En dépit de décennies de surveillance et d’ interventions (pharmacologiques et non-pharmacologiques), les virus de la grippe saisonnière continuent de causer de lourdes épidémies dans le monde chaque année. Sous nos latitudes, les affections dues aux virus influenzae A/H1N1, A/H3N2 et influenzae B surviennent chaque hiver. Le processus clé qui sous-tend ces épidémies récurrentes est la capacité évolutive des virus à échapper à la mémoire immunitaire induite par les contacts antérieurs (infection et/ou vaccination).
Bien que nous commencions à comprendre les mécanismes qui sous-tendent cette dynamique, le moment et la nature de l’ émergence de nouvelles souches demeurent encore pour la plupart imprévisibles (1). Trop souvent considérée comme une affection bénigne, la grippe est très contagieuse et à l’ origine de 1000 à 5000 hospitalisations et 1500 décès chaque année en Suisse (www.bag.admin.ch). Sa prévention par la vaccination est actuellement la mesure la plus efficace (2-5) et les recommandations ont d’ ailleurs peu changé depuis 2013 (www.infovac.ch). Le vaccin est recommandé annuellement chez toutes les personnes dites à risque accru de complications (Tableau 1, 2) sans qu’ il y ait d’ évidence d’ une réduction de l’ efficacité de protection avec ce schéma de vaccination. En Suisse, les affections grippales sont surveillées par le système de déclaration Sentinella et toutes les infections confirmées en laboratoire sont enregistrées dans un système de déclaration obligatoire.
Cette surveillance et les données collectées permettent aux chercheurs de surveiller les tendances épidémiques des virus grippaux et d’ accumuler les séquences virales dans les bases de données publiques. Une meilleure sélection des virus candidats aux vaccins et la détection précoce des virus résistants aux médicaments en est une résultante directe tout comme les avancées prometteuses en matière de prévention et de traitement.


La saison grippale 2018/19 en chiffres
En Suisse, les virus qui ont circulé pendant la dernière saison étaient très majoritaire des virus influenzae A de type A/H1N1pdm09 et A/H3N2. La résultante a été que la couverture vaccinale était excellente (99.5%). Le vaccin quadrivalent n’ a présenté qu’ un très faible avantage, car les virus de la lignée influenzae B-Yamagata n’ ont que très peu circulé. Selon les études, l’ efficacité vaccinale chez les personnes non hospitalisées a été estimée à 32-68 %. Elle était nettement plus élevée vis-à-vis des virus A/H1N1pdm09 (45-72 %) qu’ A/H3N2 (-39 à 45 %).
Sur l’ ensemble de la saison 2018/19, la surveillance entre du 30 septembre 2018 au 20 avril 2019, a été estimé que 209 200 personnes (2.5% de la population Suisse) ont consultés un médecin de premier recours pour une affection grippale, soit une incidence globale de 2466 premières consultations pour 100 000 habitants. Ce chiffre est de 13% plus bas que l’ incidence saisonnière globale moyen sur les dix dernières saisons (2846/100 000). Le seuil épidémique pour la saison 2018/19 se situait à 68 cas de suspicion de grippe pour 100 000 habitants. L’ incidence des consultations hebdomadaire a dépassé ce seuil de la mi-janvier (semaine 2/2019) à la mi-mars (semaine 12/2019) pour une durée totale de 11 semaines avec un pic épidémique atteint à la sixième semaine de 2019 (306 consultations / 100 000 habitants) qui était le plus bas mesuré depuis 2012/13. Si l’ incidence était maximale chez les enfants de 0-4 ans (4993 consultations / 100 000 habitants), les 65 ou plus étaient la catégorie de la population qui a été la moins infectée avec tout de même 1426 consultations / 100 000 habitants (www.bag.admin.ch).
Cette catégorie d’ âge par contre, le nombre de décès a très légèrement dépassé les valeurs attendues au début mars 2019. Chaque année, cette surmortalité témoigne de la gravité de l’ épidémie dans cette population et du risque d’ évolution grave chez les personnes vulnérables. Parmi l’ ensemble des cas de grippe déclarés, 7.6% appartenait au groupe des personnes présentant un risque accru de complication et 36.6% aux 65+. La proportion la plus élevée d’ hospitalisation pour suspicion de grippe était aussi enregistrée dans cette population (4.7%) et le plus faible chez les 5-29 ans (0%). Une pneumonie a été diagnostiquée chez 3,4 % des cas de suspicion de grippe déclarés, le plus souvent chez les plus de 64 ans (10.5 %), le plus rarement chez les enfants de 0 à 4 ans (1.5 %) (www.bag.admin.ch).
