiken eines erneuten Anfalls oder der antikonvulsiven Behandlung sowie die pharmakodynamischen und pharmakokinetischen Veränderungen im Alter berücksichtigt werden.
Both the incidence and prevalence of epilepsy increase after the age of 65. In view of the ageing population, the management of epilepsy in the elderly is becoming a public health issue. The management of these patients implies taking into account the etiology, comorbidities, the risks inherent in a seizure recurrence or anticonvulsant treatment, as well as the pharmacodynamic and pharmacokinetic changes that occur in old age. Key Words: epilepsy, elderly,public health, anticonvulsant treatment
Epilepsie ist die dritthäufigste neurologische Erkrankung im Alter von 65 Jahren, nach Schlaganfall und Demenz, die beide das Epilepsie-Risiko erhöhen (1). Dieser Trend wird sich angesichts der Alterung der Bevölkerung noch verstärken, da die Zahl der Personen >65 Jahre in den nächsten 15 Jahren um schätzungsweise 60% steigen wird (2). Die Behandlung von Epilepsie bei älteren Menschen erfordert aufgrund der verschiedenen Ätiologien, Komorbiditäten, des erhöhten Risikos von Nebenwirkungen der Behandlung, der Komedikation sowie der pharmakodynamischen und pharmakokinetischen Veränderungen besondere Überlegungen. Darüber hinaus sind ältere Menschen im Falle eines epileptischen Anfalls besonders verletzlich: Es besteht die Gefahr traumatischer Verletzungen, des Vertrauensverlusts und der Einschränkung der Selbstständigkeit. Trotz all dieser Faktoren wird Epilepsie bei älteren Menschen häufig verspätet diagnostiziert und die Patienten haben weniger leicht Zugang zu spezialisierten Zentren (3).
Epidemiologie
Wenn wir von epileptischen Anfällen bei älteren Menschen sprechen, sollten wir drei Szenarien klar definieren:
1) Provozierte (oder akut symptomatische) epileptische Anfälle, die in der Frühphase einer Hirnschädigung oder als Folge toxisch-metabolischer Störungen auftreten. Die Inzidenz steigt ab der dritten Dekade linear an und das Risiko wird auf 3,6% im Alter von 80 Jahren geschätzt (4). Nach einem ersten Anfall sollte eine breite biologische Untersuchung, eine Bildgebung des Gehirns und eine detaillierte Anamnese auf der Suche nach einem begünstigenden Faktor durchgeführt werden. Diese Art von Anfall führt nicht automatisch zu einer Epilepsiediagnose. Epilepsie ist nämlich definiert durch das Auftreten von ≥2 unprovozierten Anfällen im Abstand von 24 Stunden oder einem einzelnen Anfall mit einem geschätzten 10-Jahres-Rückfallrisiko von ≥60% (durch zusätzliche Untersuchungen wie EEG, Bildgebung) (5).
2) Epilepsie, die in jungen Jahren begonnen hat und bis ins hohe Alter andauert.
3) Epilepsie, die de novo im hohen Alter (>60 Jahre) auftritt. Die Inzidenz von Epilepsie ist in sehr jungen Jahren und im höheren Alter erhöht (allmählicher Anstieg ab 40-50 Jahren) und wird im Alter von 65 Jahren auf 90-150/100.000 geschätzt. Dieses bimodale Muster wurde sowohl in Industrie- als auch in Entwicklungsländern wiederholt nachgewiesen (3, 6). Angesichts der Alterung der Bevölkerung und der verbesserten Pflege mit längeren Überlebenszeiten für Epilepsiepatienten in jungen Jahren folgt die Prävalenz der Epilepsie demselben bimodalen Muster (5,4% bei älteren Menschen und bis zu 7,5% bei Patienten in Pflegeheimen) (7, 8). Die Inzidenz des Status epilepticus (SE), d. h. eines längeren Anfalls von mehr als 5 bis 10 Minuten, wird bei älteren Menschen auf 86/100’000 geschätzt, d.h. 5x höher als bei jüngeren Menschen (9). Darüber hinaus steigt die mit dem SE verbundene Mortalität mit zunehmendem Alter. Sie beträgt 38% bei > 60 Jahre (9, 10) und stellt einen der wichtigsten prognostischen Faktoren dar (11). Diese Daten untermauern die Tatsache, dass Epilepsie bei älteren Menschen nicht als harmlose Krankheit angesehen werden sollte.
Ätiologie und Risikofaktoren
Zerebrovaskuläre Erkrankungen sind für mehr als ein Drittel der Epilepsien bei älteren Menschen verantwortlich (12), wobei die Inzidenz von Epilepsie nach einem Schlaganfall bei 6,4-15% liegt (13). Neurodegenerative Erkrankungen und unbestimmte Ätiologien machen etwa ein Viertel der Fälle aus (6). Tumorätiologien sind für etwa 10% verantwortlich (14). Mehrere Studien berichten über eine Assoziation zwischen zerebrovaskulären Risikofaktoren und dem Auftreten von Epilepsie (15-18). In der ARIC-Kohorte (Atherosclerosis Risk in Communities) mit mehr als 10.000 Patienten, die über ein Jahrzehnt beobachtet wurden, wurde das Auftreten von Epilepsie mit Bluthochdruck, Diabetes und Schlaganfällen in Verbindung gebracht (18). Diese Beobachtung eröffnet die Möglichkeit eines ganzheitlichen Ansatzes, mit dem die Epileptogenese möglicherweise durch die Behandlung zerebrovaskulärer Risikofaktoren reduziert werden kann.
Diagnostische Herausforderung
Epileptische Anfälle bei älteren Menschen sind überwiegend fokal und können aufgrund ihrer weniger “motorischen” Semiologie als bei jungen Menschen unbemerkt bleiben. Sie können sich nur in paroxysmalen Episoden äußern, in denen es zu einem Stillstand der Aktivität, Kontaktverlust, Sturz oder Verwirrung kommt (3). Bei 26% der älteren Patienten mit einer endgültigen Epilepsiediagnose wäre eine Epilepsie zunächst nicht in Betracht gezogen worden (19). Bis zu 70% der Anfälle bei der Alzheimer-Krankheit sollen rein mit Bewusstseinsstörungen auftreten (20). Darüber hinaus ist die Differenzialdiagnose sehr breit gefächert und umfasst Fluktuationen im Rahmen einer neurodegenerativen Erkrankung, Synkopen, TIAs oder toxisch-metabolische Störungen.
Die Anamnese beim Patienten und bei den Angehörigen ist von entscheidender Bedeutung. Dabei sollte aktiv nach dem stereotypen Charakter der Episoden, den Umständen des Auftretens und möglichen Begleiterscheinungen wie Automatismen gesucht werden (Tab. 1). Ältere Patienten neigen zu postkritischer Verwirrung oder Toddscher Lähmung, die über einen längeren Zeitraum (mehrere Stunden oder sogar Tage) andauern und zu einer Fehldiagnose von Demenz oder Schlaganfall führen können (21). Die Diagnose einer funktionellen Episode (PNES; Psychogenic Non epileptic Seizures) wird bei älteren Menschen oft wenig beachtet. In einer Kohorte von 94 Patienten im Alter von >60 Jahren, die unter EEG-Überwachung zur Charakterisierung von Episoden standen, zeigten 27 Patienten nicht-epileptische Episoden, hauptsächlich PNES (22). Häufig handelt es sich um Personen mit schwerer somatischer Komorbidität (23).
Das EEG kann natürlich bei der Diagnose helfen. Es ist jedoch zu betonen, dass eine interiktale Aufzeichnung weder eine perfekte Spezifität noch Sensitivität aufweist. Bei etwa einem Drittel der älteren Epilepsiepatienten werden im EEG interiktale Abnormalitäten (irritative Zone) festgestellt. Umgekehrt werden interiktale Anomalien, hauptsächlich im Schlaf, bei 2-6% der Patienten mit neurodegenerativen Erkrankungen berichtet, wobei es nicht notwendigerweise zu klinischen Episoden kommt, die mit Anfällen vereinbar sind (20). Die Anamnese und das klinische Urteil sollten daher Vorrang haben. Bei starkem klinischem Verdacht kann im Einzelfall eine Testbehandlung oder eine längere EEG-Aufzeichnung, die auch den Schlaf einschließt, diskutiert werden.
Behandlung
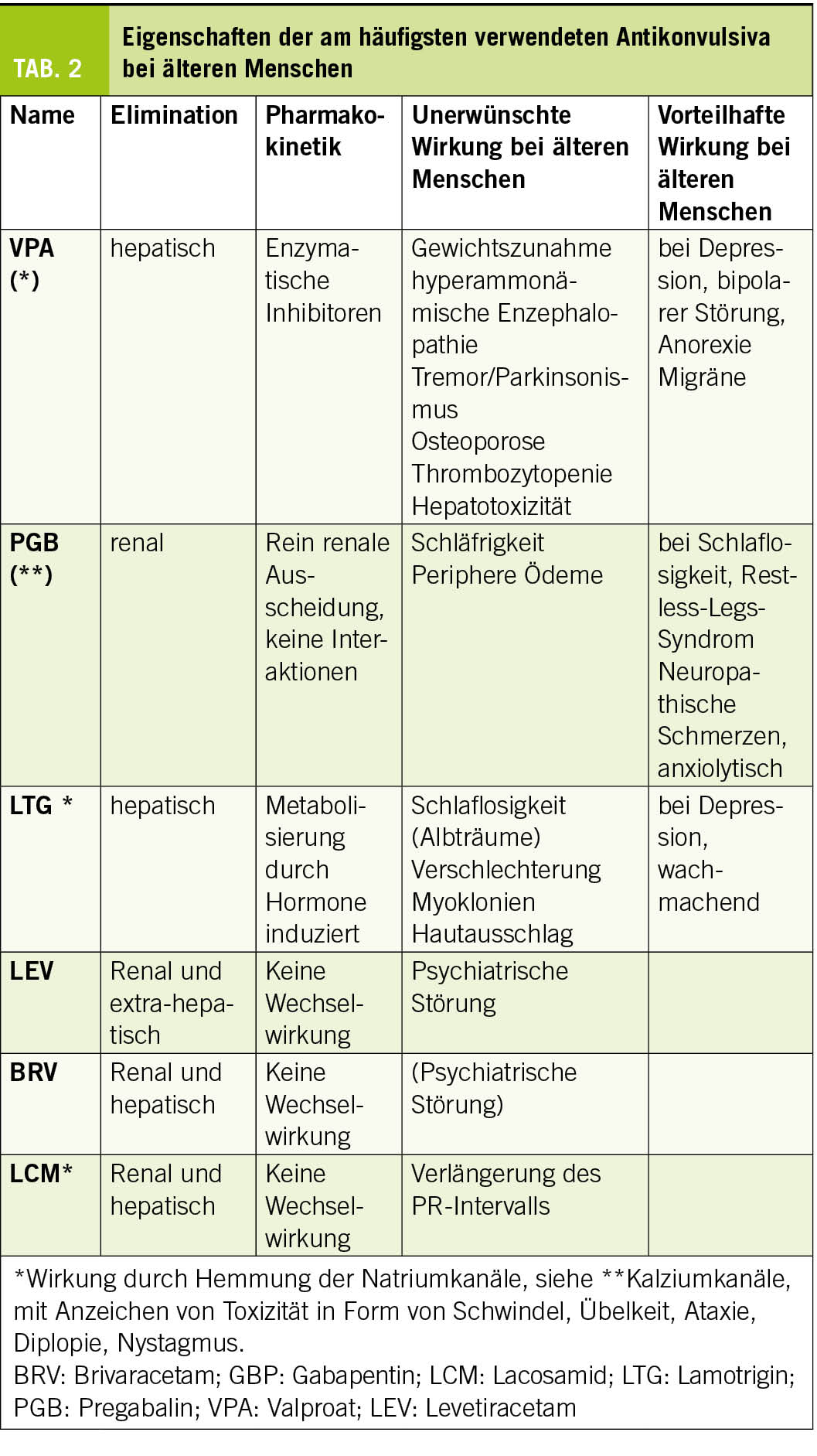
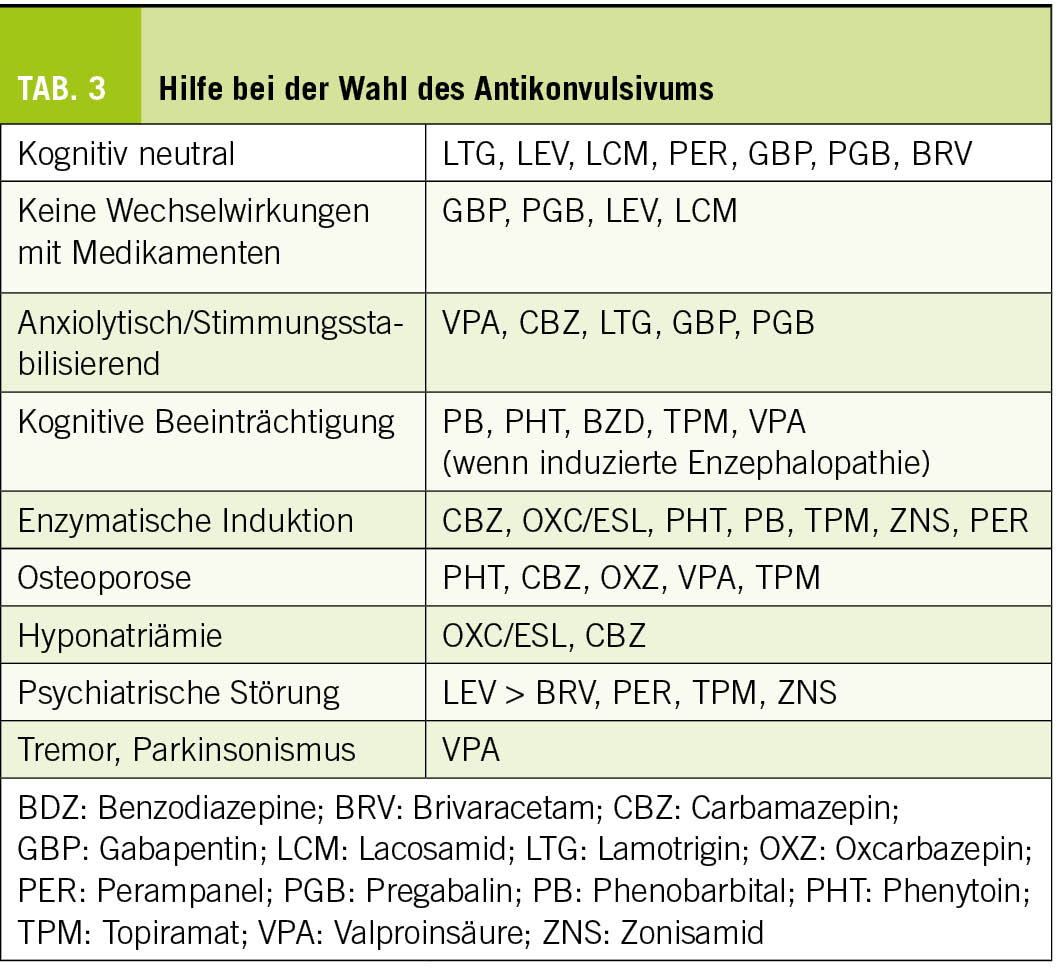
Obwohl die meisten älteren Menschen nach einem Jahr unter einer Monotherapie anfallsfrei sind (6), ist die Einführung einer Behandlung potenziell komplex und erfordert die Berücksichtigung altersbedingter pharmakodynamischer und pharmakokinetischer Veränderungen, Komedikationen und patientenspezifischer Komorbiditäten. Die Wahl der Medikation muss von Fall zu Fall unter Abwägung von Verträglichkeit und Wirksamkeit getroffen werden. Eine spezialisierte Beratung, zumindest in der Anfangsphase, ist daher empfehlenswert. Lamotrigin und Levetiracetam (24, 25) werden am häufigsten verwendet und sind zusammen mit Gabapentin/Pregabalin die Mittel der ersten Wahl bei älteren Menschen. Da Levetiracetam zu Verhaltensstörungen führen kann, sollten der Patient und seine Angehörigen darüber informiert und aktiv nach ihnen gesucht werden. Lamotrigin hat eine wachmachende und stimmungsstabilisierende Wirkung, die vorteilhaft sein kann. Diese Behandlung kann jedoch Myoklonien verstärken und ihr langsames, schrittweises Einleitungsschema angesichts des Risikos schwerer Hautreaktionen kann bei kognitiven Beeinträchtigungen schwierig zu befolgen sein. Generell sollten Enzyminduktoren aufgrund ihrer Auswirkungen auf den Knochenstoffwechsel und ihrer Wechselwirkungen mit anderen Medikamenten möglichst vermieden werden. In den Tabellen 2 und 3 sind die Eigenschaften der gängigsten Antikonvulsiva zusammengefasst. Bei der Einführung einer Behandlung sollte ein langsames Titrationsschema angewendet und eine niedrigere Dosis als bei jungen Menschen angestrebt werden.
