Les patients ayant subi une intervention bariatrique ont besoin de contrôles réguliers afin de détecter et de traiter les complications et les progressions défavorables à un stade précoce. Comme le nombre de patients recevant une chirurgie bariatrique pour traiter l’ obésité et ses comorbidités augmente rapidement, il devient de plus en plus important pour les médecins d’ avoir une connaissance pratique des principales conséquences de la chirurgie bariatrique. Les thèmes importants lors de ce suivi comprennent la perte de poids, la détection précoce d’ une évolution défavorable, la détection et le traitement des carences en macro- et micronutriments, les symptômes gastro-intestinaux tels que la douleur, le dumping, etc. L’ article suivant donne un aperçu de ces sujets et décrit les options de traitement.
L’ augmentation de l’ obésité au cours des dernières décennies a mené, entre autres, à une utilisation accrue des opérations bariatriques. L’ étude SOS (Swedish Obese Subjects) (1) a documenté de manière impressionnante les succès continus en matière de perte de poids, d’ amélioration de la comorbidité et de réduction de la mortalité chez les patients ayant subi une chirurgie, ce qui a conduit à une utilisation encore plus importante de ces opérations. Il est donc de plus en plus important que les médecins en pratique se familiarisent avec les questions et les problèmes pouvant survenir après une chirurgie bariatrique. L’ article suivant donne un aperçu des questions relatives à la pratique et vise à fournir des conseils sur la façon de procéder dans des situations typiques.
