Die diesjährige Jahrestagung der Schweizerischen Gesellschaft für Allgemeine Innere Medizin stand unter dem Motto Kreative Medizin, Erneuern und Weitergeben. Der folgende Beitrag beleuchtet das immerfort
aktuelle Thema der Raucherentwöhnung.
Eine Studie an einer Million Frauen, über die Gefahren des Rauchens im 21. Jahrhundert und die Vorteile des Rauchstopps im United Kingdom war der Einstieg in den Vortrag von Prof. Dr. med. Jacques Cornuz, Centre universitaire de médecine préventive et santé publique, Lausanne. Der Referent zeigte, dass schon eine Zigarette pro Tag das relative Risiko steigert; bei 20 Zigaretten steigt es auf 37 %. Die spezifische Mortalität umfasst Lungenkrankheit, Lungenkrebs und koronare Herzkrankheit. Die Lebenserwartung wird im Mittel um 11 Jahre verkürzt. Die Mortalität bei Frauen veränderte sich über die Zeit von 2001 bis 2018 von anfänglich einem Total von über 25/100 000 Frauen mit Brustkrebs und ca. 16 pro 100 0000 Frauen mit Lungenkrebs auf etwa gleich viel (ca. 17 pro 100 000) Fälle bei beiden Krebsarten.
rof. Dr. med. Jacques Cornuz Centre universitaire de médecine préventive et santé publique, Lausanne
Ein Vergleich der Jahre 1922 und 2022 des BAG zeigt, dass Männer weniger rauchen, insbesondere die über 65-Jährigen, aber dass im Jahre 2022 mehr über 65-jährige Frauen rauchten als 1922. Frauen rauchen aber generell immer noch weniger als Männer.
30 Jahre Erfahrung
1993–2004 erste Entwicklungen, 2004–2024 wichtigste Errungenschaften
– Ambulante Konsultationen (Patienten in städtischen Arztpraxen)
– Konsultationen für hospitalisierte Patienten
– Ausbildung von Praktikanten und Fachkräften
– 2014–2024 Konsolidierung und … Schwächung
Wirksamkeit der ambulanten Konsultation
Ein Schulungsprogramm zur Raucherentwöhnung, das Ärzten verabreicht wurde und auf Verhaltenstheorie und -praxis mit standardisierten Patienten basierte, erhöhte die Qualität der ärztlichen Beratung, die Motivation der Raucher, mit dem Rauchen aufzuhören, und ging mit einer signifikanten Abstinenzraten nach einem Jahr einher (Cornuz J et al. Annals of Internal Medicine 2002; 136, 6).
Die Wirksamkeit einer niedrig intensiven Rauchstopp-Intervention bei hospitalisierten Patienten ergab einen Rauchstopp bei 23.9 % der Interventionsgruppe gegenüber 9.7 % in der Kontrollgruppe.
Ausbildung
2002 Nationales schweizerisches Ausbildungsprogramm Programm für Ärzte zur Tabak-Entwöhnung «Leben ohne Tabak», Referenzdokument «Tabak-Entwöhnung».
Ungefähr 12 000 Ärzte wurden seit 2002 ausgebildet, ungefähr 800 Assistenzärzte in der Allgemeinmedizin (Policlinique médicale Unisanté Lausanne).