Bidirektionale Beziehung zwischen Epilepsie und Demenz
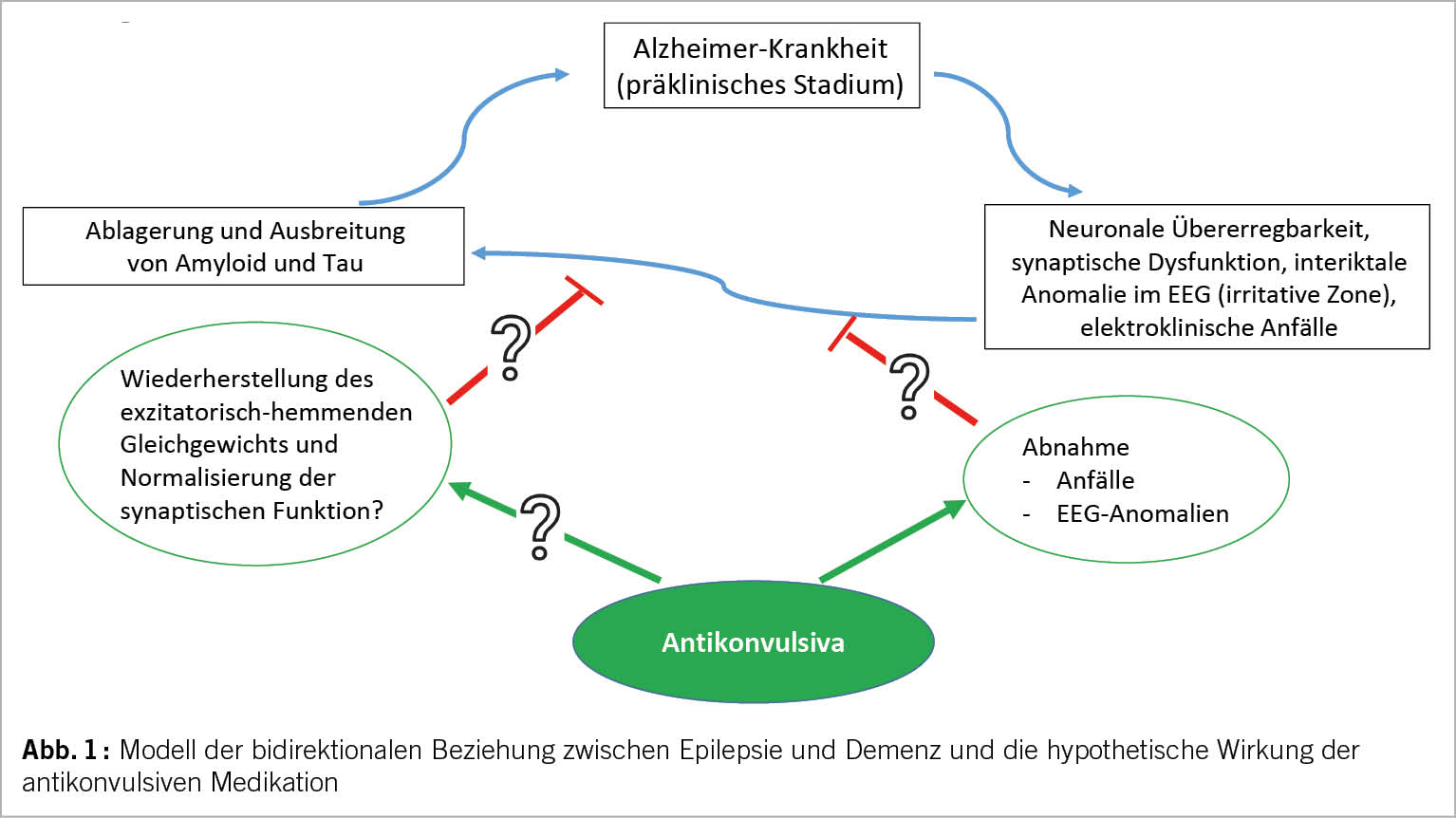
Nach einem ersten unprovozierten Anfall bei einem Patienten mit Alzheimer-Demenz wird das Risiko eines erneuten Anfalls auf 70% geschätzt (26), womit die diagnostischen Kriterien für eine Epilepsie erfüllt sind. Ein erhöhtes Risiko für epileptische Anfälle bei Demenz ist seit langem bekannt (27). Angesichts von Tierstudien, die beschrieben, dass die Akkumulation von β-Amyloid das Auftreten elektrischer Anfälle begünstigt und dass die Anfälle wiederum zu hippokampaler Dysfunktion und damit zu Gedächtnisstörungen beitragen (28), hat das Interesse an dieser bidirektionalen Beziehung in letzter Zeit zugenommen. In der Framingham-Kohorte wird von einem verdoppelten Demenzrisiko bei Patienten mit Epilepsie berichtet, und in ähnlicher Weise von einem erhöhten Epilepsie-Risiko bei Demenz (29). In ähnlicher Weise wurde in der ARIC-Kohorte ein 3x höheres Demenzrisiko bei Patienten mit Spätepilepsie (30) und ein 3x höheres Risiko für das Auftreten von Epilepsie bei Demenzpatienten (18) beobachtet. Die zugrunde liegende Pathophysiologie ist nach wie vor ungeklärt. Patienten mit Epilepsie, die in der Kindheit begonnen hatte, wiesen nach über 50 Jahren mehr kognitive Störungen und ein stärker verändertes Amyloid-PET auf als die Kontrollgruppe (31). In einer neueren Studie wurde kein Zusammenhang zwischen Antikonvulsiva und kognitiven Störungen festgestellt (32), sondern eher zwischen der Anfallshäufigkeit und dem kognitiven Verfall. Umgekehrt weisen mehr als 50% der Patienten mit Spätepilepsie zum Zeitpunkt der Diagnose ein MCI (mild cognitive impairment) auf (33, 34). Mehrere pathologisch-anatomische Studien an Patienten ohne Demenz, bei denen zur Behandlung von Epilepsie eine temporale Lobektomie durchgeführt wurde, beschreiben bei Patienten mit Epilepsie mehr Akkumulation von β-Amyloid oder Tau-Hyperphosphoryl als bei Kontrollpatienten (20, 35, 36). Schließlich werden interkritische EEG-Anomalien (irritative Zone) mit einer schlechten kognitiven Prognose bei der Alzheimer-Krankheit in Verbindung gebracht (29, 37). Es ist noch nicht geklärt, ob dies auf infraklinische Anfälle zurückzuführen ist, die die Kognition beeinträchtigen, ob diese EEG-Anomalien an sich schädlich sind oder ob sie auf eine schwerere zugrunde liegende Pathologie hindeuten. Dies hat dazu geführt, dass die Möglichkeiten einer prophylaktischen antikonvulsiven Behandlung untersucht wurden (Abb. 1). Tierstudien haben gezeigt, dass Levetiracetam durch die Verringerung der glutamatbedingten Exzitotoxizität und die Unterdrückung der hippokampalen neuronalen Hyperaktivität zu einer Wiederherstellung der synaptischen Funktion und einer Verringerung der β-Amyloidablagerungen führen kann. In einer neueren Studie wurden Alzheimer-Patienten randomisiert zwischen Placebo und sehr niedrig dosiertem Levetiracetam: Bei der kleinen Untergruppe mit irritativer Aktivität im EEG (9 Patienten!) verbesserten sich einige kognitive Funktionen nach Levetiracetam (38).
Schlussfolgerung
Epilepsie ist eine häufige Erkrankung bei älteren Menschen und kann zahllose körperliche, soziale und psychiatrische Folgen haben. Angesichts der bidirektionalen Beziehung zwischen Epilepsie und Demenz sollte ein rigoroses Screening nach klinischen Phänomenen durchgeführt werden, die auf epileptische Anfälle bei Demenzpatienten oder auf kognitive Beschwerden bei Epilepsiepatienten hindeuten. Die Diagnose und die Einleitung einer Behandlung erfordern die Hilfe eines Spezialisten.
Übersetzung aus «la gazette médicale» 02-2023
Copyright bei Aerzteverlag medinfo AG
Dr. med. Isabelle Beuchat
MD, CHUV, NLG, BH07
Centre Hospitalier Universitaire Vaudois (CHUV)
und Universität Lausanne; Abteilung für klinische
Neurowissenschaften, Abteilung für Neurologie
Abteilung für Epileptologie
Rue du Bugnon 46
1011 Lausanne
Isabelle.beuchat@chuv.ch
Prof. Dr. med. Andrea O Rossetti
MD, FAES, CHUV, NLG, BH07
Centre Hospitalier Universitaire Vaudois (CHUV)
und Universität Lausanne; Abteilung für klinische
Neurowissenschaften, Abteilung für Neurologie
Abteilung für Epileptologie
Rue du Bugnon 46
1011 Lausanne
Andrea.rossetti@chuv.ch
Die Autoren haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
◆ Epilepsie ist bei Menschen über 65 Jahren häufiger als in jeder
anderen Altersgruppe.
◆ Die Semiologie der epileptischen Anfälle ist anders, mit weniger
motorischen Manifestationen oder Generalisierung.
◆ Die antikonvulsive Therapie bei älteren Menschen muss angepasst werden, wobei Wirksamkeit und Verträglichkeit abgewogen und die spezifischen Komorbiditäten des jeweiligen Patienten berücksichtigt werden müssen.
◆ Es besteht eine bidirektionale Beziehung zwischen Epilepsie und Demenz; Demenz ist ein unabhängiger Risikofaktor für das Auftreten von Epilepsie und Epilepsie ein unabhängiger Risikofaktor für das
Auftreten von Demenz.
1. WHO. Epilepsy: a public health imperative. 2019 [online]. Available at: www.who.int/mental_health/neurology/epilepsy/report_2019/en/.
2. Available at: https://www.census.gov/content/dam/Census/%20library/publications/2016/demo/p95-16-1.pdf.
3. Stephen LJ, Brodie MJ. Epilepsy in elderly people. The Lancet 2000;355:1441-1446.
4. Annegers JF, Hauser WA, Lee JR, Rocca WA. Incidence of acute symptomatic seizures in Rochester, Minnesota, 1935-1984. Epilepsia 1995;36:327-333.
5. Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 2014;55:475-482.
6. Sen A, Jette N, Husain M, Sander JW. Epilepsy in older people. The Lancet 2020;395:735-748.
7. Beghi E, Giussani G, Nichols E, et al. Global, regional, and national burden of epilepsy, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. The Lancet Neurology 2019;18:357-375.
8. Birnbaum AK, Leppik IE, Svensden K, Eberly LE. Prevalence of epilepsy/seizures as a comorbidity of neurologic disorders in nursing homes. Neurology 2017;88:750-757.
9. DeLorenzo RJ, Hauser WA, Towne AR, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 1996;46:1029-1035.
10. Sveinsson O, Andersson T, Carlsson S, Tomson T. The incidence of SUDEP: A nationwide population-based cohort study. Neurology 2017;89:170-177.
11. Sutter R, Kaplan PW, Ruegg S. Outcome predictors for status epilepticus–what really counts. Nat Rev Neurol 2013;9:525-534.
12. Liu S, Yu W, Lu Y. The causes of new-onset epilepsy and seizures in the elderly. Neuropsychiatr Dis Treat 2016;12:1425-1434.
13. Galovic M, Ferreira-Atuesta C, Abraira L, et al. Seizures and Epilepsy After Stroke: Epidemiology, Biomarkers and Management. Drugs Aging 2021;38:285-299.
14. Hernandez-Ronquillo L, Adams S, Ballendine S, Tellez-Zenteno JF. Epilepsy in an elderly population: Classification, etiology and drug resistance. Epilepsy Res 2018;140:90-94.
15. Leppik IE. Epilepsy in the elderly: scope of the problem. Int Rev Neurobiol 2007;81:1-14.
16. Ramsay RE, Pryor F. Epilepsy in the elderly. Neurology 2000;55:S9-14; discussion S54-18.
17. Choi H, Pack A, Elkind MS, Longstreth WT, Jr., Ton TG, Onchiri F. Predictors of incident epilepsy in older adults: The Cardiovascular Health Study. Neurology 2017;88:870-877.
18. Johnson EL, Krauss GL, Lee AK, et al. Association Between Midlife Risk Factors and Late-Onset Epilepsy: Results From the Atherosclerosis Risk in Communities Study. JAMA Neurol 2018;75:1375-1382.
19. Rowan AJ, Ramsay RE, Collins JF, et al. New onset geriatric epilepsy: a randomized study of gabapentin, lamotrigine, and carbamazepine. Neurology 2005;64:1868-1873.
20. Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, Miller BL. Epileptic activity in Alzheimer’s disease: causes and clinical relevance. Lancet Neurol 2017;16:311-322.
21. Sheth RD, Drazkowski JF, Sirven JI, Gidal BE, Hermann BP. Protracted ictal confusion in elderly patients. Arch Neurol 2006;63:529-532.
22. McBride AE, Shih TT, Hirsch LJ. Video-EEG monitoring in the elderly: a review of 94 patients. Epilepsia 2002;43:165-169.
23. Duncan R, Oto M, Martin E, Pelosi A. Late onset psychogenic nonepileptic attacks. Neurology 2006;66:1644-1647.
24. Lattanzi S, Trinka E, Del Giovane C, Nardone R, Silvestrini M, Brigo F. Antiepileptic drug monotherapy for epilepsy in the elderly: A systematic review and network meta-analysis. Epilepsia 2019;60:2245-2254.
25. Lezaic N, Gore G, Josephson CB, Wiebe S, Jette N, Keezer MR. The medical treatment of epilepsy in the elderly: A systematic review and meta-analysis. Epilepsia 2019;60:1325-1340.
26. Voglein J, Ricard I, Noachtar S, et al. Seizures in Alzheimer’s disease are highly recurrent and associated with a poor disease course. J Neurol 2020;267:2941-2948.
27. Hesdorffer DC, Hauser WA, Annegers JF, Kokmen E, Rocca WA. Dementia and adult-onset unprovoked seizures. Neurology 1996;46:727-730.
28. Palop JJ, Chin J, Roberson ED, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007;55:697-711.
29. Stefanidou M, Beiser AS, Himali JJ, et al. Bi-directional association between epilepsy and dementia: The Framingham Heart Study. Neurology 2020;95:e3241-e3247.
30. Johnson EL, Krauss GL, Kucharska-Newton A, et al. Dementia in late-onset epilepsy: The Atherosclerosis Risk in Communities study. Neurology 2020;95:e3248-e3256.
31. Joutsa J, Rinne JO, Hermann B, et al. Association Between Childhood-Onset Epilepsy and Amyloid Burden 5 Decades Later. JAMA Neurol 2017;74:583-590.
32. Foster E, Malpas CB, Ye K, et al. Antiepileptic drugs are not independently associated with cognitive dysfunction. Neurology 2020;94:e1051-e1061.
33. Witt JA, Werhahn KJ, Kramer G, Ruckes C, Trinka E, Helmstaedter C. Cognitive-behavioral screening in elderly patients with new-onset epilepsy before treatment. Acta Neurol Scand 2014;130:172-177.
34. Nardi Cesarini E, Babiloni C, Salvadori N, et al. Late-Onset Epilepsy With Unknown Etiology: A Pilot Study on Neuropsychological Profile, Cerebrospinal Fluid Biomarkers, and Quantitative EEG Characteristics. Front Neurol 2020;11:199.
35. Thom M, Liu JY, Thompson P, et al. Neurofibrillary tangle pathology and Braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: a post-mortem study. Brain 2011;134:2969-2981.
36. Tai XY, Koepp M, Duncan JS, et al. Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: a study of temporal lobe resections. Brain 2016;139:2441-2455.
37. Voglein J, Noachtar S, McDade E, et al. Seizures as an early symptom of autosomal dominant Alzheimer’s disease. Neurobiol Aging 2019;76:18-23.
38. Vossel K, Ranasinghe KG, Beagle AJ, et al. Effect of Levetiracetam on Cognition in Patients With Alzheimer Disease With and Without Epileptiform Activity:
A Randomized Clinical Trial. JAMA Neurol 2021;78:1345-1354.
Ein häufiger und oft banaler Befund einer Thrombozytenzahl-Erhöhung im Blut gibt hilfreiche Information zu Grunderkrankung, aber auch einen wichtigen Hinweis für eine schwere zu Grunde liegende Erkrankung. Diese müssen heute rasch diagnostiziert werden, da spezifische und auch wirksame Therapien zu Verfügung stehen. Wir wollen mit unserem Artikel auf Pathomechanismen, Symptome, Ursachen, Differenzialdiagnosen und Therapien hinweisen.