En Suisse, des directives pour le traitement chirurgical de l’ obésité et les soins de suivi après une chirurgie bariatrique ont été élaborées par la SMOB (Swiss Society for the Study of Morbid Obesity and Metabolic Disorders), sont disponibles sur la page d’ accueil www.smob.ch et sont régulièrement actualisées. Selon ces directives, les centres bariatriques sont tenus d’ assurer le suivi de leurs patients tout au long de leur vie. Ce suivi est assuré par les centres bariatriques en collaboration avec les médecins de famille. Selon les directives du SMOB, les centres doivent pouvoir démontrer un taux de suivi d’ au moins 75 % dans les 5 premières années postopératoires. Les contrôles de suivi doivent être effectués deux, quatre, huit et 12 semaines après l’ opération, puis tous les trois mois, dans la deuxième année postopératoire tous les six mois, puis tous les ans, avec dans chaque cas un contrôle en laboratoire de la situation des micronutriments, ou plus fréquemment en cas de problèmes particuliers. Les consultations doivent inclure une évaluation et des conseils concernant l’ évolution du poids, la situation nutritionnelle et de l’ activité physique, ainsi que des comorbidités, en plus de questions individuelles qui doivent aussi être abordées.
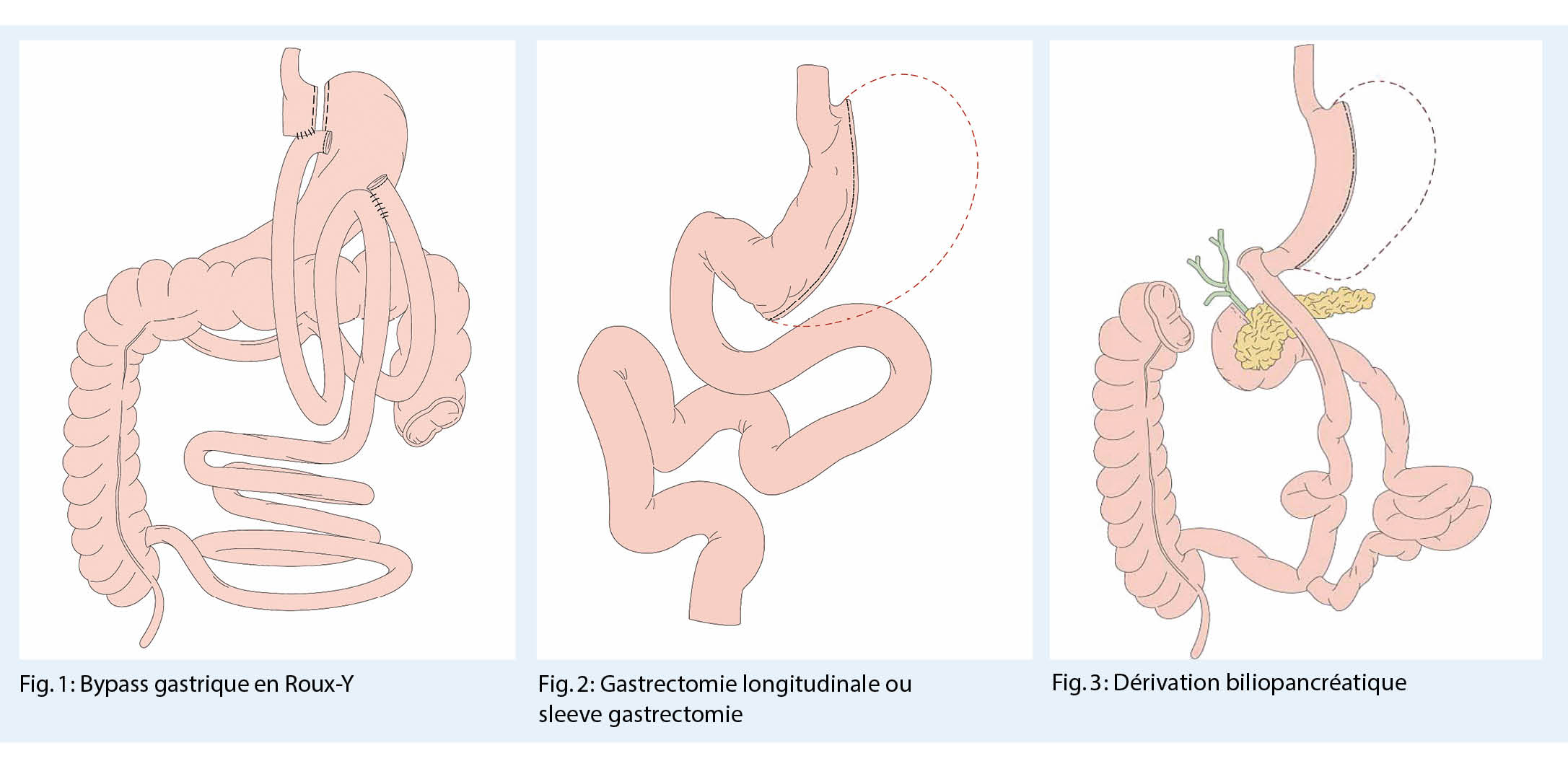
L’ opération bariatrique la plus fréquente est le bypass (ou pontage) gastrique, généralement sous la forme du pontage gastrique proximal Roux-Y (Fig. 1), qui agit à la fois de manière restrictive et malabsorptive. L’ effet restrictif est particulièrement marqué après la gastrectomie longitudinale ou gastrectomie sleeve (Fig. 2) ou après l’ implantation d’ un l’ anneau gastrique. Cette dernière était l’ opération standard jusqu’ en 2005 mais n’ est plus utilisée aujourd’ hui en raison de l’ intolérance, de la dysmotilité œsophagienne et des symptômes de reflux. La perte de poids la plus importante survient après une opération de diversion biliopancréatique (DBP) (Fig. 3). Cette opération entraîne une