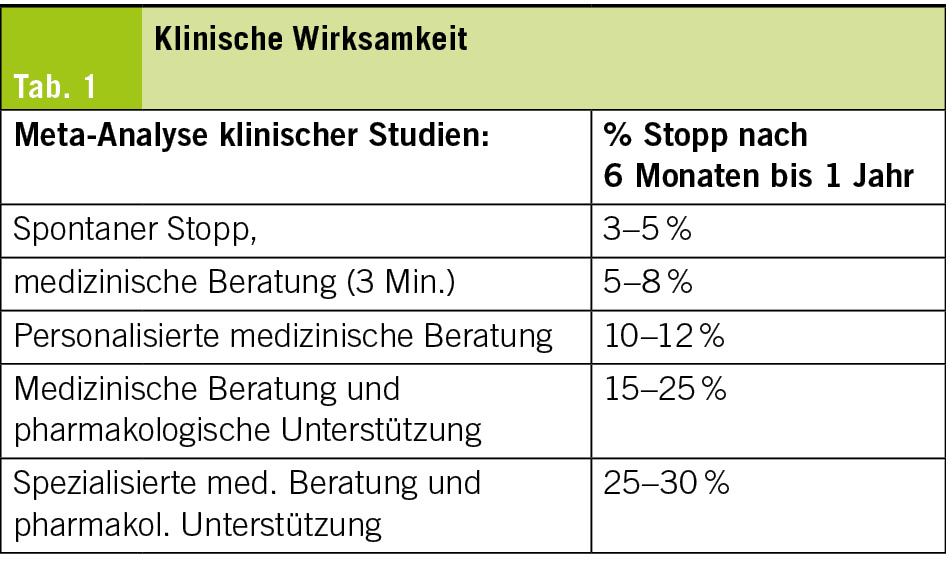
Klinische Wirksamkeit
Wirksamkeit der anderen Methoden
– Unwirksam
– Benzodiazepine (mehrere RCT), aversive Methoden
– Kontroverse Methoden, wahrscheinlich ineffektiv
– Niedrige Qualität: Hypnose
– Placebo-Effekt: Akupunktur (Sham-Akupunktur)
– Körperliche Aktivität: Nicht besser als die Kontrollen
Warum rauchen Raucher?
Die Persönlichkeit, die Umgebung, aber auch das Rauchprodukt spielen eine Rolle
Warum mit dem Rauchen aufhören? Die gängigsten und die erstaunlichsten Gründe sind Gesundheit; Ökonomie; Druck der Umgebung; Bild, das man vor Kindern abgibt; Krebs bei seinem… Hund; Auswirkungen auf die Umwelt; mit neuen Motivationen arbeiten.
Motivationsinstrumente:
Hervorheben der Auswirkungen des Rauchens, Spirometrie? Menge von CO? Genetische Marker? Stenose der Karotisarterie, Cochrane Analyse «biomedical risk assessment as an aid for smoking cessation». Eine Cochrane Analyse fand keine Belege dafür, dass es den Rauchern hilft, mit dem Rauchen aufzuhören, wenn sie eine Rückmeldung über ihre Rauchexposition, ihr genetisches Risiko für raucherbedingte Krankheiten oder die Auswirkungen des Rauchens auf ihren Körper erhalten. Am vielversprechendsten waren die Ergebnisse, wenn man den Menschen eine Rückmeldung darüber gab, wie schädlich das Rauchen für ihren Körper ist. In den Studien wurde nicht über die Nebenwirkungen des Feedbacks berichtet. Angesichts der Art der Messungen (Lungen- oder Bluttests) ist jedoch davon auszugehen, dass das Risiko einer Schädigung gering ist.
Risikowahrnehmung
Tendenzen zur Verringerung von freiwillig eingegangenen Risiken (Rauchen) im Vergleich zu erlittenen, aufgezwungenen Risiken (Militärdienst). Grössere Akzeptanz eines früheren Risikos (Rauchen). Das wahrgenommene Risiko ist wesentlich geringer als das aktuelle relative Risiko bezüglich Lungenkarzinom, wie eine Studie mit 6369 Erwachsenen, wovon 124 Raucher waren, zeigte (Weinstein ND et al. Smokers’ unrealistic optimism about their risk. Tobacco Control 2005;14:55–59).
Bias bei Rauchern und Raucherinnen
Optimismus-Bias, Bias der Verfügbarkeit (Mediatisierung von Ereignissen), Bias für die Präferenz der Gegenwart (kurzfristige Sichtweise), Bias der kognitiven Dissonanz.
Vorsorgeuntersuchungen bei Raucherinnen und Rauchern
– Brustkrebs
– Mortalität um ca. 15 % erhöht
– Screening wahrscheinlich wirksam bei Raucherinnen
– Kolonkarzinom
– Mortalität um ca. 10 % erhöht. Screening wirksam!