An often acquired result of elevated thrombocytes in blood turns out to be non-specific and with little consequences. However, it might be an indicator of severe conditions, that need immediate attention and specific, highly effective treatments, that reduce morbidity and mortality significantly. This article focuses on the pathomechanisms, symptoms, causes, differential diagnoses and therapies. Key Words: thrombocytes, blood, treatment
Definition und Regulation der Thrombozytenzahl
Thrombozytose bezeichnet den Zustand von >450 G/L = 109/L = 450’000 /μL echten Blutplättchen im peripheren Blut. Unter Thrombozytose wird eine reaktive (sekundäre) Ursache bezeichnet, während die Thrombozythämie für clonale (primäre) Ursachen reserviert sein muss. Allerdings differenziert die reine Thrombozytenzahl schlecht über die Ätiologie primär vs. sekundär (bei <500 G/L 88% sekundär, 12% clonal, bei <1000 G/L 82% sekundär, 18% clonal)! Mit einem einfachen (heute fast immer maschinellen) Hämatogramm erhält man Auskunft über die gemittelte Grösse (Volumen) und Grössen-Verteilung der vorliegenden Thrombozytenpopulation. Eine Pseudothrombozytose wird durch Partikel im Blut verursacht, die vom Analysengerät besonders wegen der ähnlichen Grösse und täuschender Struktur falsch klassifiziert werden. Darunter gehören: Veränderungen durch altes Probenmaterial, Fragmentierung von Erythrozyten bei Hämolysen oder Fragmentierung der Leukozyten bei Leukämien und Lymphomen, Patienten mit Verbrennungen und andere eher seltene Krankheiten wie Kryoglobulinämie. Ein simpler Blutausstrich und ein erfahrener Analytiker können das Problem meist rasch erkennen.
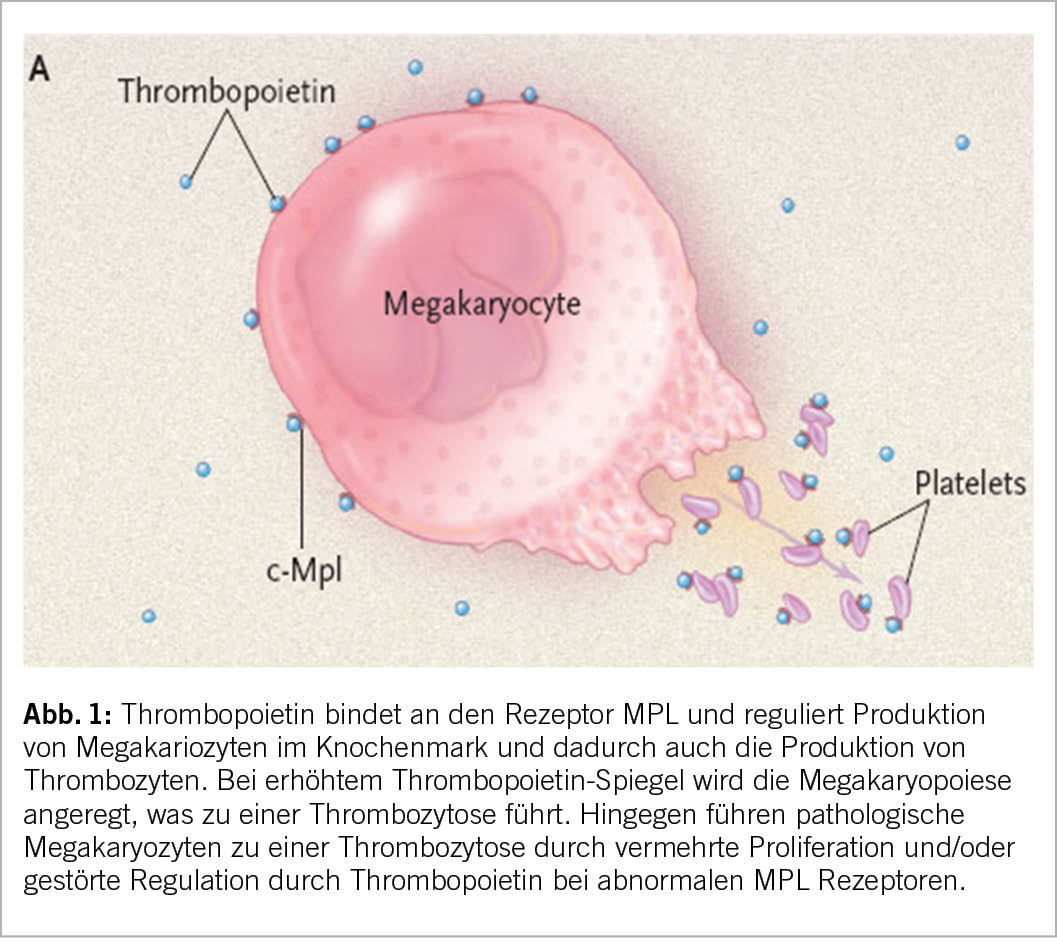
Thrombozyten entstehen aus Megakaryozyten im Knochenmark unter einem gut regulierten, primär entzündungs-hormonell gesteuerten Prozess (Abb. 1), bei welchem v.a. dazu gehören: Thrombopoetin und sein Rezeptor, aber auch Interleukine (IL-6, IL-1) und teils auch Erythropoietin. Ein erhöhter Verbrauch wird durch eine Steigerung der Megakaryozytenzahl und Verbesserung der Fragmentierung der Megakaryozyten kompensiert. Prinzipiell unterscheiden sich zwei Mechanismen der Entstehung: erhöhtes Thrombopoietin oder andere Interleukine oder eine gestörte Funktion des MPL-Rezeptors an den maligne entarteten Megakariozyten und Knochenmarks Vorstufen (Blasten).
Symptome
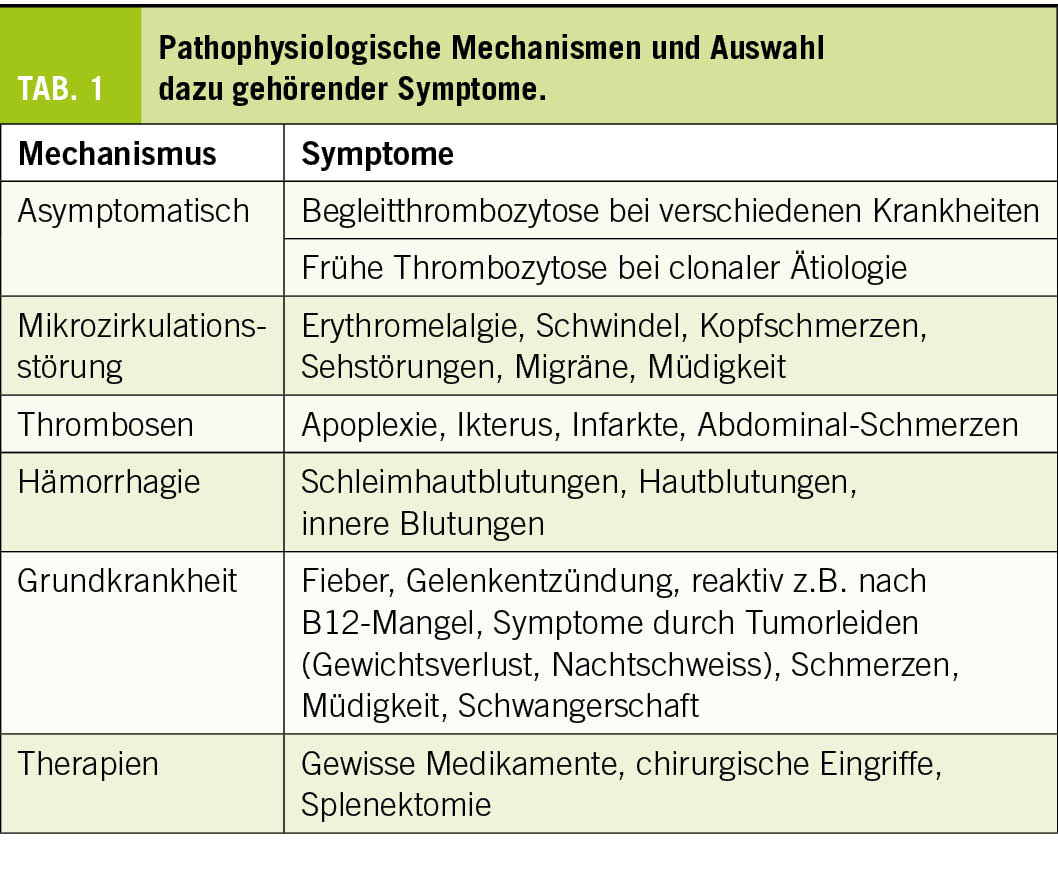
Thrombozytosen sind oft Zufallsbefunde und somit asymptomatisch. Symptome können aber auch durch Mikrozirkulationsstörungen, intravasale Thrombosen (Venen und/oder Arterien) und Blutungen bei gestörter Thrombozytenfunktion (pathologische Plättchen) entstehen. Dazu kommen Symptome und Befunde, die durch eine Grundkrankheit verursacht werden (Tab. 1).
Ursachen
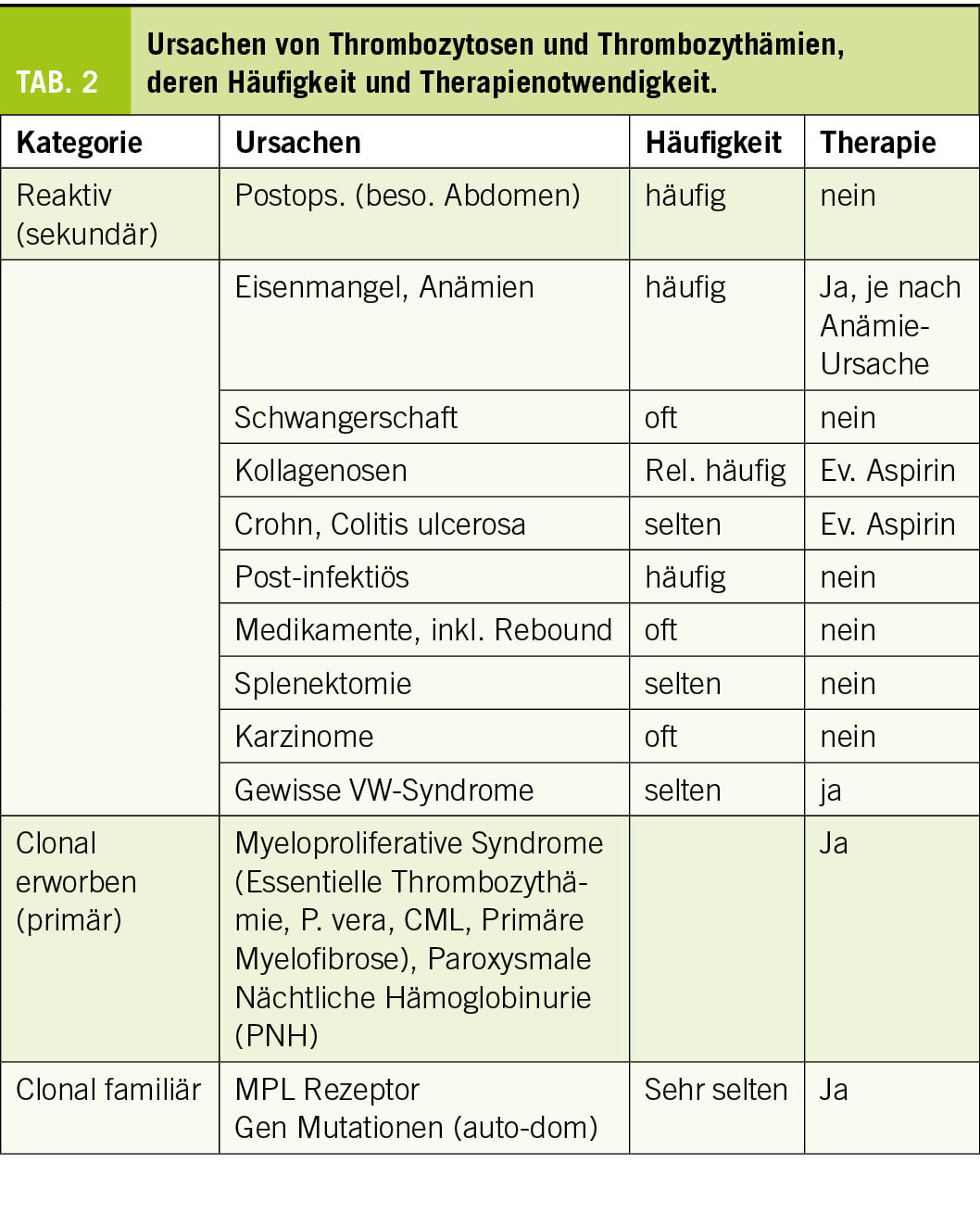
Wenn immer möglich sollte die Ursache gefunden werden, um eine Prognose stellen zu können und eine Voraussage des Nutzens einer kausalen Therapie zu ermöglichen (Tab. 2). Anamnese (allenfalls auch Familienanamnese) und klinischer Untersuch haben eine grosse Bedeutung. Ernährung, Blutungen, Entzündungen und Infekte, Begleiterkrankungen (Rheuma, Krebs, Darmerkrankung), Therapien und zeitlicher Verlauf (besonders Rebound) sind zu erwägen. Zusammen mit der Kenntnis über Prävalenzen lassen sich Posttest-Wahrscheinlichkeiten abschätzen.
Medikamentös verursachte Thrombozytosen werden öfters durch folgende Substanzen ausgelöst: Corticosteroide, Erythropoietin und als Rebound bei B12-Präparaten, Chemotherapeutika, Antibiotika. Spezifische Medikamente zur Erhöhung der Thrombozytenzahl zwecks Vermeidung von Blutungen werden unter Therapie aufgeführt.
Abklärungen zur Diagnose
Zum einen muss die Thrombozytose verifiziert werden (Ausstrich nach pathologischem Hämogramm bei Erstdiagnose) und Einbindung in ein hämatologisches Gesamtbild. Die Unterscheidung in primäre und sekundäre Ätiologie ist sehr wichtig, da ganz unterschiedliche Abklärungen und anschliessende Therapien erfolgen müssen. Bei den Abklärungen von sekundären Thrombozytosen liegt der Fokus mehr auf der Ursache und deren Behandlung als auf der Beeinflussung der Thrombozytenzahl.