forte malabsorption. En plus des effets restrictifs et malabsorbtifs, ces opérations agissent toutes en modifiant les composantes neuroentéro-humorales, c’ est-à-dire en modifiant les hormones peptidiques gastro-intestinales et en influençant les boucles de contrôle centrales pour réguler la saturation et la récompense. La modification du microbiote après l’ intervention chirurgicale joue également un rôle dans la perte de poids.
Courbe de poids et évolutions défavorables
La perte de poids moyenne 5 ans après le pontage gastrique est de 70 % de perte du poids excessif (LEF), d’ environ 60 % après la gastrectomie longitudinale et d’ environ 80 % après la dérivation biliopancréatique, le nadir étant atteint 12-18 mois après la chirurgie. Par la suite, le but devrait être la stabilisation du poids. Un gain de poids secondaire d’ environ 5 % à 10 % est considéré comme normal et multifactoriel. Toutefois, il convient de noter qu’ il n’ y a pas de limite claire à ce qui constitue une perte de poids suffisante. L’ évolution du poids doit également être évaluée dans le contexte des comorbidités.
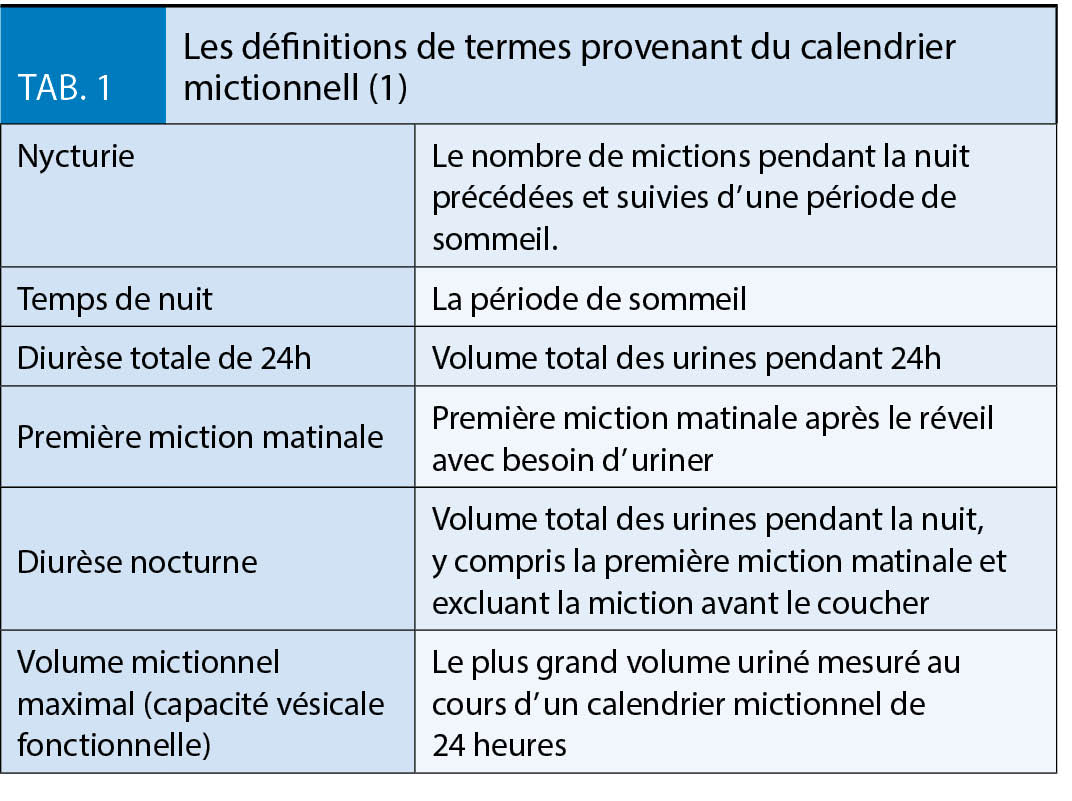
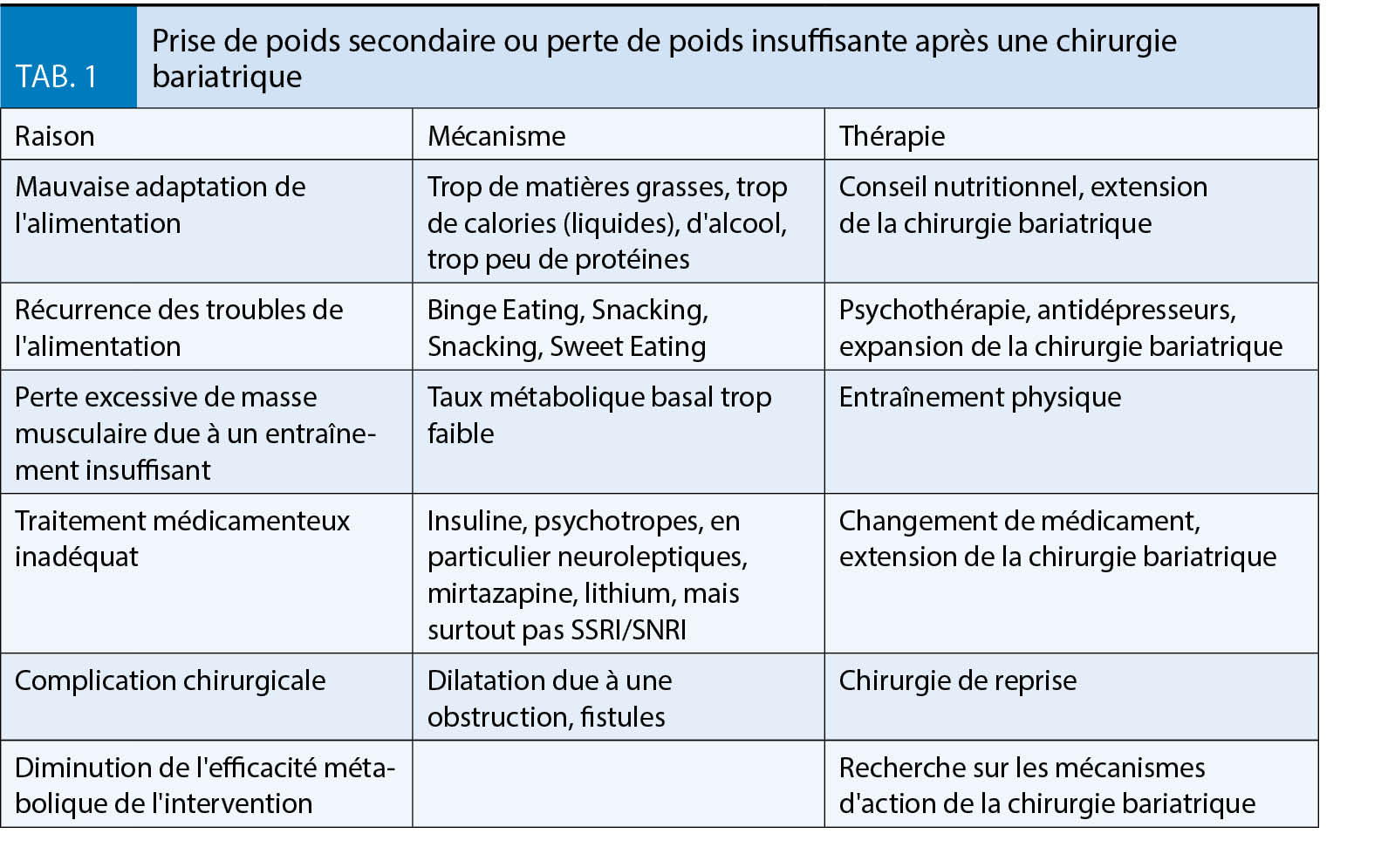
Le tableau 1 donne un aperçu des facteurs et des approches thérapeutiques en cas de perte de poids insuffisante ou d’ augmentation du gain de poids secondaire.
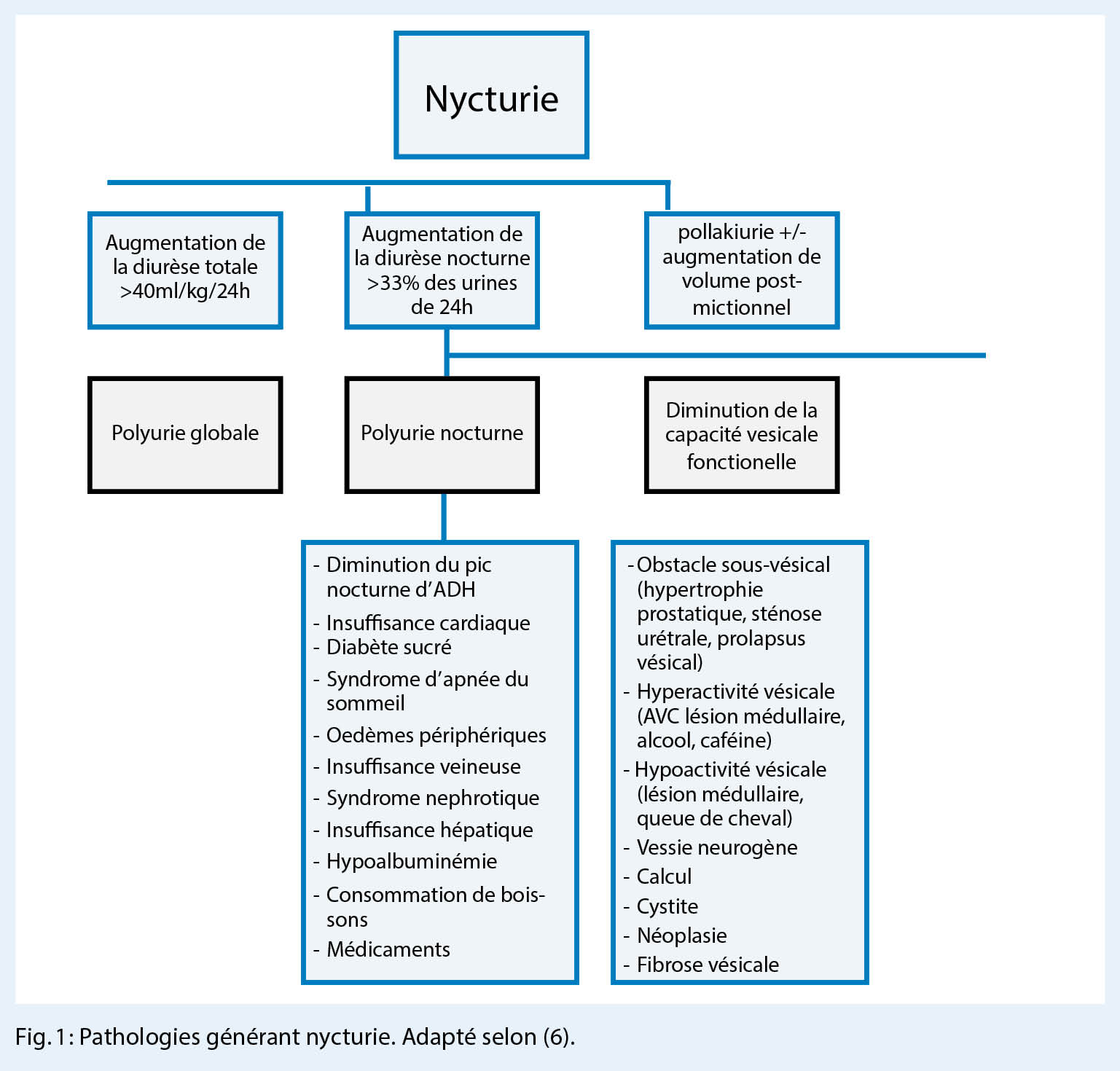
Un risque accru de perte de poids supérieure à la moyenne avec détérioration de l’ état général, de la masse musculaire et une perte de force peut survenir dans le cadre d’ une dépression, d’ une adhésion insuffisante aux recommandations alimentaires, en particulier un apport insuffisant en protéines, du développement d’ un trouble alimentaire anorexique secondaire, d’ un abus d’ alcool, d’ une toxicomanie, de tumeurs ou de maladies chroniques, en particulier la BPCO. Un risque accru de malnutrition et de développement d’ une insuffisance pondérale existe surtout après les interventions fortement malabsorptives, surtout après le détournement biliopancréatique.