– Screening auf Lungenkrebs
– Verringerung der Mortalität um ca. 20–25 %.
Neue Motivationswege
Zur Erinnerung: Informationen sind notwendig, aber nicht ausreichend. Überwindung der Optimismus-Verzerrung durch Nutzung der Verfügbarkeitsverzerrung. Neue Gesundheitsgefahren durch Tabak! Neue Wege der Informationsaufnahme und der Behandlung eröffnen, so der Referent.
Studie TABARAD (Tabak und Polonium)
Der kanzerogene Effekt von Polonium ist bekannt. Aber vergessen.
Der Terminus «sleeping giant» ist aus den Archiven von Philip Morris seit 1978 verschwunden. «Politische» Vergiftungen (Alexander Litvinenko 2006).
Studie von Unisanté bei 25 Rauchern: Messung von Polonium-210 im Urin und Vergleich mit den Werten von Nichtrauchern.
Auswirkung des CO2-Ausstosses der Tabakindustrie
84 Mio. Tonnen CO2 pro Jahr = 1/3 der jährlichen Emissionen durch Frankreich.
Konsum und Abfall
Zigarettenstummel: Zwei Drittel werden in die Umgebung geworfen, bis zu 10 Milliarden jeden Tag.
Viele toxische Substanzen: Kohlenwasserstoffe, Quecksilber, Blei. Eine Zigarettenkippe verschmutzt bis zu 500 Liter Wasser.
Filter: Azetatzellulose, Mikroplastik: mehrere Jahre.
Neue Produkte
Vaporetten (elektronische Zigaretten)
Erhitzter Tabak
Oraler Tabak
Neue Nikotinverabreichungen oder Vaporetten
Flüssigkeit ist Propylenglykol und Glycerin (> 95 %) aromatisiert und mit Nikotin versetzt Generation: (cig-a-like), 2. Generation Cleararomizers), 3. Generation (Mods),
4. Generation (Pods), 5. Generation (Puff-Bars).
Zusammensetzung des Aerosols.
Elektronische Zigaretten
– Batterie zum Erhitzen einer Flüssigkeit, die in der Regel Niktoin enthält
– E-Liquid = Propylenglykol und Glycerin (> 95 %), Aromen und Nikotin (0–20 mg/ml).
– Sehr starke Verringerung der Anzahl und Konzentration toxischer Substanzen im Vergleich zum Zigarettenrauch. Vorhandensein einiger giftiger Substanzen. (Acetaldehyd, Formaldehyd, Acrolein, …) in unterschiedlicher Quantität entsprechend
– Zusammensetzung der Flüssigkeit, Wartung des Geräts (Widerstand alle 2–3 Wochen zu erneuern)
– Heiztemperatur, Spannung.
Sekundäreffekte des Vaporisierens
Langzeit-Sekundäreffekte sind keine bekannt. Kurzzeit: geringfügige Nebeneffekte: Husten, Mundtrockenheit, bukko-pharyngeale Reizung.
Seltene schwere Sekundärwirkungen: Pneumopathien aufgrund des Vitamin E Azetats in den Flüssigkeiten.
Nikotin-E-Zigaretten als Werkzeug für den Rauchstopp, Empfehlungen für Fachkräfte des Gesundheitswesens
Gebrauch ≥1mal in den letzten 30 Tagen, elektronische Zigaretten 25 %, herkömmliche Zigaretten 15 %.
Puffs führen zu grosser Umweltverschmutzung. Verbot der Puffs ist auch gut für die Umwelt. Die Bundesversammlung stimmt für ein Verbot der Puffs.
Erhitzte Tabakprodukte
Batteriebetriebenes Erhitzungssystem und spezielle Zigaretten oder Tabakkapseln. Der Tabak wird auf 250 bis 350 Grad erhitzt, thermochemischer Abbau des Tabaks (Pyrolyse). Immer noch Exposition gegenüber den wichtigsten toxischen Verbindungen des herkömmlichen Zigarettenrauchs, jedoch in deutlich geringeren Konzentrationen.