Durant la saison 2018/19, environ 7.9% des personnes déclarées pour suspicion de grippe avec un statu vaccinal connu étaient vaccinés. Cette proportion était plus importante dans les groupes chez qui l’ OFSP recommande la vaccination (Tableau 1) avec 33.5% chez les 65+ et 40.1% avec un risque accru de complications. Un traitement antiviral, dans la plupart des cas par un inhibiteur de la neuraminidase a été administré chez 2.2% des personnes ayant déclaré une grippe ; 10.4% ont reçu un traitement antibiotique probablement en raison d’ une surinfection
bactérienne (www.bag.admin.ch).
La grippe est contagieuse avant les symptômes et parfois même asymptomatique
La grippe se transmet par contact direct avec une personne infectée (éternuement, toux jusqu’ à 1 mètre), notamment dans des espaces clos. Mais, les virus grippaux peuvent aussi rester vivants jusqu’ à 48 heures sur des surfaces inertes. Comme il a été estimé qu’ un individu adulte peut avoir jusqu’ à 40 contacts facial par heure avec ces mains, les contacts avec des objets et des surfaces inertes « contaminés » (table, poignées de portes, bouton d’ ascenseur, rampe d’ escalier, billet de banque, etc.) sont une voie de transmission à ne surtout pas banaliser (6). Les personnes contaminées peuvent transmettre les virus de la grippe à d’ autres même si elles ne se sentent pas (encore) malades (6). De plus, près d’ un tiers des personnes infectées ne présente aucun des symptômes spécifiques et ne se sent même pas malade (7). Ces personnes peuvent être des vecteurs de transmission qui s’ ignorent.
La vaccination contribue fortement à diminuer le risque de transmission chez les personnes vaccinés, mais aussi chez les non vaccinés lorsque le taux de couverture vaccinale est suffisant (≥ 75% de la population) par le biais de l’ immunité de groupe (8). Les professionnels de la santé sont parmi les personnes les plus fortement exposées au risque de contracter la grippe (9). De plus, les arrêts de travail pour maladie qui en résultent impliquent souvent une charge de travail supplémentaire pour les collègues en période épidémique et/ou des contraintes de réorganisation en rapport avec le recours à du personnel intérimaire notamment dans les EMS et les hôpitaux (10).
La grippe en clinique
Après contamination, les symptômes grippaux apparaissent généralement en un à trois jours. La grippe saisonnière se manifeste par une sensation de malaise général, une brusque poussée de fièvre, des frissons, des maux de tête, des arthro-myalgies, une perte d’ appétit et de vertiges. La seconde phase se caractérise par l’ intensification des symptômes respiratoires (toux sèche, maux de gorge, enrouement, rhinite). La fièvre dure en générale 3 à 8 jours et la convalescence 7 à 15 jours mais peut se prolonger au-delà (11). Cependant chez les personnes âgées et/ou celles présentant des affections chroniques, la grippe est loin d’ être une maladie bénigne et peut s’ accompagner des complications (12). Les complications les plus fréquentes sont les pneumonies infectieuses. Primaires, elles sont dues à la virulence directe du virus de la grippe ; secondaires, à une surinfection bactérienne (12).
La vaccination, recommandée chaque année reste la prévention la plus efficace
La vaccination reste la prévention la plus simple, efficace et économique chez les personnes à risque accru de complication et/ou de transmission de l’ infection grippale (Tableau 1). La diversité antigénique des virus grippaux humains représente cependant encore un défi pour le développement de vaccins dotés d’ une protection immunitaire durable (1).