Carences en micronutriments
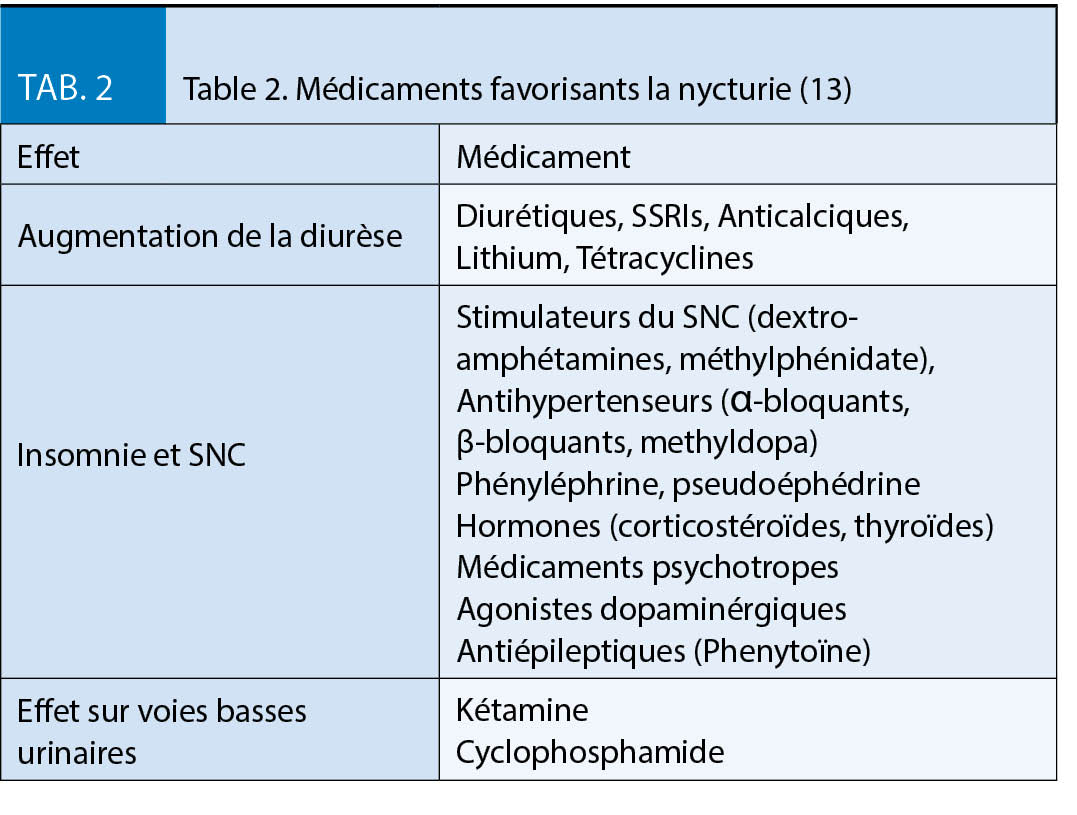
Après la chirurgie bariatrique, il existe un risque de carence en micronutriments dû aux faibles quantités d’ aliments ingérées et à la malabsorption, c’ est pourquoi une supplémentation en micronutriments à vie est essentielle (2, 3, 4). Pour répondre aux besoins des patients obèses, des préparations spécialement développées pour les patients obèses (par exemple WLS forte® de FitForMe ou Multi® de Bariatric Advantage) ou une préparation multivitaminée telle que Supradyn® peuvent être prises en alternance avec une vitamine complexe B. Des contrôles réguliers en laboratoire sont toujours indiqués afin de détecter et de traiter les défauts à un stade précoce. Il est important de noter que le risque de symptômes de carence dépend du type d’ opération, c’ est-à-dire que les opérations avec une forte composante malabsorptive, en particulier la chirurgie de dérivation biliopancréatique, mais aussi les opérations de dérivation excluant des sections plus longues de l’ intestin grêle (OAGB, BGRY avec une boucle bilio-pancréatique extra longue) ont un risque considérablement accru de carences en micronutriments. En particulier, le risque de carence en vitamines liposolubles (vitamines A, D, E, K) et de carences en sélénium ou en cuivre par ailleurs rares augmente considérablement. Après une intervention chirurgicale avec des composants purement restrictifs, comme par exemple une gastrectomie longitudinale ou une gastroplastie, le risque est nettement plus faible quoique toujours présent.