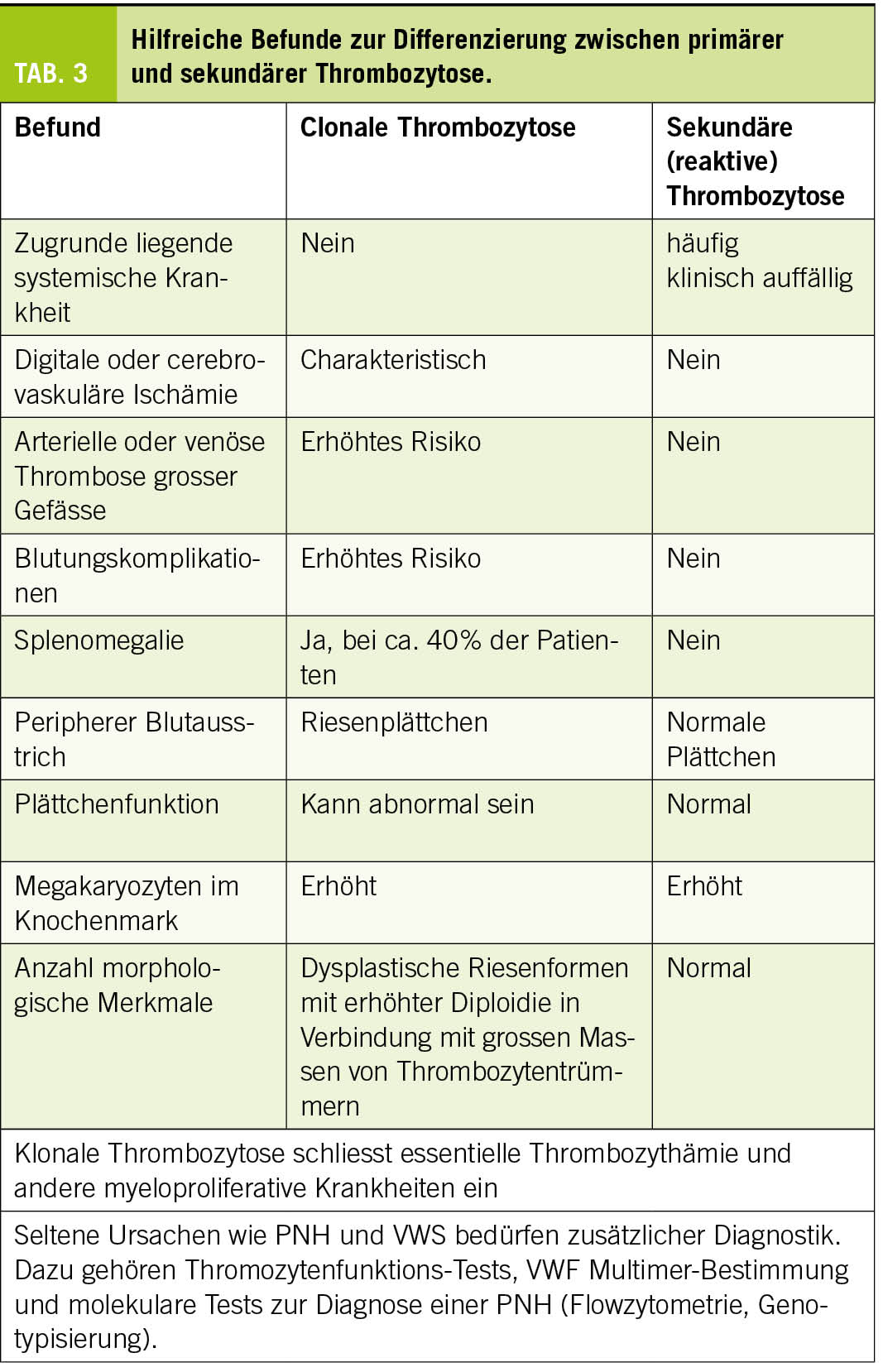
Dazu gehören komplettes Hämogramm, Ferritin, Gerinnungsmarker wie D-Dimere, Fibrinogen, Entzündungsmarker wie CRP, IL-6, Leber- und Nierenfunktion. Je nach dem auch zum Nachweis vermuteter Krankheiten spezifische Tests wie ANA, Eiweisselektrophorese, Rheumafaktoren, Stuhl Calprotectin und Infektionsmarker. Wenn eine Abklärung auf eine verdächtige Ursache hinweist, sind oft auch bildgebende Verfahren wie Röntgen, CT und Ultraschall angezeigt. Zum anderen muss die Diagnose von primären Thrombozythämien unbedingt erhärtet und differenziert werden. Es muss der clonale Defekt wie JAK-2 Mutationen, CALR, MPL, BCR-ABL Varianten und weitere Mutationen nachgewiesen oder ausgeschlossen werden. Entsprechende Therapien verbessern so Morbidität und Mortalität, da diese Krankheiten heute teils sehr effektiv behandelt werden können. Wichtige Befunde zur Differenzierung zeigt die Tab. 3.
Therapien
Asymptomatische, zufällig entdeckte sekundäre und im Rahmen einer gut definierten Grundkrankheit (postoperativ, St. n. Infekt, medikamentöse Therapie, Anämien v.a. Eisenmangel, Schwangerschaft) meist transienten Thrombozytosen bedürfen keiner Plättchen regulierender Therapie. Thrombozytenzahlen bis 1’000 g/L und darüber stellen hier keine zusätzliche Gefahr für eine Thrombose dar. Hingegen müssen primäre Plättchenerhöhungen, selbst wenn nicht so ausgeprägt, früher therapiert werden. Hierbei können spezifische antithrombozytäre Medikamente oder auch andere anti-clonale Medikamente (z.B. Tyrosin-kinase-Hemmer bei CML oder Immun- oder Chemotherapeutika bei Leukämien/Lymphomen) eingesetzt werden.
Spezifische Therapien / Medikamente
Interferone (IFN-α) und v.a. Anagrelid (Xagrid®) wie auch Hydroxyurea (Litalir®) sind bewährte Thrombozytenzahl senkende Medikamente, wobei Anagrelid auch die Funktion der Thrombozyten verbessert. Bei myeloproliferativen Syndromen, besonders der Essentiellen Thrombozythämie, werden Hydroxyurea und Anagrelid erfolgreich angewendet. Damit werden v.a. thrombotische Ereignisse und somit die Hauptursache von Mortalität und Morbidität reduziert.
Zusammenfassung
Thrombozytenzahlerhöhungen im Blut sind häufig aber sehr oft asymptomatisch und nicht entscheidend. Sie erscheinen meist transient im Rahmen einer Grunderkrankung und bedürfen keiner antithrombozytären Therapie. Sie verschwinden nach Behebung der Erkrankung. Im Gegensatz haben clonale Thrombozythämien eine klar negative Auswirkung auf Morbidität (Thrombosen und Blutungen) und Mortalität. Da sehr gute Therapien zu Verfügung stehen, muss eine Diagnose rasch erfolgen. Auch stehen gute diagnostische Methoden zu Verfügung. Bei primären Thrombozythämien sollte initial rasch ein Hämatologe beigezogen werden.
Copyright bei Aerzteverlag medinfo AG
Prof. Dr. med. Andreas Huber
Private Universität im Fürstentum Liechtenstein
Dorfstrasse 24
FL-9495 Triesen
andreas.huber@ufl.li
Der Autor hat keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
◆ Eine Thrombozytenzahl-Erhöhung wird oft nachgewiesen
◆ Die labormässige Erhebung ist heute sehr einfach, günstig und
spezifisch
◆ Es muss dringend zwischen sekundärer und primärer Thrombozytenerhöhung unterschieden werden
◆ Es stehen gute diagnostische Hilfsmittel zu Verfügung
◆ Eine frühe Diagnose einer clonalen Thrombozythämie ist äusserst wichtig, da sehr gute therapeutische Möglichkeiten bestehen
◆ Bei Thrombozythämien muss ein Hämatologe früh involviert werden
Autoimmune Lebererkrankungen gehen über die weitläufig bekannten Entitäten hinaus. Die sogenannten autoimmun-ähnlichen Hepatitiden (AIH-like) umfassen die arzneimittelinduzierte AIH-like Hepatitis sowie die Immun-Checkpoint-Inhibitor Hepatitis. Beide stellen Kliniker aufgrund Überschneidung klinischer Merkmale mit anderen Lebererkrankungen vor eine diagnostische Herausforderung. Infolge einer Polypharmazie unserer alternden Gesellschaft, respektive dem Einsatz moderner, (vor allem) onkologischer Therapieansätze, werden AIH-like Hepatitiden in Zukunft von zunehmender Bedeutung sein. Im folgenden Artikel wird deshalb besonderes Augenmerk auf die AIH-like Hepatitiden, insbesondere hinsichtlich des klinischen Erscheinungsbildes und der Diagnostik sowie der grundlegenden Therapieansätze gelegt.
Autoimmune liver diseases go beyond the widely known entities. The so-called autoimmune-like hepatitis (AIH-like) include drug-induced AIH-like hepatitis and immune checkpoint inhibitor hepatitis. Both present diagnostic challenges to clinicians due to overlap of clinical features with other liver diseases. As a result of a polypharmacy of our aging society, respectively the use of modern, (mainly) oncological therapy approaches, AIH-like hepatitis will be of increasing importance in the future. Therefore, in the following article, special attention will be paid to AIH-like hepatitis, especially with regard to clinical presentation and diagnostics as well as basic therapeutic approaches. Key Words: liver disease, hepatitis, autoimmune-like hepatitis
Unter autoimmunen Lebererkrankungen wird gemeinhin die Autoimmune Hepatitis (AIH), die primär biliäre Cholangitis (PBC) und die primär sklerosierende Cholangitis (PSC) verstanden (ein Abkürzungsverzeichnis findet sich am Ende des Artikels). Durch einen unbekannten Trigger wird ein chronisch verlaufender Entzündungsprozess ausgelöst. Hier werden im klassischen Sinne einer Autoimmunerkrankung Antikörper vermittelte Abwehrreaktionen gegen eigene Leberzellen ausgelöst. Die chronische Entzündung führt unbehandelt zu einer fortgeschrittenen Hepatopathie mit dem Bild einer Zirrhose inklusive portaler Hypertension. Ebenso durch autoimmune Prozesse vermittelt, kommt es bei der PBC zu einer chronischen Entzündung vornehmlich der kleinen Gallenwege. Die PBC ist von allen autoimmunen Hepatopathien am häufigsten mit anderen autoimmunen Erkrankungen wie Sjögren-Syndrom, Thyreoiditis, Sklerodermie und rheumatoider Arthritis vergesellschaftet.
Ebenso ist die primär sklerosierende Cholangitis (PSC) im Formenkreis der autoimmunen Hepatopathien anzusiedeln, wobei hier der Mechanismus der immun-vermittelten Erkrankung ebenso unklar ist. Diskutiert werden genetische und exogene Faktoren. Auffällig oft sind diese Erkrankungen zudem mit chronisch-entzündlichen Darmerkrankungen (CED) assoziiert.
Aus dem Formenkreis der autoimmunen Lebererkrankungen ist die am wenigsten bekannte, seltene und erst in jüngster Zeit beschriebene Entität die autoimmune Cholangiopathie, welche der PSC zwar ähnelt, aber zur Autoimmunpankreatitis Typ I/II zu zählen ist.
Obige Entitäten sind weitgehend bekannt und definiert in Hinblick auf Diagnostik, Therapie, Remissions-Erhaltung, Begleiterkrankungen, Überwachungsstrategien und eine allfällige Lebertransplantationsabklärung. Unzählige Übersichtsartikel in deutscher und englischer Sprache sind verfügbar, sodass wir in diesem Artikel den Focus auf «neuere» Differentialdiagnosen gelegt haben.
Auf Grund der ständig wachsenden Anzahl von Medikamenten und neuen Wirkmechanismen, v.a. in der Tumortherapie und bei der Behandlung von extrahepatischen Autoimmunerkrankungen, gewinnt die Gruppe der AIH-like Hepatitiden als Differentialdiagnose zunehmend an Bedeutung.
In der folgenden Übersicht möchten wir für diese Erkrankungen sensibilisieren. Auf der nächsten Seite wird eine Auswahl der verschiedenen Subgruppen zusammengefasst: Immun-Checkpoint-Inhibitor (ICI)-Hepatitis, autoimmune arzneimittelinduzierte (drug induced) Hepatitis (DI-AIH), Drug Induced Liver Injury (DILI) sowie die möglicherweise durch SARS-CoV-2-Vakzine induzierte Hepatitis.
Fallvorstellung
Ein 76-jähriger Patient mit einem metastasierten nicht-kleinzelligen Lungenkarzinom wird seit 3 Jahren mit Pembrolizumab therapiert. Das Karzinom konnte mit diesem monoklonalen Antikörper gegen PD-1-Rezeptoren innert 24 Monaten in eine komplette bildmorphologische Remission ohne metabole Aktivität im PET gebracht werden. In der Folge entwickelte sich eine Hepatitis mit einem Anstieg der Transaminasen (AST 204 U/l, ALT 325U/l) und vor allem der γ-GT (832 U/l). Klinisch bestand Fatigue, jedoch präsentierte sich der Patient nicht ikterisch und die Lebersynthesefunktion (INR, Albumin) war immer normal. Die Hepatopathie-spezifische Abklärung schloss akute und chronische virale Hepatitiden (A, B, C und E) aus. Gesamt-IgG/M zeigte normale Werte und das Autoimmunhepatitis-Panel (ANA, Anti-SMA, -LKM-1, -SLA) war ebenfalls negativ. Das Labormuster entsprach einer autoimmunen Hepatitis, bei jedoch fehlenden AK und normaler IgG-Konzentration. Eine perkutane Leberbiopsie zeigte nur subtile Veränderungen, vermutlich bei bereits seit 2 Wochen bestehender oraler Kortikosteroidtherapie von 50mg/d. Die Histologie zeigte wenig Interphasen-Hepatitis, ein Entzündungsinfiltrat und eine makrovesikuläre Steatose von 60%. Die Transaminasen normalisierten sich prompt innert Wochen, wobei es nach Stoppen der Steroidtherapie immer wieder (insgesamt 3-malig) zu neuerlichen Transaminasen-Anstiegen (=Flair) kam. Letztlich wurde eine sogenannte ICI-Hepatitis als Nebenwirkung von Pembrolizumab postuliert und das Medikament bei persistierender Tumor-Remission gestoppt. Anschliessend kam es zu keinem weiteren Flair, so dass erst durch das Stoppen von Pembrolizumab eine Antikörper-negative AIH von einer Immune Checkpoint-Inhibitor Hepatitis (ICI-Hepatitis) diskriminiert werden konnte.
Autoimmunhepatitis
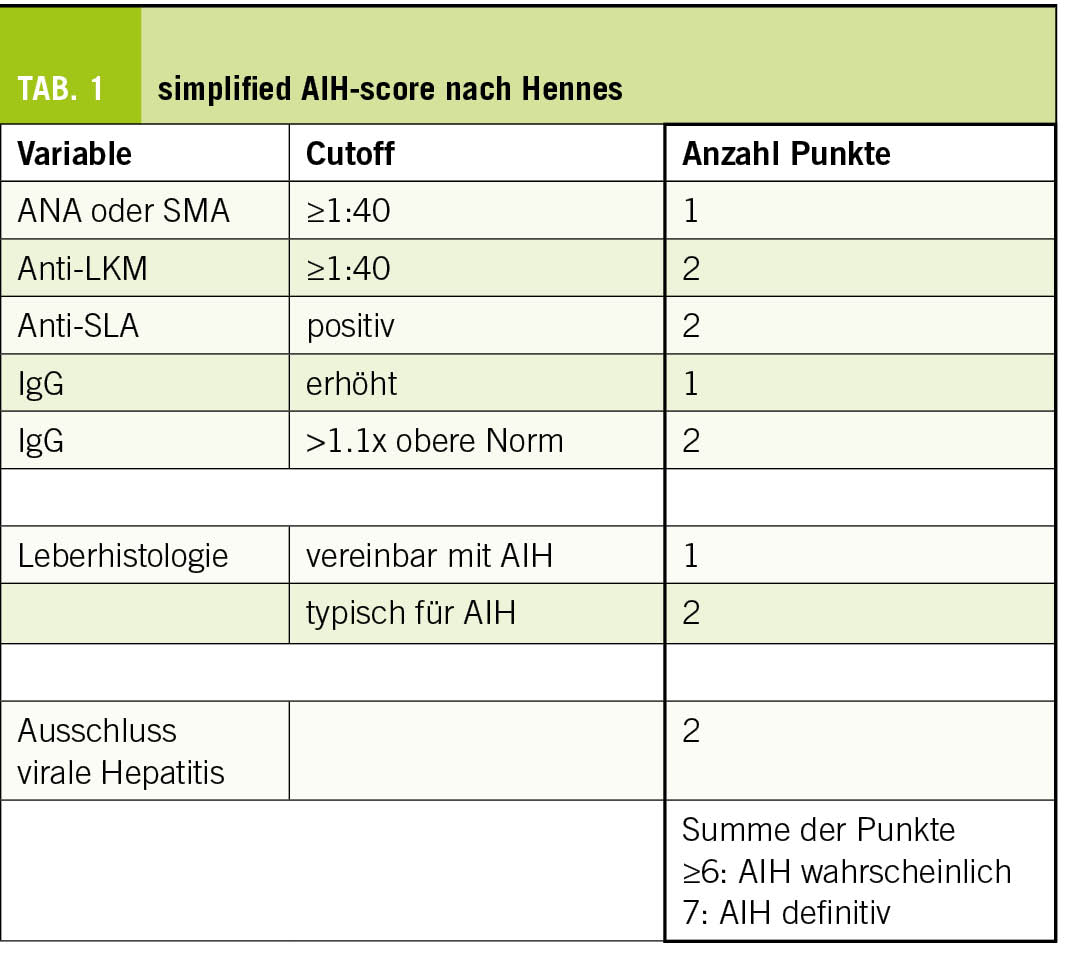
Die klassische Autoimmunhepatitis betrifft häufiger Frauen (w:m = 4:1) und weist zwei Altersgipfel auf: in der Pubertät und in der 5. - 7. Lebensdekade. Die klinischen Symptome reichen von einem asymptomatischen Verlauf bis zu schwerer Hepatitis. Je nach Antikörperprofil können drei Subtypen definiert werden, obwohl die Diagnostik und Therapie identisch sind. Eine Leberbiopsie gehört zur diagnostischen Aufarbeitung, obwohl die Histologie nicht pathognomonisch ist. Notabene gibt es seltener auch Mischbilder, sogenannte Overlap-Syndrome, mit der PBC oder PSC. Die Diagnose wird anhand von Labor und Histologie mit Hilfe eines Scores gestellt. Meist wird der vereinfachte AIH-Score nach Hennes verwendet (Tab. 1) (1). Die Therapie besteht aus einer Remissionsinduktion mit Kortikosteroiden, gefolgt von einer Remissionserhaltung mit primär Azathioprin. Zweite Wahl bei Unverträglichkeit kann Mycophenolat-Mofetil sein. Diese Therapie muss langfristig (häufig lebenslang), aber mindestens 4-5 Jahre eingenommen werden. Ein Relapse ist sehr häufig (>80%) nach Absetzen. AIH-Patienten bedürfen somit einer lebenslangen hepatologischen Betreuung mit 2-4 Kontrollen pro Jahr in Remission.