Les alternatives à la vaccination – Les autres moyens de lutter contre la grippe, notamment les mesures d’ hygiène (même si elles sont indispensables) restent un complément à la vaccination et ne peuvent la remplacer. En l’ absence de vaccin ou de traitement spécifiques des autres infections respiratoires hivernales, les masques, les appareils de protection respiratoire et l’ hygiène des mains ainsi que les mesures barrières (isolement «’ gouttelettes’ », éloignement social) restent de ce fait les seules armes efficaces (13-15). De façon intéressante, les effets immunomodulateurs de la VitD ont été considérés dans la prévention de la grippe et des infections respiratoires saisonnières (16). Dans essai randomisé contrôlé en long séjour, il a été montré qu’ une supplémentation par 100 000 UI/mois de VitD réduisait l’ incidence des infections respiratoires aiguës (2) comparativement à une supplémentation standard (400-1000 UI/jour) (17). Si les effets anti-infectieux de la VitD sont de mieux en mieux documentés, aucune donnée actuellement ne confirme un effet de la VitD sur l’ immunogénécité des vaccins antigrippes (18).
Les vaccins actuellement disponibles et autorisés pour les adultes – Ils contiennent par dose de 0,5 ml, 15 µg d’ hémagglutinine (HA) de chacune des souches virales constitutives. Il existe des vaccins trivalent (3 souches grippales = A/H1N1pdm2009, A/H3N2, et B-Victoria – Agrippal®, Fluarix®, Influvac® et Mutagrip®) et un vaccin quadrivalent (4 souches virales = trivalent + B-Yamagata – Fluarix Tetra®). Chez l’ adulte, il n’ y a pas d’ arguments cliniques à privilégier un vaccin trivalent ou un vaccin tétravalent. Le vaccin trivalent Fluad® a la particularité de contenir un adjuvant (MF59C) qui en renforce l’ immunogénicité et l’ efficacité (19). Il est particulièrement recommandé pour les adultes à partir de 65 ans (www.sevaccinercontrelagrippe.ch) (20). Si les vaccins sont disponibles pour tous, la priorité est la vaccination des personnes appartement à un groupe à risque accru de complications (Tableau 1) (www.infovac.ch). Tous les vaccins autorisés en Suisse sont inactivés et exempts de mercure et d’ aluminium. En mars dernier, l’ Organisation mondiale de la santé (OMS) a publié ses recommandations pour la composition des vaccins Influenza pour l’ hémisphère nord pour la saison 2019/2020. En comparaison avec les vaccins 2018/2019, la composition du vaccin trivalent a été modifiée en ce qui concerne les A/H3N2 (A/Kansas/14/2017) et A/H1N1 (A/Brisbane/02/2018) afin de mieux couvrir les virus en circulation. La souche B-Victoria (B/Colorado/06/2017) reste inchangée tout comme la souche supplémentaire influenzae B contenue dans le vaccin tétravalent (B / Phuket/3073/2013-like). Sans adjuvant, les vaccins sont disponibles depuis la fin du mois de septembre.
La controverse sur la vaccination annuelle – Plusieurs études observationnelles ont suggéré qu’ une vaccination annuelle répétée aurait un effet négatif sur la protection pendant certaines saisons. Cette interférence négative a été principalement observée pour l’ influenzae A/H3N2 (21, 22). Ce phénomène doit cependant être interprété avec prudence et ne doit pas encore conduire à modifier la pratique et la politique en matière de vaccination (23). En effet, le recul temporel est trop court et trop peu d’ études ont été réalisées. De plus, l’ hétérogénéité des résultats est très grande. Peu de travaux ont analysé l’ effet de plusieurs vaccinations annuelles sur l’ efficacité du vaccin, même si elles suggèrent que l’ efficacité antigrippale pourrait être influencée par le schéma de vaccination des saisons précédentes. Bien que l’ hypothèse de la «distance antigénique» offre un cadre théorique simplifié pour expliquer les effets d’ une vaccination répétée contre la grippe, des recherches supplémentaires sont nécessaires pour bien comprendre ce phénomène, et également dans un contexte où le vaccin serait administré sur plus de deux saisons consécutives (www.vaxinfopro.be/spip.php?rubrique28). D’ autres travaux ont confirmé que la vaccination répétée, tant chez les jeunes que les personnes âgées, contribuait à des réponses immunitaires largement réactives tant au sein de différents sous-types viraux que de réponses croisées entre sous-types antigéniques différents (24, 25, 26). Cela illustre l’ impact de l’ âge et des antécédents d’ exposition à la grippe sur la capacité d’ une personne à réagir à de futures infections grippales.