Oraler Tabak
Keine Verbrennung (kein CO, kein Teer). Vorhandensein von Nitrosaminen (kanzerogen), Schwermetallen, Nikotin mit hohem Suchtpotenzial. Auswirkungen auf die Gesundheit: Zunahme des Krebsrisikos des Mundes, der Speiseröhre, der Pankreasdrüse. Risiko für Herz-Kreislauf-Erkrankungen, Risiko der Frühgeburtlichkeit.
Snus
Oraler Tabak skandinavischer Herkunft mit reduzierter Menge an Nitrosaminen, Verkaufsverbot in der Europäischen Union ausser in Schweden, in der Schweiz seit 2019 zugelassen. Gewisse Produkte mit hohem Nikotingehalt (>30g/Tüte).
Nikotinbeutel
Produkte ohne Tabak aber mit Nikotin, seit 2019 im Verkauf. Keine Daten bezüglich Wirkung auf die Gesundheit. Wirksamkeit zur Tabakentwöhnung?
Und Morgen?
Das Spiel ist aus?
– UK: Endspiel für 2009 geborene Kinder.
– Neuseeland: Gesetz für Neugeborene seit 2008. Abschaffung dieses Gesetzes!
– Rolle von Zigaretten bei neuen Nikotinprodukten?
– WHO-Bericht: Non-Nikotin Tabak Alkaloide – synthetisches Nikotinanalogon (Jordt, Tab. Control 2024). CH: Sich nicht entmutigen lassen!
Prävention des Tabakkonsums in Europa:
Warum erhält die Schweiz innerhalb Europas einen tiefen Score? Präsenz der Tabakmultis, schwache Gesetzgebung gegen Tabakwerbung, neues Gesetz 2024 ohne vollständiges Werbeverbot. WHO-Rahmenübereinkommen zur Eindämmung des Tabakkonsums unterzeichnet, aber nicht ratifiziert. Keine Erhöhung der Tabaksteuer seit 2013, keine Fortschritte hinsichtlich der Unterstützung bei der Raucherentwöhnung.
Der Referent schloss mit dem Zitat «Hate the smoke, love the smoker» (Steve Schroeder, Univ. California, San Francisco).
Ce numéro de la Gazette médicale rassemble des articles ayant lien avec la psychiatrie de la personne âgée. Ce numéro s’est construit comme se construit une connaissance professionnelle, au hasard des rencontres et des lectures.
Ces rencontres ont été rendues possibles par la grande générosité des auteurs ayant pris le temps d’écrire sur un sujet qui leur tenait à cœur, et de la grande ouverture d’esprit du comité éditorial. Cela donne une série d’articles qui semblent éloignés mais qui ont en commun le sens du partage, et de la volonté de mettre la clinique au centre des préoccupations.
Ainsi à l’heure d’une accélération du volume des connaissances et de l’innovation pédagogique, ce numéro témoigne d’une pratique faite d’ouverture à l’autre et à toutes les formes de savoir issu de la clinique auprès des sujets âgés.
J’invite ici le lecteur à lire l’ensemble des articles de ce numéro, et pas seulement l’éventuel article dont le titre correspondrait à ses attentes ou à son domaine d’expertise ou de pratique. Espérons que ce numéro nourrisse ainsi la curiosité des lecteurs, et réaffirme le maintien de l’énigme que constitue l’autre.