D’ autres raisons pour lesquelles les carences surviennent fréquemment après les opérations bariatriques sont les aversions gustatives, la mauvaise compliance des patients dans la prise de suppléments, l’ information insuffisante des patients, peut-être aussi un besoin accru pour d’ autres causes, par exemple une perte supplémentaire de fer due à l’ hyperménorrhée. Un autre problème est le fait que les suppléments ne sont pas pris en charge par l’ assurance maladie obligatoire. Les carences en micronutriments sont déjà fréquentes avant les opérations bariatriques et doivent être traitées déjà avant l’ opération.
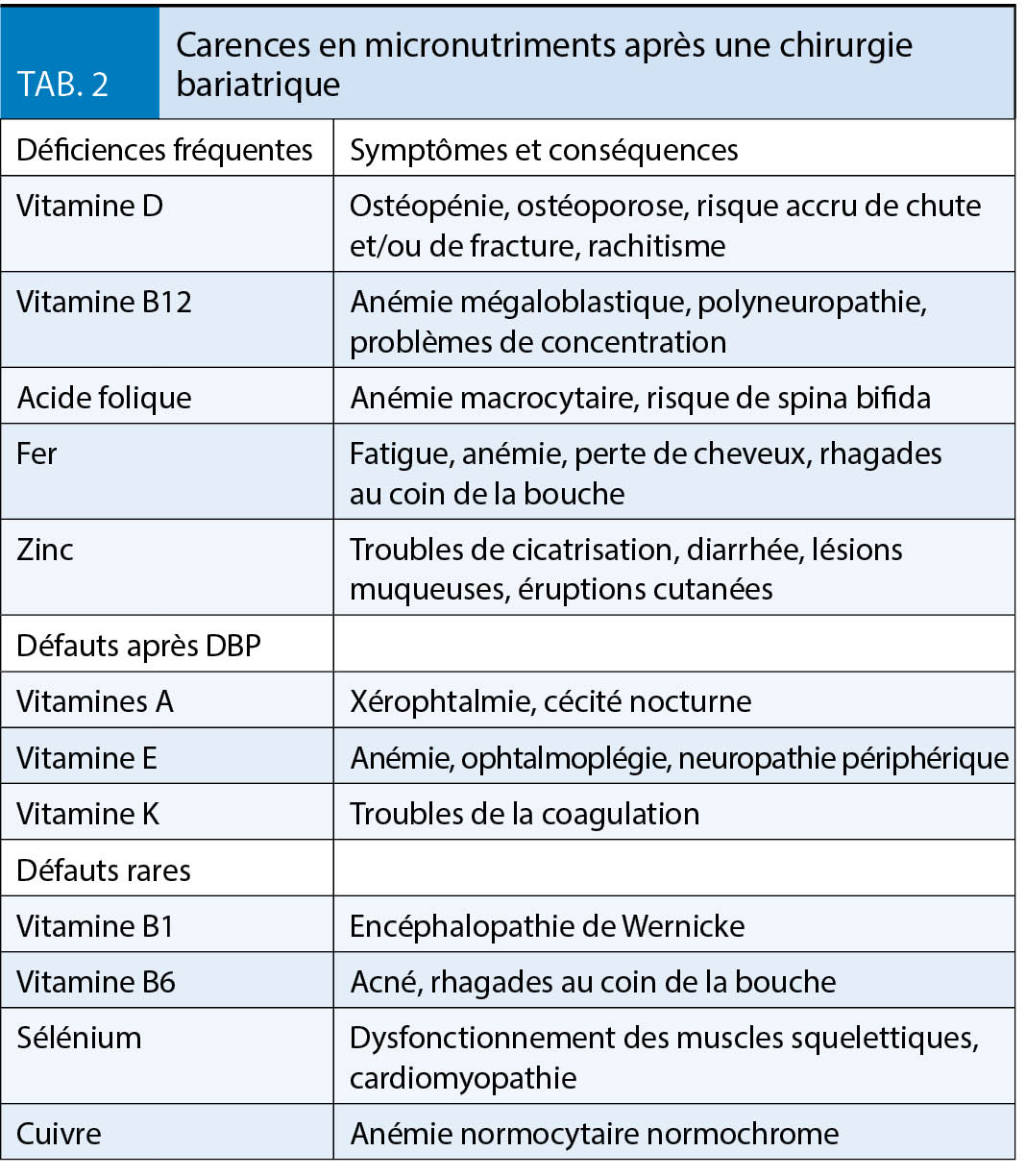
Les carences les plus importantes après une opération bariatrique, y compris les symptômes et les conséquences, sont énumérées au tableau 2.
L’ obésité est en soi associée à une carence en vitamine D, de sorte qu’ une carence doit déjà être recherchée et traitée déjà avant l’ opération. L’ absorption du calcium chute fortement après un pontage gastrique. Pour la prophylaxie de l’ ostéoporose après un pontage gastrique et une chirurgie de dérivation biliopancréatique, une supplémentation en calcium-D3 est nécessaire pour couvrir le besoin accru et éviter le développement d’ une hyperparathyroïdie secondaire. Selon les directives de l’ American Society of Metabolic and Bariatric Surgery (ASMBS), un apport total en calcium de 1500 mg/jour après une gastrectomie sleeve, de 1500 à 2000 mg/jour après un pontage gastrique et de 1800 à 2400 mg après une dérivation biliopancréatique est recommandé, étant bien entendu que l’ apport alimentaire devrait continuer à couvrir autant que possible les besoins (2, 3). Il est important que la supplémentation en calcium D3 soit prise avec un certain décalage par rapport aux autres suppléments, en raison du risque d’ une inhibition mutuelle de l’ absorption.