Quel est le futur en matière de vaccination ? – Les vaccins actuellement disponibles permettent en théorie de réduire de 70 % le risque de grippe chez un adulte en bonne santé lorsque les souches vaccinales correspondent bien aux souches circulantes (ce qui n’ a pas été le cas notamment durant la saison 2015/16 par exemple) (4). Cette réponse immunitaire spécifique aux souches vaccinales présente parfois une efficacité sous-optimale. Si l’ âge et les capacités immunitaires du vacciné (20) contribuent à expliquer pourquoi la protection vaccinale s’ abaisse à 30-40 % chez les seniors (2, 3), la qualité de la protection virale est dépendante aussi de la qualité de la reformulation annuelle du vaccin (27). L’ ajout d’ un adjuvant est un moyen simple et efficace d’ améliorer l’ immunogénicité, mais cela augmente de facto la réactogénicité. Si cela se résume le plus souvent à des réactions au point d’ injection plus intenses (28), cela induit surtout un rejet de la vaccination au sein des populations (29). De nouveaux vaccins dits «universels», sont actuellement en cours de développement. Ils devraient permettre de surmonter les problèmes liés à la forte variabilité des virus grippaux nécessitant la mise à jour annuelle de la composition des vaccins saisonniers et la revaccination. Ces vaccins sont actuellement principalement élaborés à partir des épitopes hautement conservés du domaine HA, NA ou extracellulaire de la protéine M2 de la grippe, ainsi que ceux basés sur les protéines internes telles que NP et M1. Ces vaccins devraient pouvoir induire une protection contre les souches homologues, dérivées et celles issues d’ un glissement antigénique du virus grippal en évitant ainsi la nécessaire reformulation annuelle et surtout atténuer le fardeau de la maladie. Si ces vaccins démontraient leur immunogénicité, efficacité et leur capacité à conférer une immunité durable, ils pourraient être intégrés à la composition des vaccins actuels voir les remplacer (29).
La place et l’ efficacité des antiviraux dans la lutte antigrippe
Des antiviraux contre la grippe sont disponibles et leur utilisation contribue en cas d’ infection à éviter des complications sévères dans les situations à risque. Ils doivent idéalement être administrés au plus tôt après le début des symptômes grippaux. Le traitement empirique des patients suspects d’ avoir une grippe n’ est habituellement pas recommandé. Un traitement antiviral est indiqué pour les patients dont la maladie respiratoire est sévère, durant la période d’ épidémie avec des symptômes grippaux de moins de 48 heures (30).
Les principaux antiviraux utilisés actuellement sont les inhibiteurs de la neuraminidase représentés par l’ oseltamivir, le zanamivir et le peramivir (non disponible en Suisse) (31). Ils limitent la diffusion des virus en dehors des cellules infectées. Les inhibiteurs de la protéine M2 tels que l’ amantadine et la rémantadine limitent la pénétration du virus dans la cellule. Ils réduisent efficacement les complications et plus généralement l’ évolution des symptômes. Si la grande majorité des virus y sont encore sensibles, certaines mutations conduisent à des résistances (neuraminidase : H275Y et E119V ; gène de la protéine M2 : Ser31). Les taux de résistance pour les virus grippaux en circulation sont sous étroite surveillance. L’ OMS peut fournir en temps réel les informations relatives à l’ utilisation possible dans la prise en charge thérapeutique ou prophylactique (par ex. épidémie en communautés fermées, institution, etc.) (30, 32). Durant la saison 2018/19, un seul frottis a montré une résistance contre l’ oseltamivir (www.bag.admin.ch).
Même si les cas de résistance sont rares (Europe < 0.3% et USA : 1% des A/H1N1pdm09, et 0% pour les autres virus) en raison de la possibilité de mutations virales et de résistance, une certaine énergie est engagée dans le développement d’ antiviraux dotés de différents mécanismes, surtout dans le cas d’ une nouvelle souche pandémique.
Les nouveaux antiviraux – Plusieurs nouveaux antiviraux en sont à divers stades de développement et peuvent représenter de nouvelles classes de traitements qui pourraient réduire les symptômes et les complications chez les patients à risque élevé (Tableau 1). Par exemple, le baloxavir est une molécule dotée d’ un nouveau mécanisme d’ action qui vient juste d’ être approuvée par la Food and Drug Administration aux États-Unis (31). Il est le premier agent d’ une nouvelle classe que sont les inhibiteurs de l’ endonucléase du virus influenza qui est nécessaire pour la réplication du virus dans la cellule hôte. D’ autres cibles sont encore à l’ étude, notamment les kinases virales, l’ endocytose et la fusion virale.