Dr. Jean-Pierre Schuster
Dr Jean-Pierre Schuster
Service universitaire de psychiatrie de l’âge avancé
Centre hospitalier universitaire vaudois (CHUV)
Route de Cery 60
1008 Prilly
Les soins aux résidents en EMS souffrant de démence sont complexes et doivent prendre en compte les besoins médicaux, cognitifs, émotionnels, psychologiques et sociaux de chaque individu. L’utilisation d’animaux dans le cadre de soins aux personnes âgées atteintes de démence suscite un intérêt croissant, avec des approches variées telles que l’activité assistée par animal et la thérapie assistée par animal. Bien que certaines études aient suggéré des effets positifs sur le bien-être des résidents, il reste des limites méthodologiques à la démonstration de l’efficacité de ces techniques. Les avantages de la présence d’un animal en EMS doivent être équilibrés avec les risques pour les résidents et le personnel. Il est essentiel d’établir des règles institutionnelles pour assurer le bien-être de tous les animaux et personnes impliqués.
The care of residents in medical-social establishments suffering from dementia is complex and must take into account the medical, cognitive, emotional, psychological and social needs of each individual. There is growing interest in the use of animals in the care of older people with dementia, with varied approaches such as animal-assisted activity and animal-assisted therapy. Although some studies have suggested positive effects on the well-being of residents, there remain methodological limitations to demonstrating the effectiveness of these techniques. The potential benefits of having an animal in an EMS must be balanced against the risks to residents and staff, such as allergies or bites. It is essential to establish regulations to ensure the well-being of everyone involved. Keywords: dementia, animal-assisted therapy, psyhchology, medical-social establishments
Ces dernières années, les médias ont régulièrement rapportés des pratiques de soins médiés par des animaux en établissement médicaux sociaux (EMS). Cette pratique est l’occasion d’interroger ce qui fait soins auprès des sujets souffrant de démence vivant en institution, quels sont nos relations avec les animaux, et ce que veut dire vivre et travailler en EMS.
Les soins auprès des résidant.e.s en EMS souffrant.e.s de démences sont particulièrement complexes. Ils reposent sur des principes de respect de la dignité, de respect de l’autonomie, de franchise et de respect. Ils doivent considérer la personne dans son ensemble et répondre à ses besoins médicaux, cognitifs, émotionnels, psychologiques et sociaux. Ils ont pour objet de préserver la qualité de vie et le bien être du patient souffrant de démence reconnu comme une personne. Ainsi, les soins vont être différents et uniques pour chaque personne, et auprès de chaque personne ils vont se modifier à travers le temps (1).
Les axes de soins sont multiples et combinés. Ils englobent les traitements psychosociaux (visant par exemple l’optimisation de la communication, et l’adaptation de l’environnement), l’accompagnement social, la gestion des risques, les interventions cognitives (approches visant à maintenir ou à améliorer la cognition par le biais d’activités stimulantes sur le plan mental), l’accompagnement des proches aidants, la prise en charge des maladies intercurrentes, de la douleur, la prise en charge des symptômes psycho comportementaux de la démence, les soins palliatifs … (2, 3).
Se développe ces dernières années un intérêt concernant le soin par le contact animalier auprès de sujets souffrant de démence vivant en institution. La co existence d’animaux et d’humain dans les institutions n’est pas nouvelle. On pourrait distinguer plusieurs modalités différentes de ces liens : animal privé en visite, animal privé séjournant avec le résidant, animal conduit en visite par une association, animal appartenant à la résidence (4).
Dans un certain cadre, les animaux peuvent participer, médier, contribuer à une activité avec un objectif de produire un bénéfice pour un sujet dément. Ces approches sont particulièrement hétérogènes et s’appuient sur des constructions différentes, selon l’espèce animal employé, le cadre pensé pour la technique, le rapport entre humain et animal, la formation du soignant, la conception de ce qui fait soin… Il est classique cependant de répartir ces approches en activité assistée par animal et thérapie assistée par animal. La première a pour objet d’améliorer la qualité de vie, à travers des activités/animations avec un animal, et la seconde conçoit l’animal comme un auxiliaire de thérapie conventionnelle avec un objectif de soin planifié, où l’animal est en lien avec un.e thérapeute qualifié.e et une personne démente. Les méthodes d’action de la thérapie assistée par l’animal sont diverses et comprennent : la stimulation multisensorielle, le contact physique, le jeu… Cette thérapie peut être développée en groupe ou en individuel. Les animaux les plus employés dans ce cadre sont les chiens et les chevaux.