AIH-like Hepatitiden
a. Arzneimittelinduzierte autoimmunähnliche Hepatitis
Die arzneimittelinduzierte (drug induced) autoimmunähnliche Hepatitis (DI-AIH) kann das vorherrschende Muster einer idiosynkratischen medikamentösen Leberschädigung (Englisch: Drug induced liver injury, DILI) sein (2). Es wird geschätzt, dass 2% bis 18% der AIH-Fälle und rund 3% bis 9% aller DILI auf eine DI-AIH zurückzuführen sind (3, 4). Bei einer idiosynkratischen DILI handelt es sich um eine unerwünschte Reaktion der Leber, welche aufgrund der pharmakologischen Wirkung des verabreichten Arzneimittels unerwartet ist. Die Latenzzeit ist variabel und beträgt im Falle der DI-AIH oft zwei Monate oder mehr (5). Vom idiosynkratischen DILI unterschieden wird die intrinsische DILI, bei der man von einer direkten Hepatotoxizität ausgeht. Das auslösende Agens verursacht dabei reproduzierbar und vorhersehbar eine dosisabhängige DILI. Die Latenzzeit ist dabei kurz und oft unmittelbar. Ein Beispiel hierfür ist Paracetamol. Diese Einteilung ist nicht nur akademischer Natur, da sich hierdurch eine Einschätzung der Plausibilität über ein mögliches auslösendes Agens machen lässt. Hilfreich sind hierbei Datenbanken, z.B. LiverTox® (5).
Prädisponierende Faktoren und auslösende Medikamente
Es wird davon ausgegangen, dass reaktive Metaboliten, die beim hepatischen Stoffwechsel einiger Arzneimittel entstehen, an zelluläre Proteine wie Cytochrom P450 binden. Diese können dann vom Immunsystem als Neoantigene erkannt werden und eine DI-AIH auslösen (4). Begünstigende Faktoren sind die Dosierung und Pharmakokinetik des auslösenden Medikamentes, genetische Polymorphismen (z.B. CYP, HLA-Allele usw.), Interaktionen mit anderen Medikamenten, Alkohol, demographische Faktoren (Alter, Geschlecht) und zugrunde liegende Lebererkrankungen (2-4, 6).
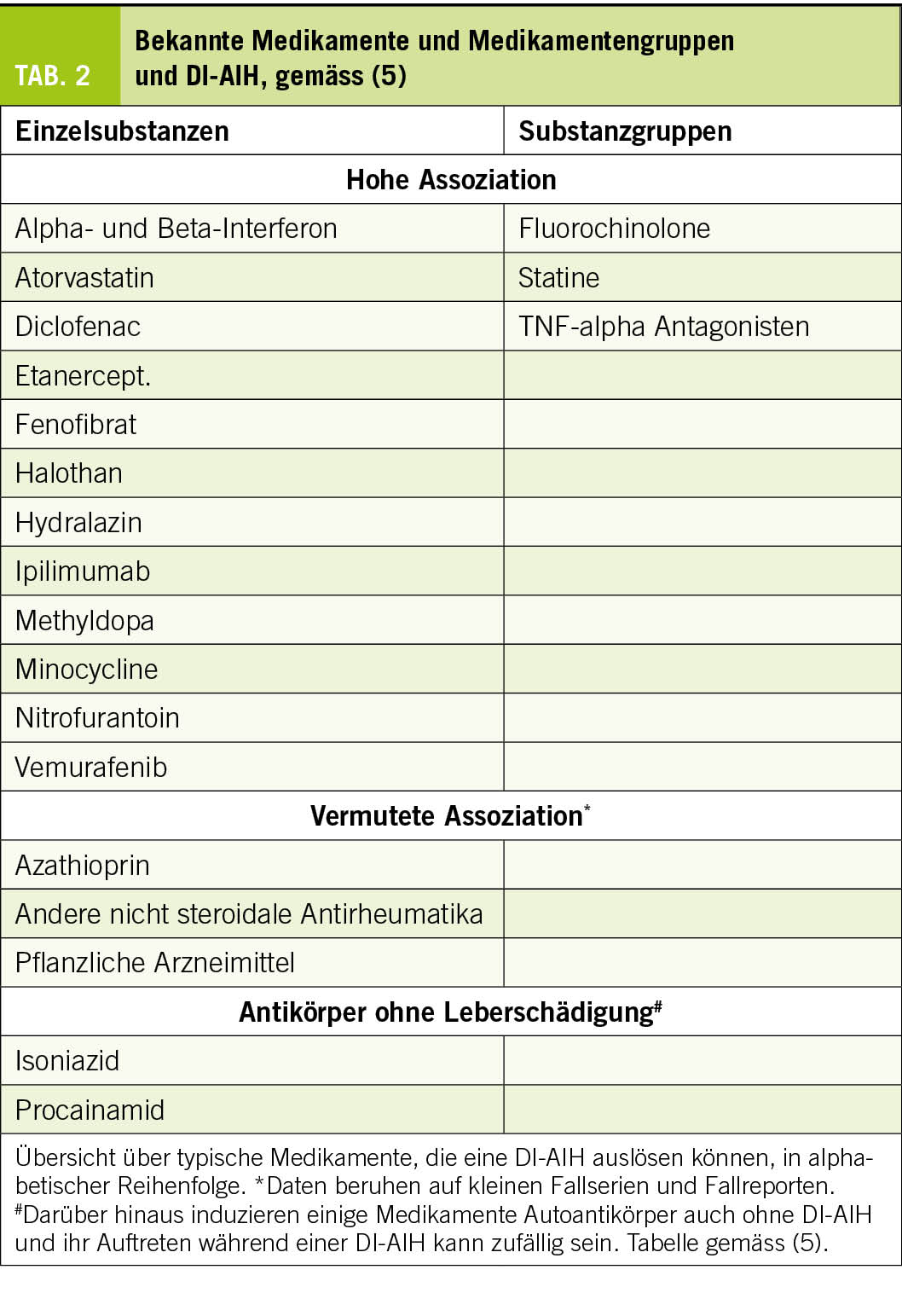
Eine steigende Anzahl an Medikamenten wird mit einer DI-AIH in Verbindung gebracht. Dabei wird zwischen Medikamenten mit hoher Assoziation, z.B. Minocyclin, Nitrofurantoin, Infliximab, Atorvastatin, Diclofenac, vermuteter, z.B. Rosuvastatin, Etanercept und möglicher Assoziation unterschieden (Tab. 2) (5, 7).
Symptomatik
Eine DI-AIH ist anhand der Klinik oder Serologie nicht von einer «klassischen» AIH zu unterscheiden (4). Bei mehr als der Hälfte der DI-AIH liegt eine akute Leberschädigung vor, die in 70-75% der Fälle mit einem Ikterus einhergeht. Die Patienten weisen erhöhte Aminotransferasewerte (ALT, AST) häufig zusammen mit erhöhten Gammaglobulinwerten (Gesamt IgG) auf. In 70-80% der Fälle finden sich antinukleäre Antikörper (ANA) und/oder Antikörper gegen glatte Muskulatur (anti-SMA, 50% der Fälle) oder Antikörper gegen Leber-Nieren-Mikrosomen (anti-LKM) (4). Letztlich unterscheidet nur das Ausbleiben eines Rückfalls (bei klassischer AIH >80% der Fälle) nach Abklingen der Leberschädigung mit oder ohne immunsuppressive Therapie die DI-AH von der AIH, wobei auch bei der DI-AIH, wenn auch seltener, Rückfälle auftreten (siehe Fallvorstellung zu Beginn des Artikels).
Komplizierend kommt hinzu, dass einzelne Wirkstoffe ein oder mehrere Schädigungsmuster hervorrufen können. Die Schädigungsmuster werden anhand des R-Wertes in ein hepatozelluläres (R>5), cholestatisches (R<2) und ein gemischtes Schädigungsmuster unterschieden. Der R-Wert beschreibt das Verhältnis der ALT zur alkalischen Phosphatase (AP) im Verhältnis zum jeweiligen oberen Grenzwert (ULN) und wird nach der Formel: (ALT/ALT ULN) ÷ (AP/ AP ULN), berechnet. Diese Formel kann in der Praxis angewandt werden, um ein Schädigungsmuster bei einem Patienten mit dem zu erwartenden Muster eines bestimmten Medikamentes zu vergleichen und damit eine Kausalität zu erhärten. Bei Patienten, bei denen eine DI-AIH plausibel ist, aber mehrere differentialdiagnostische Erklärungen existieren, kann mit dem R-Wert zudem die Differentialdiagnose eingegrenzt werden (8).
Die DI-AIH bzw. AIH sollten bei Patienten mit einem hepatozellulären Muster (R>5) als Differenzialdiagnose in Betracht gezogen werden, da eine DI-AIH nur in zirka 8% der Fälle mit einem cholestatischen oder gemischten Schädigungsmuster verbunden ist (Tab. 3) (9).
Differentialdiagnostisches Vorgehen
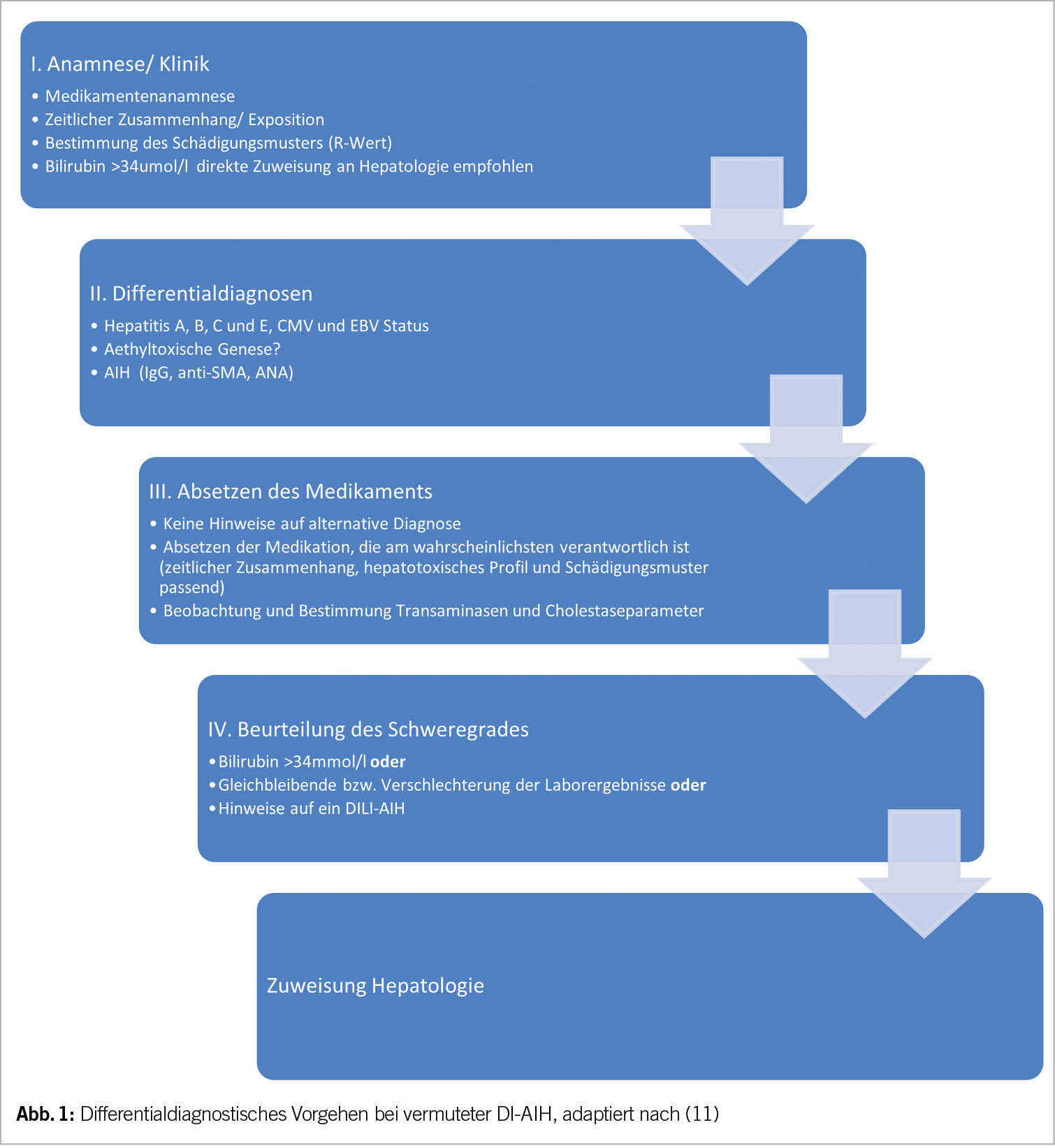
Es existieren keine etablierten diagnostischen Kriterien für die DI-AIH. Aufgrund der Ähnlichkeit mit der AIH sollen jedoch die folgenden Elemente für die Diagnose hervorgehoben werden (Tab. 3 und Abb. 1).
Eine ausführliche Medikamentenanamnese mit Schwerpunkt auf die jüngste Medikamentenexposition, einschliesslich komplementärer alternativer Arzneimittel, ist unerlässlich. Die Latenzzeit der Medikamentenexposition bei DI-AIH ist in der Regel länger als bei anderen Arten von DILI, in einigen Fällen mit einer Latenzzeit von mehr als einem Jahr, z.B. bei Nitrofurantoin und Minocyclin.
Eine Seropositivität für ANA, anti-SMA, anti-LKM und/oder ein erhöhtes IgG im Serum deutet auf eine DI-AIH hin. Allerdings trifft dies nicht für alle Patienten zu, da ein Teil der Patienten mit DI-AIH keine nachweisbaren AIH-Antikörper aufweist (3, 9). Bei Vorliegen von AIH-Antikörpern oder erhöhtem IgG ist ein AIH-Scoring, z.B. mittels «simplified AIH score» sinnvoll (Tab. 1).
Bewertungsinstrumente für die Kausalität können verwendet werden, um die Stärke des Zusammenhangs zwischen Arzneimittelexposition und klinischer Manifestation zu ermitteln, z.B. Roussel Uclaf Causality Assessment Method (RUCAM) (10).
Die Leberbiopsie ist relevant für die Diagnose der DI-AIH, v.a. um diese gegen eine DILI abzugrenzen. Sie sollte in Betracht gezogen werden, wenn sich eine nicht abklingende oder sich verschlimmernde Leberschädigung trotz Absetzen des Medikamentes zeigt oder wenn eine Seropositivität für AIH-Antikörper, ein erhöhtes IgG oder eine mögliche AIH auf der Grundlage des «simplified AIH score» besteht (7). Gegenüber der AIH ist eine histologische Abgrenzung schwierig. Der histologisch fehlende Nachweis einer Fibrosierung oder Zirrhose kann Hinweis auf das Vorliegen einer DI-AIH sein.
Therapie
Der erste Schritt bei der Behandlung der DI-AIH ist zunächst das Absetzen des ursächlichen Medikaments. In den bisher veröffentlichten Publikationen zu diesem Thema war dies neben dem rückfallfreien Verlauf das wichtigste Unterscheidungsmerkmal zwischen DI-AIH und AIH (7). Sollte dies nicht zum Abklingen der Symptomatik und zum Rückgang der Transaminasen und AIH-Seropositivität führen oder ein schwerwiegender Verlauf vorliegen, werden in der Regel Kortikosteroide analog zur Behandlung der AIH eingesetzt (11).