Les alternatives thérapeutiques – Au cours de la dernière décennie, un certain nombre d’ anticorps monoclonaux humains ont démontré leur capacité à se lier à une vaste gamme de virus grippaux A et B et surtout à les neutraliser. La plupart de ces anticorps monoclonaux sont dirigés contre la tige de l’ hémagglutinine virale et certains ont maintenant été évalués dans le cadre d’ essais cliniques de stade précoce à intermédiaire (33). Une conclusion importante de ces études cliniques est que ces anticorps sont sûrs et réduisent les symptômes de la grippe. Des anticorps antigrippaux bi- et multi-spécifiques ont également été identifiés, mais n’ ont par contre pas encore fait l’ objet d’ essais cliniques. À l’ avenir, les thérapies à base d’ anticorps pourraient faire partie intégrante de notre arsenal pour prévenir et traiter la grippe (33).
Conclusion
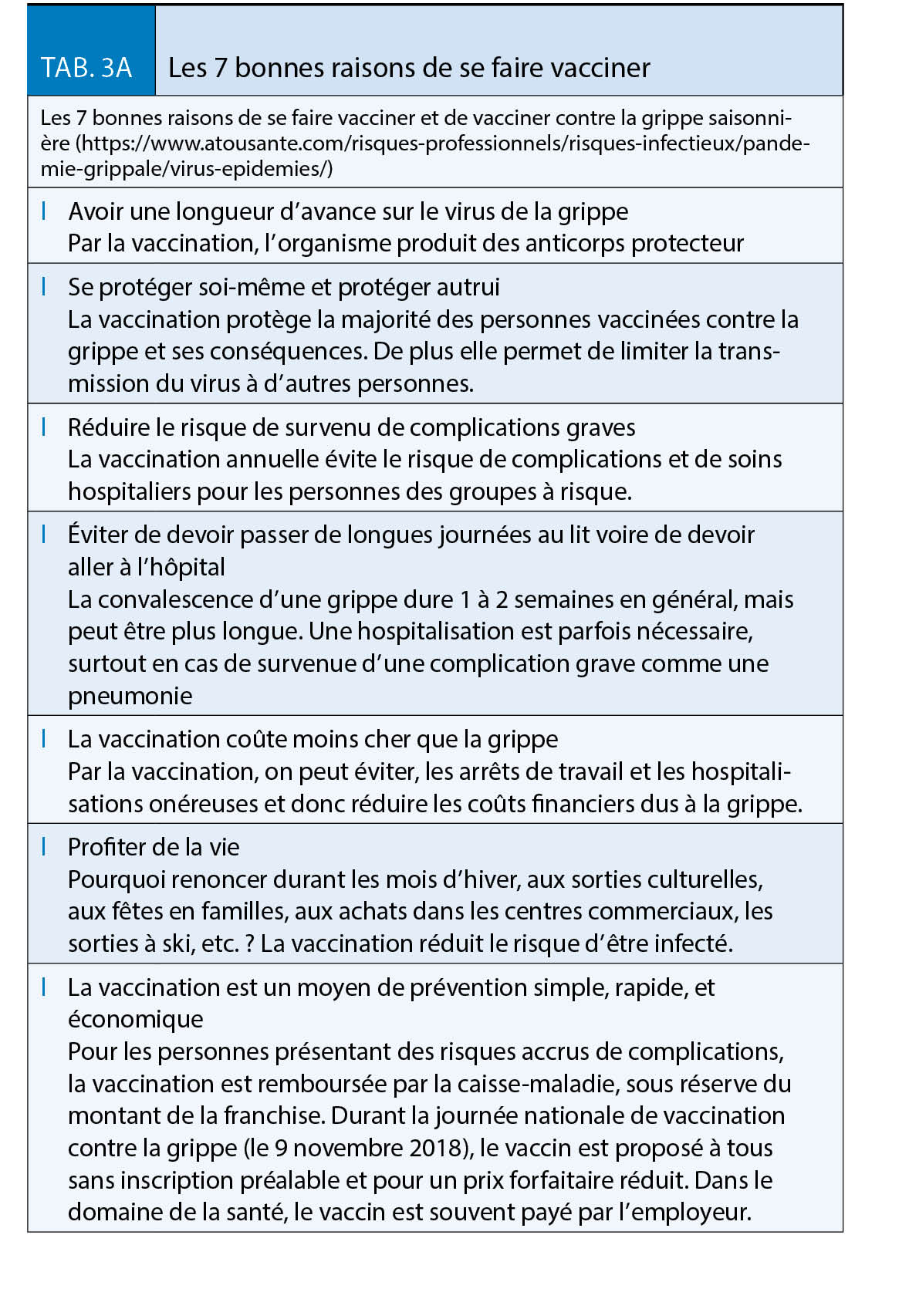
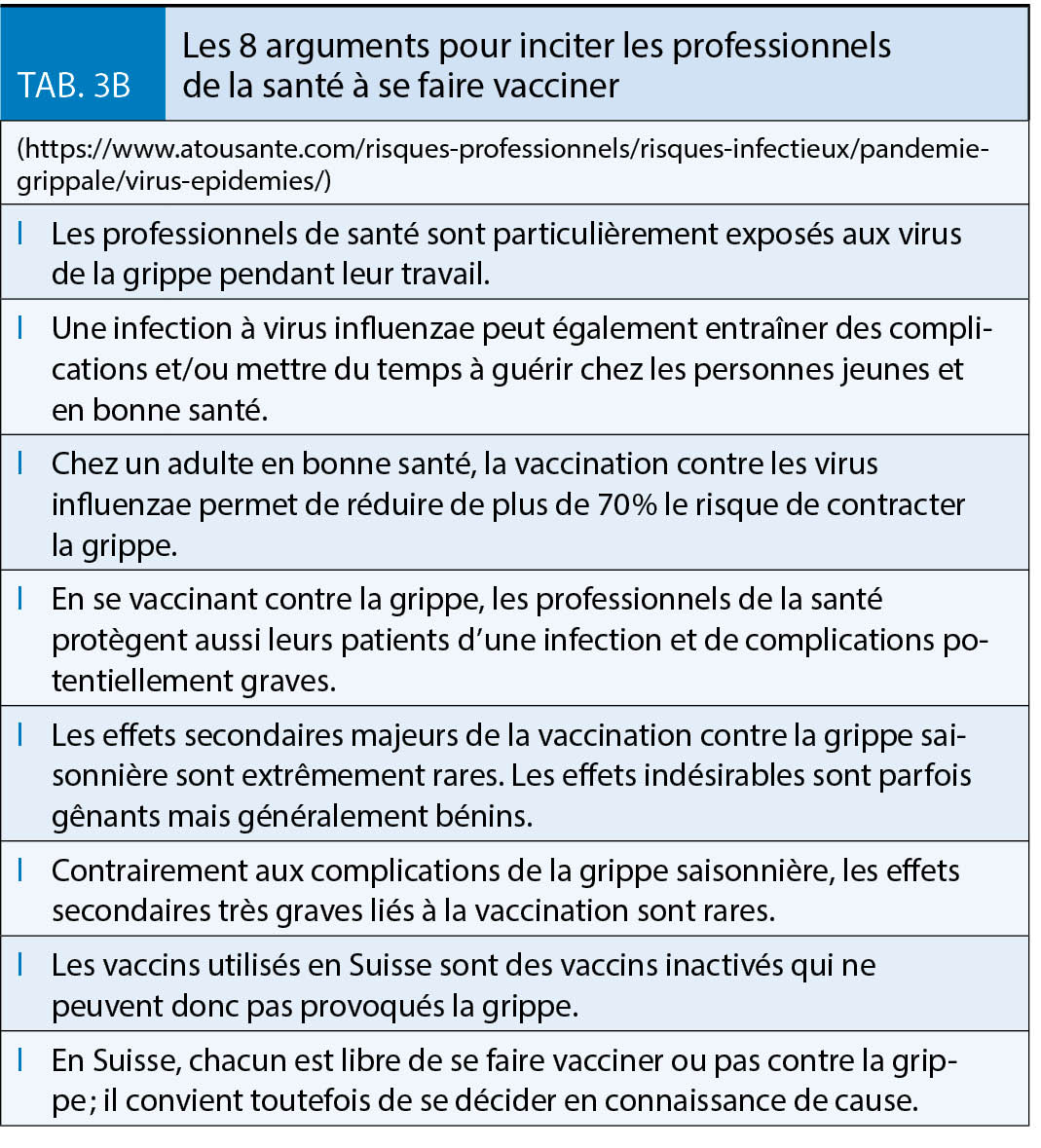
La grippe est l’ infection qui, en Suisse, cause chaque année le plus de décès et notamment parmi les plus vulnérables. Bien que les antiviraux et les vaccins contribuent à réduire le fardeau sanitaire et économique de la grippe, les épidémies continuent de faire des ravages. Si les mesures de protection individuelles (port de masque et hygiène des mains) sont un bon complément, la vaccination reste le pilier en matière de prévention. Il faut continuer à redoubler d’ effort pour améliorer les taux de couverture vaccinale chez les patients à risque et les professionnels de santé
(Tableaux 1, 3A et 3B).