La carence en zinc est fréquente après une chirurgie bariatrique. Dans une étude, 9 % des 324 patients présentaient déjà une carence en zinc en phase préopératoire, contre 42.5 % 12 mois après l’ opération. Les raisons en étaient, d’ une part, le manque de compliance à la supplémentation et, d’ autre part, une absorption de zinc fortement réduite. L’ absorption fractionnée du zinc diminue après pontage de 32,3 % en préopératoire à 13,6 % 6 mois en postopératoire et à 21 % 12 mois après l’ opération. Pour cette raison, des contrôles réguliers en laboratoire doivent également déterminer la teneur en zinc comme marqueur de l’ apport en oligo-éléments. Il convient de noter que le taux de zinc dans le sérum n’ est pas une méthode fiable pour diagnostiquer une carence en zinc, car seulement 0,1 % de la teneur totale en zinc est dissous dans le sérum et la concentration sérique en zinc peut également être affectée par une réaction de phase aiguë (5).
La carence en vitamine B1 mérite une attention particulière. Les réserves de vitamine B1 sont faibles, c’ est pourquoi une carence en vitamine B1 peut survenir après seulement 2 semaines environ si l’ apport est insuffisant et en cas de vomissements à répétition. La triade classique de Wernicke avec ataxie motrice, parésie des muscles oculaires et confusion n’ est pas toujours entièrement présente, mais une carence en vitamine B1 non traitée peut provoquer des déficits neurologiques irréversibles. Si une carence en vitamine B1 est suspectée, le traitement approprié (thiamine 100 mg iv) doit donc être initié avant que les résultats de laboratoire soient disponibles (6).
Carences en macronutriments
Une instruction nutritionnelle détaillée est indispensable après la chirurgie bariatrique, en particulier en ce qui concerne l’ apport en protéines, car une perte de masse musculaire supérieure à la moyenne est à craindre en cas d’ apport insuffisant. L’ objectif est un apport protéique de 1 g de protéines/KG de poids normal. La prise de boissons protéinées (shakes) est généralement nécessaire dans les premiers mois postopératoires afin d’ atteindre cet objectif et de répondre à la demande. Surtout après des opérations avec une malabsorption accrue, une malnutrition protéique sévère avec perte de force, une diarrhée chronique et un œdème généralisé peuvent encore survenir des années après l’ opération. Une hypalbuminémie est souvent observée en laboratoire. Sur le plan thérapeutique, un apport protéique à forte dose peut être obtenu, selon la situation clinique, par des suppléments alimentaires, des compléments protéiques, dans les cas graves également par l’ administration de protéines par une sonde gastrique, ou, si nécessaire, par voie parentérale (4).
Symptômes gastro-intestinaux après une chirurgie bariatrique
Les symptômes gastro-intestinaux après une chirurgie bariatrique sont fréquents, le plus souvent avec plus d’ une diarrhée par semaine chez 23 % des patients, le dumping chez 13 %, des douleurs abdominales chez 10 %, une dysphagie chez 5 %, des vomissements chez 4 %. Une fréquence > 1 x mois est rapportée pour la diarrhée chez 24 % des patients, le dumping chez 27 %, les douleurs abdominales chez 15 %, les vomissements chez 15 %, la dysphagie chez 7 % (7). Les causes particulières de la douleur après une chirurgie bariatrique et le syndrome de dumping sont brièvement expliquées ci-dessous.
Douleurs
Des douleurs survenant après une chirurgie bariatrique peuvent avoir des causes multiples. Une anamnèse précise et l’ examen clinique est généralement utile et capitale. Une clarification précise est importante afin de ne pas rater d’ éventuelles complications graves. Le tableau 3 montre le diagnostic différentiel des douleurs abdominales après une chirurgie bariatrique.
Sur la base de la clinique et de l’ anamnèse, un diagnostic suspect est posé, ce qui détermine la suite de la clarification (tableau 4).
La cause la plus dangereuse de douleurs abdominales est l’ apparition d’ une obstruction des parties exclues de l’ intestin grêle avec accumulation dans l’ estomac exclu, ce qu’ on appelle l’ obstruction du pontage. Les patients éprouvent d’ importantes nausées, mais ne peuvent pas vomir, de même que des douleurs dans la partie supérieure de l’ abdomen ou du dos. Les patients sont extrêmement stressés et donc tachycardes. Le diagnostic est fait par CT abdominal, une radiographie abdominale conventionnelle pouvant être faussement négative en raison d’ un manque de niveau liquides. Dans cette situation une intervention chirurgicale est indiquée sans délai. Une cause beaucoup plus fréquente de douleur abdominale est la hernie interne. Cette complication est favorisée par la réduction de la graisse mésentérique et ne se produit donc généralement qu’ après une perte de poids importante. Une anse de l’ intestin grêle est herniée au niveau des lacunes mésentériques, soit entre le mésocôlon transverse et le méso de l’ anse montée, soit au niveau de l’ espace dit de «Petersen», ce qui peut entraîner une obstruction et une ischémie de l’ intestin grêle. Les symptômes typiques sont des douleurs sévères épigastriques ou de l’ abdomen moyen avec exacerbation postprandiale, dans certains cas des vomissements; en phase précoce les symptômes sont souvent spasmodiques, évoluant par la suite dans une douleur permanente. Une hernie interne peut cependant également se présenter de manière atypique, c’ est-à-dire avec seulement une douleur intermittente. La fréquence indiquée dans la littérature est d’ environ 2,5 à 10 % (8). Le CT abdominal est utile pour le diagnostic, avec une attention particulière accordée à une composante de rotation des vaisseaux mésentériques («whirl sign»). Une intervention chirurgicale rapide par un chirurgien expérimenté en bariatrie est indiquée.