Prognose
Die Prognose einer DILI-AIH ist gut, wobei prinzipiell eine cholestatische DILI eine bessere Prognose hat. Bei einem Bilirubinwert >2 ULN sollte hepatologischer Rat eingeholt werden, da eine erhöhte Mortalität besteht (11). Sowohl DI-AIH als auch AIH sprechen hervorragend auf Kortikosteroide an, wobei die Remissionsrate bei mehr als 90% liegt und bei der DI-AIH ein rascheres Ansprechen innerhalb von vier Wochen mit einem Rückgang der ALT um 50% in den ersten zwei Wochen erwartet werden kann (5, 7). Bei 30-35% der Patienten mit anfänglicher AIH-Seropositivität besteht diese bei der DI-AIH unabhängig von der Behandlung mit Kortikosteroiden nach sechs Monaten nicht mehr (3). Es besteht jedoch kein Konsens über die Behandlungsdauer. Eine Studie hat gezeigt, dass das Absetzen der Immunsuppressiva innerhalb von 3-17 Monaten ohne Anzeichen eines Rückfalls erfolgreich war (7).
b. Immun-Checkpoint-Inhibitor-Hepatitis (ICI-Hepatitis)
Die Anwendung von Immun-Checkpoint-Inhibitoren (ICI) bei fortgeschrittenen Krebserkrankungen hat in den letzten zehn Jahren stark zugenommen. Die Indikationen für ICI erweitern sich ständig. Aufgrund ihrer Wirkung auf das menschliche Immunsystem führen ICI jedoch zu immunbedingten unerwünschten Ereignissen (irAE), die bei herkömmlichen Chemotherapien nicht beobachtet wurden (12). Mit der zunehmenden Anwendung haben sich diese Ereignisse, so auch die Immun-Checkpoint-Inhibitor-bedingte Hepatitis (ICH), zu einem klinischen Problem entwickelt. Die Inzidenz der ICH schwankt zwischen 1% und 20% (13).
ICI stellen die Funktion der T-Zellen gegen tumorspezifische Antigene wieder her, indem sie den «programmed death-1 receptor -1-Rezeptor» (PD-1), seinen Liganden (PD-L1) oder das zytotoxische T-Lymphozyten-assoziierte Antigen 4 (CTLA-4) blockieren. Dabei hängt die Entwicklung einer ICH von der Dosis, der Art und der Dauer der Immuntherapie ab. Die ICH tritt meist 4-6 Wochen nach dem Beginn der ICI-Therapie auf (13). Die Häufigkeit einer fulminanten Hepatitis wird mit <1% angegeben (14).
Diagnose
Ein Verdacht auf eine ICH besteht bei einer Erhöhung der Aminotransferasewerte unter ICI-Therapie. Es besteht meist ein hepatozelluläres Schädigungsmuster. In der Mehrzahl der Fälle besteht keine Symptomatik, in schwerwiegenderen Fällen entsteht jedoch eine Hepatopathie typische Symptomatik mit Fatigue, Ikterus oder unspezifischen Abdominalschmerzen und Erbrechen. Die Diagnose einer ICH kann eine Herausforderung darstellen, da sich die klinischen Merkmale mit anderen Lebererkrankungen überschneiden und somit der Ausschluss einer DILI, einer Virushepatitis, einer AIH und einer metastasierten Lebererkrankung erforderlich ist. Entsprechend wird eine serologische und bildgebende Diagnostik empfohlen (15).
Die Leberhistologie ist wichtig, um den Schweregrad der Lebergewebeschäden zu beurteilen und Differentialdiagnosen auszuschliessen (16). Dies ist z.B. im Fall von Nivolumab, das sowohl eine DI-AIH als auch eine ICH auslösen kann, sinnvoll.
Therapie
Der Schweregrad der ICH kann auf der Grundlage von zwei Skalen kategorisiert werden (15, 17), die eine Kombination von Leberenzymen, Bilirubin und Gerinnungsuntersuchungen verwenden. Je nach Schweregrad der ICH erfolgt eine Fortführung der Medikation unter engmaschiger Kontrolle der Transaminasen (Grad 1: asymptomatisch, AST o. ALT u./o. Bilirubin ≥ ULN), ein Pausieren der ICI-Therapie und eine Behandlung mit oralen Glukokortikoiden kann in Betracht gezogen werden, wenn die Leberschädigung 3-5 Tage nach Beendigung der Therapie fortbesteht oder signifikante klinische Symptome auftreten (Grad 2: asymptomatisch, AST o. ALT ≥3 ULN u./o. Bilirubin ≥1.5 ULN). Die meisten Patienten mit ICH erholen sich und können die ICI-Therapie sicher wiederaufnehmen.
Bei einer drittgradigen ICH (symptomatisch, AST o. ALT ≥5 ULN u./o. Bilirubin ≥3 ULN) müssen die ICI pausiert und eine Glukokortikoid-Therapie begonnen werden. Eine Wiederaufnahme derselben ICI-Therapie ist von Fall zu Fall möglich. Eine ICH Grad 4 (symptomatisch, AST o. ALT ≥20 ULN u./o. Bilirubin ≥10 ULN) rechtfertigt eine dauerhafte Unterbrechung der ICI-Therapie, die Verabreichung von hochdosierten Glukokortikoiden und bei steroidrefraktären Fällen die Gabe von Mycophenolat-Mofetil oder Tacrolimus. Zudem ist eine engmaschige Kontrolle auf (opportunistische) Infektion notwendig (15, 17). Ein Wechsel der ICI-Therapie auf eine andere Checkpoint-Inhibitor-Therapie ist erfolgsversprechend, da es sich bei der ICH häufig um eine substanzspezifische Nebenwirkung handelt.
Da eine schwere ICH allerdings sehr selten ist, gibt es auch Autoren, die die Meinung vertreten, dass durch diesen Ansatz medizinisch nicht indizierte hohe Dosen von Glukokortikoiden verabreicht werden (16). Eine ICH sollte daher von einem erfahrenen multidisziplinären Team behandelt werden.
c. SARS-CoV-2-Impfung induzierte Hepatitis?
Nach verschiedenen SARS-CoV-2-Impfungen wurden Neu-Manifestationen oder eine Verstärkung vorbestehender Autoimmunerkrankungen beobachtet (18). Neben den während des Zulassungsprozesses beschriebenen seltenen Nebenwirkungen, wie der immunogenen thrombotischen Thrombozytopenie, Myokarditis oder Anaphylaxie, wurde beobachtet, dass mRNA-Impfstoffe den Interferon-Signalweg aktivieren können, sodass die Vermutung besteht, dass hierüber verschiedene Immunphänomene ausgelöst werden könnten (19). Der Anteil an Patienten mit einem Anstieg der Aminotransferasen wird mit <1% angegeben. Wie bei der DILI-AIH finden sich eine Seropositivität für ANA, anti-SMA, anti-LKM und ein erhöhtes IgG (20).
Die akute Hepatitis trat im Median 24 Tage nach der Impfung und in der Mehrzahl der Fälle nach der zweiten Dosis auf. Patienten mit Status nach mRNA-Impfung wiesen dabei eine stärkere Transaminasenerhöhung auf. Eine fulminante Hepatitis wurde bei einem Patienten beobachtet. (vergleiche Abschnitt «a. Arzneimittelinduzierte autoimmunähnliche Hepatitis») (18). Die meisten Patienten wurden mit Steroiden mit oder ohne Azathioprin behandelt. In zweidrittel der Fälle zeigte sich eine Normalisierung der Leberenzyme nach sechs Monaten (18).
Bisher kann ein kausaler Zusammenhang zwischen SARS-CoV-2-Impfstoffen und Leberschäden mit autoimmunen Merkmalen weder bewiesen noch widerlegt werden. Wenn ein hoher Anteil der Weltbevölkerung innerhalb weniger Monate geimpft wird, steigt die Wahrscheinlichkeit, dass seltene Erkrankungen wie die AIH oder DI-AIH (aufgrund anderer Medikamente) zufällig in zeitlicher Nähe zu einer SARS-CoV-2-Impfung diagnostiziert werden. Durch phasenweise bestehende Überlastung der Gesundheitssysteme wird eine von der Impfung unabhängige Verschlechterung einer bestehenden Autoimmunerkrankung möglicherweise übersehen oder eine Erstdiagnose zu spät gestellt. Dies wäre z.B. eine Erklärung für die niedrigere Anzahl an neu diagnostizierten AIH während der Pandemie in Deutschland (21).
Entsprechend der Empfehlungen bei der DI-AIH und ICH ist aufgrund der aktuellen Datenlage eine gründliche und sorgfältige Nachsorge der Patienten und bei schweren Verläufen eine frühzeitige hepatologische Beurteilung indiziert.
Primär biliäre Cholangitis und primär sklerosierende Cholangitis
Die primär biliäre Cholangitis (PBC) und die primär sklerosierende Cholangitis (PSC) sind seltene autoimmune Lebererkrankungen. Pathophysiologisch kommt es bei der PBC zu einer progredienten Entzündung der kleinen, intrahepatischen Gallengängen, wohingegen bei der PSC die grösseren, sowohl intra- als auch extrahepatischen Gallengänge betroffen sind. Beiden Entitäten gemeinsam ist der chronisch-progrediente Charakter, der im Endstadium zu einer Leberzirrhose führen kann.
Die Ätiologie beider Erkrankungen ist bislang unzureichend verstanden. Im Gegensatz zur PSC (w:m = 1:2), tritt die PBC überwiegend bei Frauen mittleren Alters auf. Die PBC tritt gehäuft mit anderen Autoimmunerkrankungen, wie z.B. der Zöliakie, der rheumatoiden Arthritis oder einer Thyreopathie auf. Nach Diagnose einer PSC gilt es eine gleichzeitig bestehende chronisch entzündliche Darmerkrankung auszuschliessen.
Sowohl die PBC als auch die PSC fallen klinisch durch Hepatopathie-spezifische Beschwerden und laborchemisch durch eine cholestatische Hepatopathie auf. Bei entsprechendem Verdacht kann die PBC mittels Bestimmung der antimitochondrialen Antikörper (AMA; im Speziellen AMA-M2) diagnostiziert werden. Bei der Diagnostik der PSC ist die Bestimmung von pANCA hilfreich, wobei die Diagnose letztlich anhand typischer Kontrastmittelanreicherungen in der Magnetresonanz-Cholangiopankreatikographie (MRCP) und/oder der endoskopischen retrograden Cholangiopankreatikographie (ERCP) gestellt werden kann.
Die therapeutischen Ansätze sind sowohl bei PBC als auch bei PSC eingeschränkt. Im Fall der PBC besteht die Indikation zur gewichtsadaptierten Therapie mit Ursodeoxycholsäure (UDCA), durch welche sowohl Symptome als auch die cholestatischen Laborveränderungen positiv beeinflusst werden können. Im Gegensatz zur PSC, verzögert eine frühzeitige UDCA-Therapie bei der PBC zudem eine Fibrosierung. Im Falle der PSC werden dominante Stenosen interventionell behandelt. Im Endstadium bedarf es bei beiden Erkrankungen häufig einer Lebertransplantation.
Die Autoren haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
◆ Die klassische Autoimmunhepatitis bedarf der Bestimmung des
Antikörper-Panels, IgG und einer Leberbiopsie.
◆ Kortison bleibt das Medikament der Wahl zur Induktion der Remission.
◆ AIH-like Hepatitiden treten zunehmend häufiger auf.
◆ Immun-Checkpoint-Inhibitoren, Statine und TNF-alpha-AK sind
nachgewiesene Auslöser der AIH-like Hepatitiden.
1. Hennes, E.M., et al., Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology, 2008. 48(1): p. 169-76.
2. Aithal, G.P., et al., Case definition and phenotype standardization in drug-induced liver injury. Clin Pharmacol Ther, 2011. 89(6): p. 806-15.
3. de Boer, Y.S., et al., Features of Autoimmune Hepatitis in Patients With Drug-induced Liver Injury. Clin Gastroenterol Hepatol, 2017. 15(1): p. 103-112.e2.
4. Bjornsson, E., et al., Drug-induced autoimmune hepatitis: clinical characteristics and prognosis. Hepatology, 2010. 51(6): p. 2040-8.
5. in LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. 2012: Bethesda (MD).
6. Castiella, A., et al., Drug-induced autoimmune liver disease: A diagnostic dilemma of an increasingly reported disease. World J Hepatol, 2014. 6(4): p. 160-8.
7. Tan, C.K., et al., Drug-induced autoimmune hepatitis: A minireview. World J Gastroenterol, 2022. 28(24): p. 2654-2666.
8. Bénichou, C., Criteria of drug-induced liver disorders. Report of an international consensus meeting. J Hepatol, 1990. 11(2): p. 272-6.
9. Licata, A., et al., Clinical features and outcomes of patients with drug-induced autoimmune hepatitis: a retrospective cohort study. Dig Liver Dis, 2014. 46(12): p. 1116-20.
10. Danan, G. and C. Benichou, Causality assessment of adverse reactions to drugs–I. A novel method based on the conclusions of international consensus meetings: application to drug-induced liver injuries. J Clin Epidemiol, 1993. 46(11): p. 1323-30.
11. Leise, M.D., J.J. Poterucha, and J.A. Talwalkar, Drug-induced liver injury. Mayo Clin Proc, 2014. 89(1): p. 95-106.
12. Nishida, N. and M. Kudo, Liver damage related to immune checkpoint inhibitors. Hepatol Int, 2019. 13(3): p. 248-252.
13. Bolte, F.J., R.D. Hall, and N.L. Shah, Immune checkpoint inhibitor–related liver toxicity. Clinical Liver Disease, 2022. 20(3): p. 93-96.
14. Wang, D.Y., et al., Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol, 2018. 4(12): p. 1721-1728.
15. Schneider, B.J., et al., Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: ASCO Guideline Update. J Clin Oncol, 2021. 39(36): p. 4073-4126.
16. De Martin, E., et al., Liver toxicity as a limiting factor to the increasing use of immune checkpoint inhibitors. JHEP Rep, 2020. 2(6): p. 100170.
17. Haanen, J., et al., Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol, 2018. 29(Suppl 4): p. iv264-iv266.
18. Codoni, G., et al., Histological and serological features of acute liver injury after SARS-CoV-2 vaccination. JHEP Rep, 2023. 5(1): p. 100605.
19. Teijaro, J.R. and D.L. Farber, COVID-19 vaccines: modes of immune activation and future challenges. Nature Reviews Immunology, 2021. 21(4): p. 195-197.
20. Guardiola, J., et al., Unexplained liver test elevations after SARS-CoV-2 vaccination. J Hepatol, 2022. 77(1): p. 251-253.
21. Rüther, D.F., et al., Autoimmune hepatitis and COVID-19: No increased risk for AIH after vaccination but reduced care. Journal of Hepatology, 2022. 77(1): p. 250-251.
Die Zöliakie ist eine durch das Immunsystem vermittelte immunologisch vermittelte, lebenslange, entzündliche Erkrankung des Dünndarms. Zöliakie-Betroffene haben eine genetische Prädisposition und die Erkrankung hat mit sowohl intestinalen als auch extraintestinalen Symptomen eine sehr grosse Variabilität, weshalb die Erkrankung auch als das «Chamäleon der Gastroenterologie» bezeichnet wird. Die Erkrankung betrifft ca. 1% der Bevölkerung und bestimmte Risikopopulationen haben eine höhere Prävalenz der Zöliakie, dazu zählen u.a. Verwandte ersten und zweiten Grades mit Zöliakie, Typ-1-Diabetes, autoimmuner Thyreoiditis sowie Down- und Turner-Syndrom. Die Diagnose der Zöliakie beruht auf der Anamnese, der klinischen Untersuchung, Serologie und der Histologie aus dem Duodenum. Die einzig bisher verfügbare Therapie ist eine strikte lebenslange glutenfreie Ernährung.
Celiac disease is an immune-mediated, lifelong, inflammatory disease of the small intestine. Celiac disease sufferers have a genetic predisposition and the disease has a wide variability with both intestinal and extraintestinal symptoms which is why the disease is also referred to as the «chameleon of gastroenterology». The disease affects approximately 1% of the population, and certain at-risk populations have a higher prevalence of celiac disease, including first- and second-degree relatives with celiac disease, type 1 diabetes, autoimmune thyroiditis, and Down syndrome and Turner syndrome. The diagnosis of celiac disease is based on history, clinical examination, serology, and histology from the duodenum. The only therapy available to date is a strict lifelong gluten-free diet. Key Words: Celiac disease, gastroenterology, genetic predisposition
Die Zöliakie ist eine lebenslange, immunologisch vermittelte, chronisch entzündliche Erkrankung des Dünndarms. Gluten ist eine Proteinkomponente von verschiedenen Getreidesorten, wozu unter anderem Roggen, Weizen, Gerste und Dinkel zählen. Die Aufnahme von Gluten führt bei der Erkrankung zu einer Schädigung des Dünndarms, welche durch Schleimhautentzündung, Kryptenhyperplasie und Zottenatrophie gekennzeichnet ist. Die Erkrankung betrifft genetisch prädisponierte Individuen (1) und der Dünndarmschaden kann im Verlauf zu einer Malabsorption von Nährstoffen und damit verbundenen Komplikationen führen (2).