Genolier Klinik und Montchoisi Klinik
Route du Muids 3
1272 Genolier
plang@genolier.net
plang@genolier.net
L’ auteur n’ a déclaré aucun conflit d’ intérêts en relation avec cet article.
1. Petrova VN, Russell CA: The evolution of seasonal influenza viruses. Nat Rev Microbiol 2018, 16:47-60.
2. Lang PO, Mendes A, Socquet J, Assir N, Govind S, Aspinall R: Effectiveness of influenza vaccine in aging and older adults: comprehensive analysis of the evidence. Clin Interv Aging 2012, 7:55-64.
3. Demicheli V, Jefferson T, Di Pietrantonj C, Ferroni E, Thorning S, Thomas RE, Rivetti A.: Vaccines for preventing influenza in the elderly. Cochrane Database Syst Rev 2018, 2:CD004876. doi: 004810.001002/14651858.CD14004876.pub14651854.
4. Demicheli V, Jefferson T, Ferroni E, Rivetti A, Di Pietrantonj C.: Vaccines for preventing influenza in healthy adults. Cochrane Database Syst Rev 2018, 2:CD001269. doi: 001210.001002/14651858.CD14001269.pub14651856.
5. Jefferson T, Rivetti A, Di Pietrantonj C, Demicheli V: Vaccines for preventing influenza in healthy children. Cochrane Database Syst Rev 2018, 2:CD004879. doi: 004810.001002/14651858.CD14004879.pub14651855.
6. Aspinall R Lang PO: The Avalanche is Coming … And Just Now It’ s Starting to Snow. Front Immunol 2013, Jun 25;4:165.
7. Office fédéral de la santé publique (OFSP): Grippe saisonnière 2017/2018 : réduire le risque de maladie pour soi et ses proches. OFSP – Bulletin 2017, 41:10-13.
8. Lang PO, Samaras D, Samaras N, Govind S, Aspinall R: Influenza vaccination in the face of immune exhaustion: is herd immunity effective for protecting the elderly? Influenza Res Treat 2011, 2011:419216.
9. Maltezou HC, Theodoridou K, Ledda C, Rapisarda V: Vaccination of healthcare personnel: time to rethink the current situation in Europe. Future Microbiol 2019, 14:5-8.
10. Imai C, Toizumi M, Hall L, Lambert S, Halton K, Merollini K: A systematic review and meta-analysis of the direct epidemiological and economic effects of seasonal influenza vaccination on healthcare workers. PLoS One 2018, 13::e0198685. doi: 0198610.0191371/journal.pone.0198685. eCollection 0192018.
11. Seki M, Fuke R, Oikawa N, Hariu M, Watanabe Y: Association of influenza with severe pneumonia/empyema in the community, hospital, and healthcare-associated setting. Respir Med Case Rep 2016, 19:1-4.
12. Mauskopf J, Klesse M, Lee S, Herrera-Taracena G: The burden of influenza complications in different high-risk groups: a targeted literature review. J Med Econ 2013, 16:264-277.
13. Offeddu V, Yung CF, Low MSF, Tam CC: Effectiveness of Masks and Respirators Against Respiratory Infections in Healthcare Workers: A Systematic Review and Meta-Analysis. Clin Infect Dis 2017, 65:1934-1942.