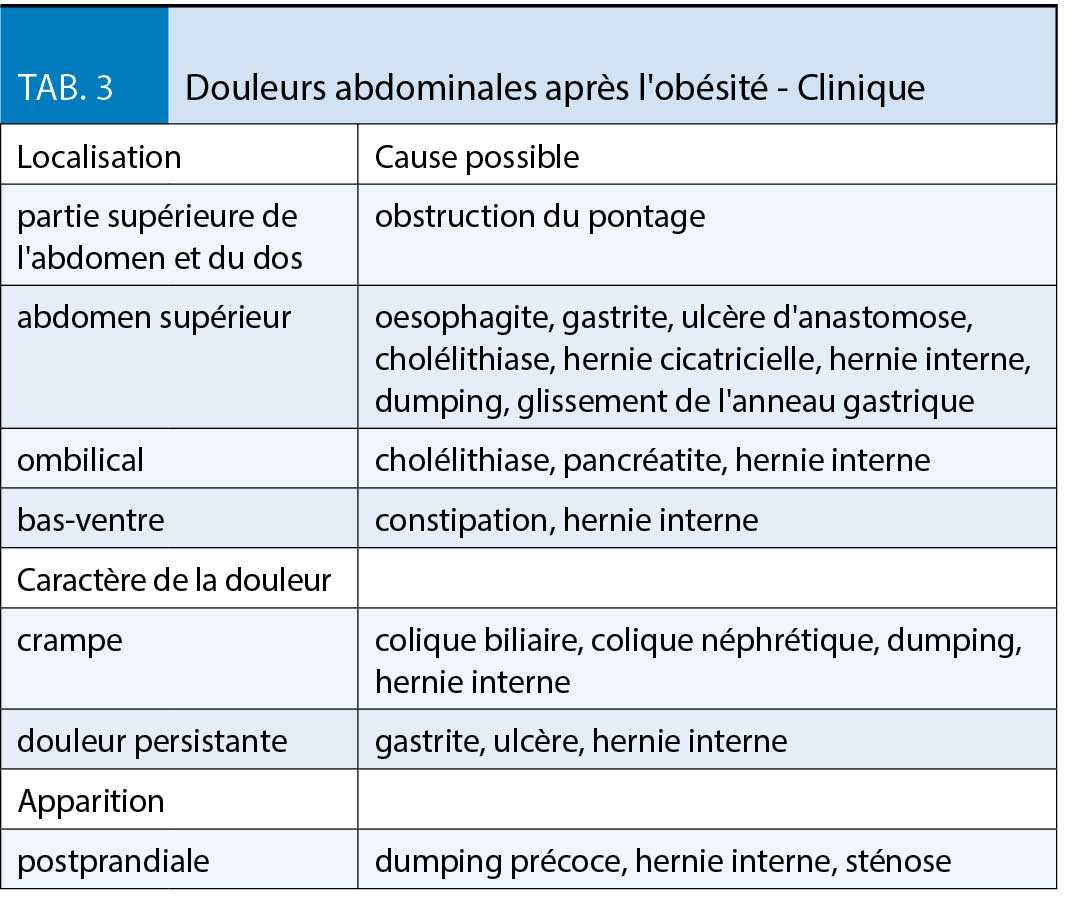
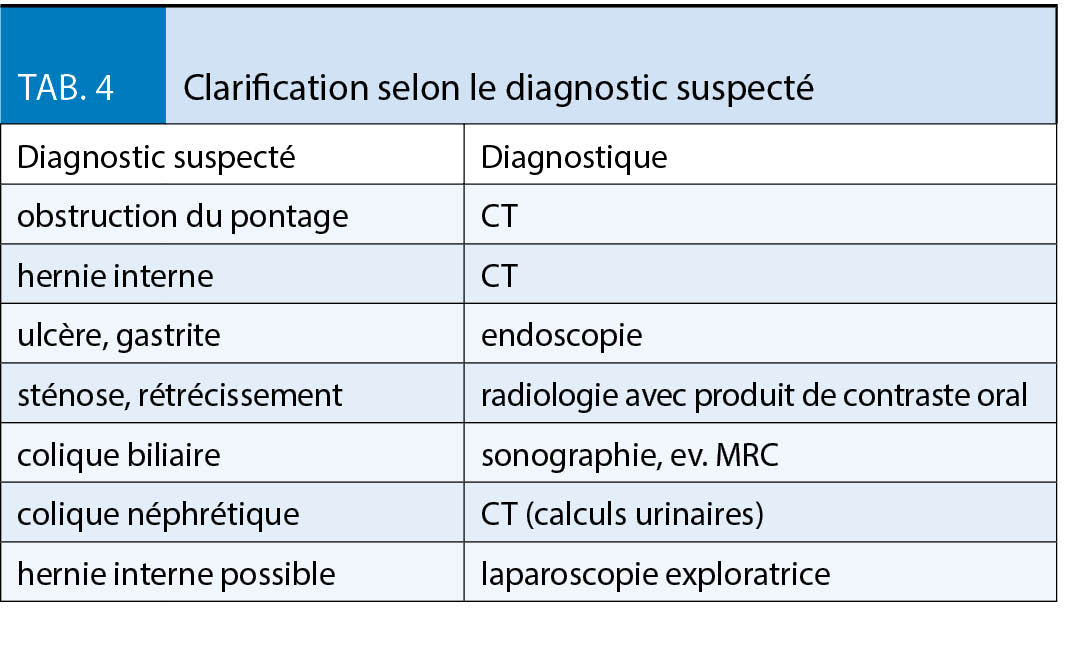
Les ulcères de la muqueuse gastrique sont divisés en ulcères précoces et tardifs (9). Habituellement, les ulcères sont situés dans la zone de l’ anastomose. Les ulcères précoces apparaissent jusqu’ à 10 mois après l’ opération. La cause la plus probable est l’ ischémie ou l’ inflammation. Les facteurs de risque pour le développement d’ un ulcère tardif sont la contamination acide du jéjunum, par exemple par l’ élargissement de la poche, l’ abus de nicotine, la prise d’ AINS et le diabète sucré. La clinique typique pour les ulcères est une douleur épigastrique sévère pendant l’ alimentation. Pour établir le diagnostic, une endoscopie doit être effectuée, le traitement consiste en un traitement par inhibiteur de la pompe à protons (IPP) pendant des mois. Une étude a montré que les formulations solubles, c’ est-à-dire les capsules ouvertes permettent une guérison plus rapide de l’ ulcère que les capsules non ouvertes (10). Une infection par Helicobacter pylori, éventuellement aussi une persistance malgré l’ éradication préopératoire, doit être recherchée et traitée si elle est présente. L’ abstinence de nicotine est fortement recommandée.