Die erste neuere Beschreibung der Zöliakie geht auf das Jahr 1888 zurück durch den englischen Arzt Samuel Gee. Erste Einblicke in die Pathogenese der Erkrankung wurden während des Zweiten Weltkrieges durch den Arzt Willem K. Dicke ermöglicht. Er beobachtete, dass sich der Zustand von Patienten mit chronischer Diarrhoe verbesserte, wenn in Zeiten der Nahrungsmittelknappheit Brot durch nicht weizenhaltige Lebensmittel wie Kartoffeln ersetzt wurde (3). Die ersten Beschreibungen des durch Gluten vermittelten Dünndarmschadens erfolgten 1954 durch JW Paulley (4). Lange Zeit galt die Zöliakie als eine seltene Kindererkrankung, im Verlauf wurde jedoch eine zunehmende Verschiebung des Diagnosealters in Richtung Erwachsenenalter beobachtet (5). Durch eine verbesserte Diagnostik der Erkrankung mit Entwicklung von Nachweismethoden für Endomysium- und Transglutaminase-Antikörpern kam es zu einer signifikanten Zunahme der Diagnose der Zöliakie in den 1980er und 1990er Jahren.
Epidemiologie
Durch die grosse Variabilität des klinischen Erscheinungsbildes und oft nur milden Symptomen kam es in der Vergangenheit zu einer Unterdiagnose der Erkrankung. Durch die Entwicklung serologischer Tests konnte die Einschätzung der Inzidenz und Prävalenz der Erkrankung jedoch stark verbessert werden (6, 7). In einer finnischen Studie konnte eine Prävalenz bei Schulkindern von 1% nachgewiesen werden (8). Bei Erwachsenen wurden in den USA und in europäischen Ländern ähnliche Prävalenzen festgestellt (9-13).
Es konnte ein deutlicher Anstieg der Prävalenz der Zöliakie in den letzten Jahrzehnten beobachtet werden. In einer finnischen Studie liess sich über ungefähr 20 Jahre eine Verdopplung der Prävalenz nachweisen. In den USA zeigte sich eine 4- bis 4,5-fache Zunahme der Prävalenz über die letzten 50 Jahre (14-15).
Galt die Erkrankung früher in Asien und Afrika als selten, kam es in diesen Regionen zu einer steigenden Prävalenz bedingt unter anderem durch die Globalisierung des Weltmarktes, welcher zu einer Änderung der Ernährungsgewohnheiten in den Entwicklungsländern geführt hat. Während früher die Ernährung in diesen Ländern auf glutenfreien Getreidesorten wie Reis und Mais beruhte, wurden mit der Zeit zunehmend weizenhaltige Lebensmittel in die Ernährung integriert (16).
Bei bestimmten Risikopopulationen wie z.B. Typ-1-Diabetes, autoimmune Thyreoiditis, Verwandte ersten und zweiten Grades mit Zöliakie sowie Down- und Turner-Syndrom ist eine höhere Prävalenz für Zöliakie zu verzeichnen (17).
Im Vergleich mit Erwachsenen kommt es bei Kindern mit Zöliakie häufiger zu Diarrhoe, Bauchschmerzen und Zeichen der Malabsorption und es ist eine Wachstumsverzögerung zu beobachten (18). Bei erwachsenen Zöliakie-Patienten kommt es eher zu extraintestinalen Manifestationen der Erkrankung wie Eisenmangelanämie und Osteoporose und seltener zu Darmsymptomen.
Bei der Geschlechterverteilung ist eine weibliche Dominanz zu verzeichnen von ca. 2-3 : 1 (19). Des Weiteren ist bei Frauen die Wahrscheinlichkeit einer symptomatischen Zöliakie höher als bei männlichen Zöliakie-Patienten (20).
Pathogenese
Der Hauptfaktor bei der Pathogenese der Zöliakie ist die Exposition gegenüber Gluten. Daneben werden weitere umweltbedingte und genetische Faktoren in der Pathogenese der Zöliakie unterschieden.
Die adaptive als auch die angeborene Immunantwort führen zu den pathologischen Veränderungen des Dünndarms, die durch intraepitheliale Lymphozytose, Kryptenhyperplasie und Zottenatrophie gekennzeichnet sind.
Bei der Pathogenese der Zöliakie spielt die angeborene Immunantwort eine entscheidende Rolle. Diese spielt sich im Epithel der Darmschleimhaut ab und führt unter anderem zu einer erhöhten Produktion von Zytokinen. Im Rahmen dieser Immunreaktion differenzieren sich die intraepithelialen Lymphozyten dann zu zytotoxischen CD8+-T-Zellen (21).
Neben der angeboreren Immunantwort spielt auch die adaptive Immunantwort eine wichtige Rolle. Dadurch, dass Gluten im Gastrointestinaltrakt nicht vollständig enzymatisch gespalten wird, kommt es zu einer adaptiven Immunantwort. Das primäre Autoantigen der Zöliakie ist das Enzym Transglutaminase (TTG), welches Gliadinmoleküle deamidiert (22). Dadurch wird die Immunogenität von Gliadin erhöht, anschliessend kommt es zu einer Bindung von Gliadin über HLA-DQ2- oder HLA-DQ8-Moleküle an antigenpräsentierende Zellen. Diese Gliadinpeptide werden dann CD4 + -T-Zellen präsentiert (23, 24). Im weiteren Verlauf der Immunkaskade sezernieren die aktivierten T-Helferzellen Zytokine wie Tumor necrosis factor α, Interferon γ und IL-2. Diese lösen unter anderem eine Expression von Matrixmetalloproteinasen in Makrophagen und Fibroblasten aus, welche dann zu einem direkten Schaden der Mukosa im Dünndarm führen (25).
Zusätzlich werden auch verschiedene Umweltfaktoren bei der Pathogenese der Zöliakie diskutiert. Es wird angenommen, dass Infektionen, das Stillen und Veränderungen der Darmflora Faktoren in der Pathogenese der Zöliakie eine Rolle spielen (26-28).
Bezüglich der Genetik wird nach aktuellem Wissensstand angenommen, dass fast 100% aller Zöliakie-Patienten Träger der Varianten der HLA-Klasse-II-Gene HLA-DQA1 und HLA-DQB1 sind. Diese kodieren für die α- und β-Kette der Heterodimer-Proteine DQ2 und DQ8 und sind mit der Zöliakie assoziiert (29). Da ca. ein Drittel der Allgemeinbevölkerung Träger von DQ2 und/oder DQ8 sind, ist der HLA-Test allein nicht für die Diagnose von Zöliakie ausreichend. Aufgrund seines sehr hohen negativen Vorhersagewerts von fast 100% kann dieser Test jedoch zum Ausschluss der Zöliakie verwendet werden.
Diagnose
Die Diagnose der Zöliakie stützt sich auf die Kombination von Anamnese, klinischer Untersuchung, Serologie und der histologischen Untersuchung von Duodenalbiopsien, welche im Rahmen einer Ösophagogastroduodenoskopie entnommen werden. Bei klinischem Verdacht auf eine Zöliakie sollte zunächst immer eine serologische Untersuchung erfolgen. Die Gewebetransglutaminase-IgA-Antikörper (tTG) (ELISA) weist eine gute Spezifität und Sensitivität auf, um eine Zöliakie zuverlässig zu dedektieren. Die Sensitivität beträgt 74% - 100% und die Spezifität 78% - 100% (30, 31). Wichtig ist jedoch zu wissen, dass die serologischen Tests nur unter einer glutenhaltigen Ernährung aussagekräftig sind, da diese sonst falsch-negativ ausfallen können. Bei der serologischen Untersuchung muss zudem auch eine Bestimmung des Gesamt-IgA erfolgen, um einen IgA-Mangel auszuschliessen, insbesondere da bei Zöliakie-Patienten ein selektiver IgA Mangel häufiger als in der Allgemeinbevölkerung anzutreffen ist. Bei Vorliegen eines IgA-Mangels sind die tTG-IgA-Antikörper nicht zuverlässig nachzuweisen.
Liegt eine positive Zöliakie-Serologie vor, sollte eine Endoskopie mit Biopsieentnahme aus dem Duodenum erfolgen, um die Diagnose zu bestätigen. Es wird empfohlen, mindestens vier Biopsien aus dem distalen Duodenum und zwei Biopsien aus dem Bulbus duodeni zu entnehmen. Bei pädiatrischen Patienten muss nicht in allen Fällen eine histologische Sicherung erfolgen: die Diagnose einer Zöliakie kann gestellt werden, wenn Symptome, die auf eine Zöliakie hinweisen und zusätzlich ein positiver EmA-Test und ein hoher tTG-IgA-Wert (≥ 10-fache der Obergrenze des Normalwerts) vorliegen (32, 33).
In besonderen Situationen kann eine HLA-Diagnostik erfolgen. Man geht davon aus, dass bei so gut wie 100% der Zöliakie-Patienten die Haplotypen HLA-DQ2 oder DQ8 vorhanden sind. Da die Haplotypen aber auch in ca. 30% der Normalbevölkerung nachweisbar sind, hat der Gentest einen niedrigen positiven Vorhersagewert von nur 12%. Daher ist die HLA-DQ2 und -DQ8 Bestimmung nur geeignet eine Zöliakie auszuschliessen (34). Die Untersuchung kann bei Verdacht auf Zöliakie bei Patienten unter bereits erfolgter glutenfreier Ernährung hilfreich sein, bei denen eine mehrwöchige Gluten-Belastung vor der serologischen Bestimmung nicht möglich oder nicht gewünscht ist, oder falls nicht-eindeutige histologische Befunde aus dem Dünndarm vorliegen.
Da bei bestimmten Risikopopulationen, wie Verwandte ersten und zweiten Grades mit Zöliakie, Patienten mit autoimmuner Thyreoiditis, Patienten mit Typ-1-Diabetes sowie Down- und Turner-Syndrom eine erhöhte Zöliakie-Prävalenz besteht (17), empfiehlt sich bei diesen zumindest einmalig eine serologische Bestimmung von Gesamt-IgA und tTG-IgA-Antikörpern.
Klinik
Zöliakie wird oft als Chamäleon der Gastroenterologie bezeichnet, da die klinischen Symptome und deren Schweregrad individuell sehr unterschiedlich ausgeprägt sein können. Dies erschwert die Definition und Charakterisierung typischer Symptome und kann zu einer Verzögerung bei der Diagnosestellung führen (35). Insbesondere bei Frauen kommt es zu einer signifikanten Verzögerung, die dem sogenannten «Doctor’s Delay» zuzuschreiben ist; bei Frauen werden die unspezifischen Symptome von Fachpersonen vermutlich häufig anderen Ursachen zugeschrieben. Während die Zöliakie früher als eine Erkrankung im Kindesalter angesehen wurde, erfolgt die Diagnosestellung heute häufiger im Erwachsenenalter (zwischen 40 und 59 Jahren) und auch bei älteren Menschen (35). Die Symptome der Zöliakie können in gastrointestinale und extraintestinale Manifestationen unterteilt werden. Tabelle 1 gibt einen Überblick über die Vielfalt der klinischen Präsentation.
Zu den klassischen gastrointestinalen Symptomen zählen Diarrhoe, Blähungen mit abdomineller Distension, Flatulenz oder Steatorrhoe, die auf eine Malabsorption hindeuten können. Diese Malabsorption kann wiederum die Anämie und ungewollten Gewichtsverlust verursachen und im Kindesalter zu Gedeihstörungen führen. Neurologische Symptome sind seltener, können aber durch einen Mangel an B-Vitaminen (insbesondere B1, B6, B12) verursacht werden und sich als unspezifische Nausea oder Kopfschmerzen bis hin zu peripherer Neuropathie oder Polyneuropathie manifestieren (36). Bis zu 50% der Patienten mit Zöliakie zeigen letztere Symptome, die sich als Kribbeln, Brennen und Taubheitsgefühl in den Händen und Füssen bemerkbar machen können (37). Mit Beginn einer glutenfreien Ernährung und der entsprechenden Supplementation mit einem hochdosierten B-Komplex können diese Symptome oft vollständig regredieren.
Die Osteopenie und Osteoporose sind häufige extraintestinale Manifestationen der Zöliakie und werden bei bis zu 75% aller Betroffenen diagnostiziert. Diese Erkrankungen sind auf eine Malabsorption von Vitamin D und Kalzium zurückzuführen und können auch ohne gleichzeitige gastrointestinale Symptome auftreten (38-40). Das Frakturrisiko bei Patienten mit Zöliakie wird um 40% höher geschätzt als bei alters- und geschlechtsangepassten gesunden Kontrollpersonen (41). Das Ausmass der Osteopenie/Osteoporose bei Erstdiagnose der Zöliakie korreliert ebenfalls mit dem Ausmass der Zottenschädigung (42). Eine DEXA nach der Diagnosestellung der Zöliakie ist deswegen bei adulten Betroffenen empfehlenswert.
Bei unbehandelter Zöliakie treten gehäuft folgende Mangelerscheinungen auf: Eisen, Vitamin B12, Folsäure, Vitamin A, B6, Vitamin D, Kupfer und Zink (43). Eisenmangel wird durch okkulten Blutverlust der Dünndarmschleimhaut und die gleichzeitige Malabsorption verursacht. Die anderen Nährstoffe werden auf Grund der verringerten Resorptionsfläche der Zottenatrophie nur mangelhaft aufgenommen. So zeigen Zöliakie-Betroffene mit Eisenmangelanämie auffallend häufig eine ausgeprägte Zottenatrophie (44). Bei Vorliegen einer Eisenmangelanämie ist immer eine Eisensubstitution empfohlen. Weiter soll die probatorische Behandlung von Mangelerscheinungen durch den Einsatz eines Multivitaminsupplements (mit oder ohne Eisen) bei Diagnosestellung erfolgen.
Als Hautmanifestationen der Zöliakie gelten die Dermatitis herpetiformis Duhring sowie orale und pharyngeale Aphthen. Die Dermatitis herpetiformis Duhring ist eine Autoimmunerkrankung, die mit Blasenbildung der Haut einhergeht und sich durch herpesartige gruppierte Bläschen zeigt. Die Betroffenen leiden häufig unter starkem Juckreiz und Brennen. Betroffen sind häufig Ellbogen, Knie, und Gesäss. Der Brustkorb und die Kopfhaut sind ebenso häufig betroffen. Männer sind häufiger von dieser Hauterkrankung betroffen als Frauen. Als Pathogenese wird eine Ablagerung von Komplexen aus epidermaler Transglutaminase (eTG) und eTG-gerichtetem IgA an der Basalmembran beschrieben (45). Bei einem Grossteil der Betroffenen mit Dermatitis herpetiformis Duhring wird eine subklinische Zöliakie als Grunderkrankung diagnostiziert. Unter einer glutenfreien Ernährung ist eine vollständige Remission dieser Hauterkrankung zu erwarten (46).
Zusätzlich zur intestinalen Manifestation kann auch eine Gelenkbeteiligung auftreten. Zöliakie-Patienten zeigen häufiger frühe Anzeichen einer Arthritis in den unteren Gliedmassen im Vergleich zur Normalbevölkerung und es treten gehäuft Arthralgien auf.
Bei Frauen mit unerklärter Infertilität ist die Prävalenz von undiagnostizierter Zöliakie erhöht. Es wurden auch Fehlgeburten bei undiagnostizierter Zöliakie beschrieben, weshalb eine Zöliakieabklärung in diesen Fällen durchgeführt werden sollte (47, 48).
Die seltenste Manifestation der Zöliakie ist die sogenannte «Zöliakie-Krise». Dabei kommt es zu einer akuten klinischen Verschlechterung mit rascher Progression von Durchfall und Erbrechen. Die damit einhergehende Dehydrierung, verschiedene Elektrolytstörungen und die deutliche Abnahme des Körpergewichts führen meist zu einer stationären Betreuung (49, 50).
Therapie
Noch immer ist eine lebensbegleitende, strikt glutenfreie Ernährung die einzige verfügbare Therapieoption bei Zöliakie. Daher sollten alle Patientinnen und Patienten nach der Diagnosestellung eine qualifizierte und auf Zöliakie spezialisierte Ernährungsberatung erhalten. Eine solche Fachperson kann über die Fachgruppe Zöliakie (https://zoeliakiernaehrungsberatung.ch) vermittelt werden.
Ziele der glutenfreien Diät sind die vollständige histologische Remission, die Verbesserung der gastrointestinalen oder extraintestinalen Beschwerden, die Behandlung als auch Prävention von Nährstoffdefiziten und deren Komplikationen sowie eine Verbesserung der ernährungsbezogenen Lebensqualität. Insbesondere Letzteres stellt in der Ernährungstherapie eine Herausforderung dar, da diese einschränkende Ernährungsform verschiedenste Lebensbereiche tangiert und auch psychosoziale Herausforderungen birgt (51).