14. MacIntyre CR, Chughtai AA: Facemasks for the prevention of infection in healthcare and community settings. BMJ 2015 Apr 9, 350:h694.
15. Prévention de la grippe et des infections respiratoires virales saisonnières (https://www.hcsp.fr/explore.cgi/avisrapportsdomaine?clefr=521)
16. Lang PO, Aspinall R: Vitamin D status and the host resistance to infections: What it is currently (not) understood. Clin Ther 2017, 39:930-945.
17. Ginde AA, Blatchford P, Breese K, Zarrabi L, Linnebur SA, Wallace JI, Schwartz RS: High-dose monthly Vitamin D for prevention of acute respiratory infection in older long-term care residents: A randomized clinical trial. J Am Geriatr Soc 2017, 65:496-503.
18. Lang PO, Aspinall R: Can we translate vitamin D immunomodulating effect on innate and adaptive immunity to vaccine response? Nutrients 2015, 7:2044-2060.
19. Aspinall R, Lang PO,: Vaccine responsiveness in the elderly: best practice for the clinic. Expert Rev Vaccines 2014, 13:885-894.
20. Aspinall R, Lang PO,: Vaccination choices for older people, looking beyond age specific approaches. Expert Rev Vaccines 2018, 17:23-30.
21. McLean HQ, Thompson MG, Sundaram ME, et al: Influenza vaccine effectiveness in the United States during 2012-2013 : variable protection by age and virus type. Clin Infect Dis 2014, 59:1375-1385
22. Skowronski DM, Chambers C, Sabaiduc S, et al: A perfect storm : Impact of genomic variation and serial vaccination on low influenza vaccine effectiveness during the 2014-2015 season. Clin Infect Dis 2016, 63:21-32.
23. Bartoszko JJ, McNamara IF, Aras OAZ, Hylton DA, Zhang YB, Malhotra D, Hyett SL, Morassut RE, Rudziak P, Loeb M.: Does consecutive influenza vaccination reduce protection against influenza: A systematic review and meta-analysis. Vaccine 2018, 36:3434-3444.
24. Carlock MA, Ungram JG, Clutter EF, Cecil NC, Ramgopal M, Zimmerman RK, Warren W, Kleanthous H, Ross TM: Impact of age and pre-existing influenza on the induction of human antibody responses against influenza B viruses. Hum Vaccin Immunother 2019, Jul10 doi: 10.1080/21645515.2019.1642056. (Epub ahead of print).
25. Ramsay LC, Buchan SA, Stirling RG, Cowling BJ, Feng S, Kwong JC, Warshawsky BF: The impact of repeated vaccination on influenza vaccine effectiveness: a systematic review and meta-analysis. BMC Med 2019, 17:9.
26. Lang PO, Bonduelle O, Benhabiles N, Combadières B: Prior contacts with the 2000-2003 seasonal vaccines extends the 2009 pandemic A/H1N1 vaccine-specific immune protection to non-humoral compartments. Eur Geriatr Med 2014, 5:136-138.
27. Trucchi C, Paganino C, Amicizia D, Orsi A, Tisa V, Piazza MF, Icardi G, Ansaldi F: Universal influenza virus vaccines: what needs to happen next? Expert Opin Biol Ther 2019, 19:671-683.
28. Lang PO, Aspinall R,: Vaccination in the elderly: what can be recommended? Drugs Aging 2014, 31:581-599.
29. Smith TC: Vaccine rejection and hesitancy: a review and call to action. Open Forum Infect Dis 2017, 4:ofx146.
30. Yen HL: Current and novel antiviral strategies for influenza infection. Curr Opin Virol 2016, 18:126-134.
31. Szollosi DE, Bill A: Potential role of endonuclease inhibition and other targets in the treatment of influenza. Curr Drug Targets 2019, Aug 1 doi: 10.2174/1389450120666190801115130. (Epub ahead of print). : .
32. Organisation Mondiale de la Santé (OMS): Composition recommandée des vaccins antigrippaux pour la saison grippale 2014-2015 dans l’ hémisphère Nord. Wkly Epidemiol Rec 2014, 89:93-104.
33. Sedeyn K, Saelens X: New antibody-based prevention and treatment options for influenza. Antiviral Res 2019, 170:104562.