Dumping
Un symptôme courant après un pontage gastrique est l’ apparition de symptômes de dumping. Nous faisons la distinction entre le dumping précoce et le dumping tardif. Les mécanismes physiopathologiques du syndrome du dumping ne sont pas bien élucidés. Un des mécanismes possibles est une vidange rapide de la poche de l’ estomac. Le transfert rapide d’ aliments hautement osmolaires, en particulier de glucides isolés, dans l’ anse jéjunale montée déclenche un afflux de liquide dans la lumière intestinale et donc une hypotension, parfois jusqu’ au collapsus, des étourdissements, de la fatigue, des crampes et de la diarrhée. Ce dumping précoce se produit de 0 à 30 minutes après le début du traitement. Le dumping tardif se produit 90 à 120 minutes après un repas contenant des glucides et est causé par une réponse insulinique excessive à la forte concentration de glucides dans l’ intestin grêle, entraînant une hypoglycémie avec les symptômes classiques de la transpiration, des tremblements, une altération de la vision et une diminution de la concentration.
Si les symptômes ne sont pas clairs, il est utile d’ obtenir un protocole de l’ alimentation et des symptômes, combiné avec mesure de glycémie. La surveillance continue de la glycémie peut également être utile en cas d’ incertitude. Sur le plan thérapeutique, il est très important de suivre les recommandations diététiques (pas de repas de glucides purs, intervalle de 30 minutes entre les repas, petits repas réguliers, augmentation de la consommation de fibres, éventuellement de fibres solubles, par ex. Optifiber®). Si les symptômes persistent, on peut essayer un traitement médicamenteux à l’ acarbose pour stabiliser la glycémie ; dans les cas de résistance au traitement, l’ utilisation de liraglutide ou d’ octréotide est également recommandée. En cas de perte de poids insuffisante et de symptômes de décharge, l’ insertion d’ un anneau en silicone, appelé anneau de Fobi, autour de la poche gastrique peut être envisagée. Il en résulte une restriction accrue, une vidange plus lente de la poche et donc une amélioration des symptômes de dumping. Dans le cas d’ un dumping résistant au traitement et d’ une perte de poids supérieure à la moyenne, les experts croient que l’ administration d’ une alimentation entérale continue par le biais d’ un cathéter de gastrostomie dans l’ estomac exclu constitue une option thérapeutique. La dernière option thérapeutique est la réversion du pontage, mais il faut écarter les rares diagnostics différentiels d’ hypoglycémie, comme la présence d’ un insulinome ou d’ une insuffisance surrénalienne. Les médicaments, en particulier la venlafaxine, peuvent également augmenter l’ incidence de l’ hypoglycémie.
Médecin-cheffe Médecine interne / Endocrinologie
Claraspital Centre de Nutrition / Clarunis Bariatric Reference Centre
Lukas Legrand-Strasse 4
4058 Bâle
martina.gebhart@claraspital.ch
L’ auteur n’ a déclaré aucun conflit d’ intérêts en rapport avec cet article.
1. Sjöström L. A Review of the key results from the Swedish Obese Subjects (SOS) trial – a prospective controlled intervention study of bariatric surgery. J Intern Med 2013;273:219-234.
2. Mechanick JI, Kushner RF, Sugerman HJ et al, American Association of Clinical Endocrinologists, Obesity Society, American Society for Metabolic and Bariatric Surgery, American Association of Clinical Endocrinologists, The Obesity Society, American Society for Metabolic and Bariatric Surgery. Medical guidelines for clinical practice for the perioperative nutritional, metabolic, and nonsurgical support of the bariatric surgery patient. Obesity 2009;17(Suppl 1):1-70.
3. Mechanick JI et al. Clinical practice guidelines for the perioperative nutritional, metabolic and nonsurgical support of the bariatric surgery patient – 2013 update: Cosponsored by american association of clinical endocrinologists, the obesity society and American society for metabolic & bariatric surgery. Endo Pract 2013;19:337-72.
4. Busetto L, Dicker D, Azran C. Practical recommendations of the Obesity Management Task Force of the European Association for the Study of Obesity for the post-bariatric surgery medical management. Obes Facts 2017;10:597-632.
5. Sallé A, Demarsy D, Poirier AL. Zinc deficiency: a frequent and underestimated complication of bariatric surgery. Obes Surg 2010;20:1660-70
6. Aasheim ET. Wernicke encephalopathy after bariatric surgery: a systematic review. Ann Surg 2008; 248: 714.
7. Edholm et al. Long-term results 11 years after primary gastric bypass in 384 patients. Surg Obes Rel Dis 2013;9:708-713.
8. Iannelli A, Facchiano E, Gugenheim J. Internal hernia after laparoscopic Roux-en-Y gastric bypass for morbid obesity. Obes Surg. 2006;16:1265-71.
9. Csendes A, Burgos AM, Altuve J et all. Incidence of marginal ulcer 1 month and 1 to 2 years after gastric bypass: A prospective consecutive endoscopic evaluation of 442 patients with morbid obesity. Obes Surg 2009;19:135-138.
10. Schulman AR, Chan WW, Devery A et all. Opened proton pump inhibitor capsules reduce time to healing for marginal ulcer after Roux-en-Y-Gastric Bypass. Clin Gastrol and Hepatol 2017;15:494-500.