Wichtig zu wissen ist der Umstand, dass es mehrere Monate dauern kann, bis sich die Symptome nach Beginn einer glutenfreien Ernährung zurückbilden. Oftmals dauert es ebenso bis zu einem Jahr, bis sich die histologischen Veränderungen im Dünndarm sowie die Serologie vollständig normalisieren. Wenn trotz adhärenter glutenfreier Ernährung unspezifische Magen-Darm-Beschwerden persistieren, lohnt es sich, auch andere Trigger für die Symptome zu evaluieren (52, 53). Bei vielen Zöliakiebetroffenen besteht anfangs eine sekundäre Laktoseintoleranz, (und interessanterweise findet sich auch bei Patienten mit einer Laktoseintoleranz überaus häufig eine Zöliakie (54)), die mittels einer lactosearmen Ernährung gut kontrolliert werden kann. Es empfiehlt sich Milch, Joghurt und Frischkäse durch lactosearme Alternativen zu ersetzen, Käse kann weiterhin uneingeschränkt konsumiert werden, da er keine klinisch relevante Lactosemenge enthält und eine gute Calciumquelle darstellt. Oftmals ist eine Regression der Beschwerden unter diesen einfachen Anpassungen möglich (55).
Auch andere funktionelle Beschwerden sind bei Betroffenen gehäuft vorhanden, die mittels des FODMAP-Konzepts ergänzend zur glutenfreien Ernährung erfolgreich behandelt werden können (56).
Innerhalb der Ernährungstherapie ist es besonders wichtig, Fragen zu unbeabsichtigten Kontaminationen mit Gluten zu besprechen sowie zu klären, ob und wann kontrolliert glutenfreier Hafer in die Ernährung eingeführt werden soll.
In den letzten Jahren hat sich in beiden Fragen ein Umdenken vollzogen: Glutenfreier Hafer wird mittlerweile generell als sicher für Zöliakiebetroffene eingestuft (57), und bereits nach der Diagnosestellung kann er in die Ernährung eingeführt werden (58). Allerdings empfehlen internationale Leitlinien eine Wartezeit von 6 Monaten nach Diagnosestellung und/oder vollständiger Remission, da es einige wenige Fallbeschreibungen gibt, die eine Sensitivität von Zöliakiebetroffenen gegenüber Hafer gezeigt haben (59). In der Schweiz ist kein Fall einer Hafer-sensitiven Zöliakie bekannt.
Aktuell geht man davon aus, dass Kontaminationen bei der Zubereitung von glutenfreien Gerichten weitaus seltener sind als bisher angenommen. Vor allem Mehlstaub kann zu relevanten Kontaminationen führen, während die Verwendung von denselben Kochutensilien unproblematisch ist (60, 61).
Schliesslich haben sich auch glutenfreie Spezialprodukte in den letzten 10 Jahren deutlich verändert. Der Zucker- und Fettzusatz wurde reduziert, und es besteht ein Trend, vermehrt auf vollwertige glutenfreie Getreide zurückzugreifen, anstelle von nährstoffarmen Stärkemehlen und Verdickungsmitteln. Diese Entwicklungen sind sehr erfreulich und erleichtern die Durchführung dieser doch einschränkenden Ernährungsform (62, 63).
Prognose
Wenn die Zöliakie rechtzeitig diagnostiziert und korrekt behandelt wird, ist die Prognose der Erkrankung sehr gut (64). Erfreulicherweise sprechen mehr als 90% der Zöliakie-Betroffenen auf eine glutenfreie Diät an. Etwa 1% aller Erkrankten entwickeln jedoch eine refraktäre Zöliakie. Diese ist gekennzeichnet durch eine fortbestehende Zottenatrophie und intestinale oder extraintestinale Symptome, trotz einer korrekt durchgeführten glutenfreien Diät über einen Zeitraum von mindestens 12 Monaten (65). Die Diagnosestellung und Behandlung der refraktären Zöliakie gestaltet sich oft herausfordernd und sollte an spezialisierten Zentren vorgenommen werden, um den Betroffenen ebenso eine gute Lebensqualität zu ermöglichen.
Insgesamt zeigt sich, dass die Zöliakie eine Systemerkrankung ist, die verschiedene Organsysteme betreffen kann und somit ein breites Spektrum an Symptomen hervorruft. Eine frühzeitige Diagnose und konsequente glutenfreie Ernährung können jedoch dazu beitragen, eine Vielzahl von Komplikationen und Folgeerkrankungen zu vermeiden.
Copyright bei Aerzteverlag medinfo AG
PD Dr. med. Jonas Zeitz
– GastroZentrum Hirslanden, Witellikerstrasse 40, 8008 Zürich
– Zöliakie Zentrum am GastroZentrum Hirslanden, Zürich
– Klinik für Gastroenterologie und Hepatologie, Universitätsspital Zürich, Rämistrasse 100, 8091 Zürich
– Ernährungstherapie Basel, Klosterberg 11, 4051 Basel
Die Autoren haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Green PH, Cellier C. Celiac disease. N Engl J Med 2007;357(17):1731-43. doi: 10.1056/NEJMra071600 [published Online First: 2007/10/26]
2. Green PH, Krishnareddy S, Lebwohl B. Clinical manifestations of celiac disease. Dig Dis 2015;33(2):137-40. doi: 10.1159/000370204 [published Online First: 2015/05/01]
3. Booth C. History of celiac disease. BMJ 1989;298:527.
4. Paulley JW. Observation on the aetiology of idiopathic steatorrhoea; jejunal and lymph-node biopsies. Br Med J 1954;2(4900):1318-21. doi: 10.1136/bmj.2.4900.1318 [published Online First: 1954/12/04]
5. Riddle MS, Murray JA, Porter CK. The incidence and risk of celiac disease in a healthy US adult population. Am J Gastroenterol 2012;107(8):1248-55. doi: 10.1038/ajg.2012.130 [published Online First: 2012/05/16]
6. Green PH, Lebwohl B, Greywoode R. Celiac disease. J Allergy Clin Immunol 2015;135(5):1099-106; quiz 107. doi: 10.1016/j.jaci.2015.01.044 [published Online First: 2015/05/10]
7. Lebwohl B, Sanders DS, Green PHR. Coeliac disease. Lancet 2018;391(10115):70-81. doi: 10.1016/S0140-6736(17)31796-8 [published Online First: 2017/08/02]
8. Maki M, Mustalahti K, Kokkonen J, et al. Prevalence of Celiac disease among children in Finland. N Engl J Med 2003;348(25):2517-24. doi: 10.1056/NEJMoa021687 [published Online First: 2003/06/20]
9. Di Sabatino A, Corazza GR. Coeliac disease. Lancet 2009;373(9673):1480-93. doi: 10.1016/S0140-6736(09)60254-3 [published Online First: 2009/04/28]
10. Dube C, Rostom A, Sy R, et al. The prevalence of celiac disease in average-risk and at-risk Western European populations: a systematic review. Gastroenterology 2005;128(4 Suppl 1):S57-67. doi: 10.1053/j.gastro.2005.02.014 [published Online First: 2005/04/13]
11. West J, Logan RF, Hill PG, et al. Seroprevalence, correlates, and characteristics of undetected coeliac disease in England. Gut 2003;52(7):960-5. doi: 10.1136/gut.52.7.960 [published Online First: 2003/06/13]
12. Fasano A, Berti I, Gerarduzzi T, et al. Prevalence of celiac disease in at-risk and not-at-risk groups in the United States: a large multicenter study. Arch Intern Med 2003;163(3):286-92. doi: 10.1001/archinte.163.3.286 [published Online First: 2003/02/13]
13. Lebwohl B, Green P. Risk of celiac disease according to HLA haplotype and country. N Engl J Med 2014;371(11):1073-4. doi: 10.1056/NEJMc1409252 [published Online First: 2014/09/11]
14. Rubio-Tapia A, Kyle RA, Kaplan EL, et al. Increased prevalence and mortality in undiagnosed celiac disease. Gastroenterology 2009;137(1):88-93. doi: 10.1053/j.gastro.2009.03.059 [published Online First: 2009/04/14]
15. Lohi S, Mustalahti K, Kaukinen K, et al. Increasing prevalence of coeliac disease over time. Aliment Pharmacol Ther 2007;26(9):1217-25. doi: 10.1111/j.1365-2036.2007.03502.x [published Online First: 2007/10/20]
16. Kearney J. Food consumption trends and drivers. Philos Trans R Soc Lond B Biol Sci 2010;365(1554):2793-807. doi: 10.1098/rstb.2010.0149 [published Online First: 2010/08/18]
17. Du Y, Shan LF, Cao ZZ, et al. Prevalence of celiac disease in patients with Down syndrome: a meta-analysis. Oncotarget 2018;9(4):5387-96. doi: 10.18632/oncotarget.23624 [published Online First: 2018/02/13]
18. Reilly NR, Aguilar K, Hassid BG, et al. Celiac disease in normal-weight and overweight children: clinical features and growth outcomes following a gluten-free diet. J Pediatr Gastroenterol Nutr 2011;53(5):528-31. doi: 10.1097/MPG.0b013e3182276d5e [published Online First: 2011/06/15]
19. Bardella MT, Fredella C, Saladino V, et al. Gluten intolerance: gender- and age-related differences in symptoms. Scand J Gastroenterol 2005;40(1):15-9. doi: 10.1080/00365520410008169 [published Online First: 2005/04/22]
20. Dixit R, Lebwohl B, Ludvigsson JF, et al. Celiac disease is diagnosed less frequently in young adult males. Dig Dis Sci 2014;59(7):1509-12. doi: 10.1007/s10620-014-3025-6 [published Online First: 2014/01/22]
21. Schuppan D, Junker Y, Barisani D. Celiac disease: from pathogenesis to novel therapies. Gastroenterology 2009;137(6):1912-33. doi: 10.1053/j.gastro.2009.09.008 [published Online First: 2009/09/22]
22. Dieterich W, Ehnis T, Bauer M, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med 1997;3(7):797-801. doi: 10.1038/nm0797-797 [published Online First: 1997/07/01]
23. Molberg O, McAdam SN, Korner R, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med 1998;4(6):713-7. doi: 10.1038/nm0698-713 [published Online First: 1998/06/12]
24. Lundin KE, Scott H, Hansen T, et al. Gliadin-specific, HLA-DQ(alpha 1*0501,beta 1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. J Exp Med 1993;178(1):187-96. doi: 10.1084/jem.178.1.187 [published Online First: 1993/07/01]
25. Daum S, Bauer U, Foss HD, et al. Increased expression of mRNA for matrix metalloproteinases-1 and -3 and tissue inhibitor of metalloproteinases-1 in intestinal biopsy specimens from patients with coeliac disease. Gut 1999;44(1):17-25. doi: 10.1136/gut.44.1.17 [published Online First: 1998/12/24]
26. Riddle MS, Murray JA, Cash BD, et al. Pathogen-specific risk of celiac disease following bacterial causes of foodborne illness: a retrospective cohort study. Dig Dis Sci 2013;58(11):3242-5. doi: 10.1007/s10620-013-2733-7 [published Online First: 2013/07/03]
27. Welander A, Tjernberg AR, Montgomery SM, et al. Infectious disease and risk of later celiac disease in childhood. Pediatrics 2010;125(3):e530-6. doi: 10.1542/peds.2009-1200 [published Online First: 2010/02/24]
28. Akobeng AK, Ramanan AV, Buchan I, et al. Effect of breast feeding on risk of coeliac disease: a systematic review and meta-analysis of observational studies. Arch Dis Child 2006;91(1):39-43. doi: 10.1136/adc.2005.082016 [published Online First: 2005/11/17]
29. Sollid LM, Lie BA. Celiac disease genetics: current concepts and practical applications. Clin Gastroenterol Hepatol 2005;3(9):843-51. [published Online First: 2005/10/20]
30. Giersiepen K, Lelgemann M, Stuhldreher N, et al. Accuracy of diagnostic antibody tests for coeliac disease in children: summary of an evidence report. J Pediatr Gastroenterol Nutr 2012;54(2):229-41. doi: 10.1097/MPG.0b013e318216f2e5 [published Online First: 2012/01/24]
31. Kurppa K, Rasanen T, Collin P, et al. Endomysial antibodies predict celiac disease irrespective of the titers or clinical presentation. World J Gastroenterol 2012;18(20):2511-6. doi: 10.3748/wjg.v18.i20.2511 [published Online First: 2012/06/02]
32. Werkstetter KJ, Korponay-Szabo IR, Popp A, et al. Accuracy in Diagnosis of Celiac Disease Without Biopsies in Clinical Practice. Gastroenterology 2017;153(4):924-35. doi: 10.1053/j.gastro.2017.06.002 [published Online First: 2017/06/19]
33. Husby S, Koletzko S, Korponay-Szabo IR, et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr 2012;54(1):136-60. doi: 10.1097/MPG.0b013e31821a23d0 [published Online First: 2011/12/27]
34. Kaukinen K, Partanen J, Maki M, et al. HLA-DQ typing in the diagnosis of celiac disease. Am J Gastroenterol 2002;97(3):695-9. doi: 10.1111/j.1572-0241.2002.05471.x [published Online First: 2002/04/02]
35. Vavricka SR, Vadasz N, Stotz M, et al. Celiac disease diagnosis still significantly delayed – Doctor’s but not patients’ delay responsive for the increased total delay in women. Dig Liver Dis 2016;48(10):1148-54. doi: 10.1016/j.dld.2016.06.016 [published Online First: 2016/07/13]
36. Kennedy DO. B Vitamins and the Brain: Mechanisms, Dose and Efficacy–A Review. Nutrients 2016;8(2):68. doi: 10.3390/nu8020068 [published Online First: 2016/02/02]
37. Chin RL, Sander HW, Brannagan TH, et al. Celiac neuropathy. Neurology 2003;60(10):1581-5. doi: 10.1212/01.wnl.0000063307.84039.c7 [published Online First: 2003/05/29]
38. Kemppainen T, Kroger H, Janatuinen E, et al. Osteoporosis in adult patients with celiac disease. Bone 1999;24(3):249-55. doi: 10.1016/s8756-3282(98)00178-1 [published Online First: 1999/03/11]
39. Shaker JL, Brickner RC, Findling JW, et al. Hypocalcemia and skeletal disease as presenting features of celiac disease. Arch Intern Med 1997;157(9):1013-6. [published Online First: 1997/05/12]
40. Mustalahti K, Collin P, Sievanen H, et al. Osteopenia in patients with clinically silent coeliac disease warrants screening. Lancet 1999;354(9180):744-5. doi: 10.1016/S0140-6736(99)01990-X [published Online First: 1999/09/04]
41. Lucendo AJ, Garcia-Manzanares A. Bone mineral density in adult coeliac disease: an updated review. Rev Esp Enferm Dig 2013;105(3):154-62. doi: 10.4321/s1130-01082013000300006 [published Online First: 2013/06/06]
42. Garcia-Manzanares A, Tenias JM, Lucendo AJ. Bone mineral density directly correlates with duodenal Marsh stage in newly diagnosed adult celiac patients. Scand J Gastroenterol 2012;47(8-9):927-36. doi: 10.3109/00365521.2012.688217 [published Online First: 2012/05/17]
43. Wierdsma NJ, van Bokhorst-de van der Schueren MA, Berkenpas M, et al. Vitamin and mineral deficiencies are highly prevalent in newly diagnosed celiac disease patients. Nutrients 2013;5(10):3975-92. doi: 10.3390/nu5103975 [published Online First: 2013/10/03]
44. Abu Daya H, Lebwohl B, Lewis SK, et al. Celiac disease patients presenting with anemia have more severe disease than those presenting with diarrhea. Clin Gastroenterol Hepatol 2013;11(11):1472-7. doi: 10.1016/j.cgh.2013.05.030 [published Online First: 2013/06/13]
45. Donaldson MR, Zone JJ, Schmidt LA, et al. Epidermal transglutaminase deposits in perilesional and uninvolved skin in patients with dermatitis herpetiformis. J Invest Dermatol 2007;127(5):1268-71. doi: 10.1038/sj.jid.5700682 [published Online First: 2007/01/06]
46. Sticherling M, Erfurt-Berge C. Autoimmune blistering diseases of the skin. Autoimmun Rev 2012;11(3):226-30. doi: 10.1016/j.autrev.2011.05.017 [published Online First: 2011/06/07]
47. Choi JM, Lebwohl B, Wang J, et al. Increased prevalence of celiac disease in patients with unexplained infertility in the United States. J Reprod Med 2011;56(5-6):199-203. [published Online First: 2011/06/21]
48. Grode LB, Agerholm IE, Humaidan P, et al. Unrecognised coeliac disease among men and women undergoing fertility treatment: A screening study. United European Gastroenterol J 2018;6(10):1477-84. doi: 10.1177/2050640618796750 [published Online First: 2018/12/24]