Des recherches ont eu pour objet d’évaluer l’efficacité le soin par contact animalier (principalement des chiens) auprès des personnes démentes. Dans le cadre de recherche avec des méthodologies quantitatives, il a été mis en avant un effet positif de l’interaction avec un animal sur les variables mesurant le sentiment de solitude, de l’anxiété et de la dépression. Ces recherches recouvrent cependant des cadres de soins très différents. Les biais méthodologiques de ces études et la limitation du nombre des études rendent aujourd’hui impossible d’avoir des conclusions avec un niveau de preuve élevé sur l’efficacité des techniques de soins avec des animaux auprès de sujet âgé. Ce point est cependant à pondérer, effectivement il s’agit d’un domaine de recherche en expansion et la limitation à prouver l’efficacité, n’équivaut pas à une conclusion d’un manque d’efficacité. Par ailleurs, les méthodologies quantitatives afin de démontrer l’efficacité d’une technique de soin, font face à un obstacle important, qui est la grande variance du rapport entre un animal et un individu.
D’autres recherches avec des méthodologies qualitatives, rapportent que la présence d’un animal peut avoir un impact significatif sur la santé et le bien-être de certains résidents de maisons de retraite. Certains résidents rapportent trouver dans les interactions avec un animal plaisir et réconfort. Ce lien, semble également pour certains un moyen de maintenir une identité (en vivant avec leur animal de compagnie et prenant soin d’un animal, en vivant des expériences sensorielles et corporelles avec les animaux…), et de pouvoir investir la maison de retraite comme un « chez soi » (5, 6).
Les bénéfices de soins de la présence d’un animal en EMS pour certains sujets déments doivent être mis en regard des risques potentiels de la présence d’un animal auprès de sujets vulnérables. Dans ce cadre sont usuellement évoqués les risques de morsures, d’allergies, de transmissions infectieuses, mais également d’angoisses pour certains résidents ou soignants. Dans ce cadre, où l’institution doit mettre en place un cadre de protection pour ses résidents et ses employés, il est utile de rappeler que l’animal (quel que soit le cadre de sa présence dans le cadre institutionnel, vivant en institution, ou conduit en visite de l’extérieur) doit également être protégé de toutes maltraitances, bénéficier d’un suivi vétérinaire et d’un cadre d’assurance de responsabilité civile.
Accueillir un animal en EMS, c’est évidemment accueillir plus de vie, plus d’interactions, ouvrir chacun à de nouvelles rencontres. L’animal est d’ailleurs accueilli comme un individu avec son caractère, et n’est jamais réduit à un représentant de son espèce. Il doit faire l’objet de bienveillance et de soins de toute un chacun, et c’est finalement la rencontre de deux individus avec leurs histoires et non d’un patient dément et d’un animal qui se produit en institution. Certaines dimensions en lien avec la présence d’un animal en institution nécessitent des explorations complémentaires, comme celle de l’impact de celle-ci sur l’institution et les soignants, mais aussi sur l’animal.
Les modalités de penser la présence d’un animal en EMS sont multiples, et doivent faire l’objet de discussions préalables entre résidents, familles, personnels, directions, personnes formées dans le cadre des soins avec médiations animal. La présence d’un animal doit être en amont de son accueil faire l’objet de réglementations qui assurent à chacun résidents, personnel, mais aussi animal toutes les mesures d’un cadre d’intervention respectueux de chacun. Ces discussions préalables entre toutes les parties sont l’occasion de réaffirmer la co-construction du projet institutionnel.
Copyright Aerzteverlag medinfo AG
Dr Jean-Pierre Schuster
Service universitaire de psychiatrie de l’âge avancé
Centre hospitalier universitaire vaudois (CHUV)
Route de Cery 60
1008 Prilly
jean-pierre.schuster@chuv.ch
Dre Beatriz Pozuelo Moyano
Service universitaire de psychiatrie de l’âge avancé
Centre hospitalier universitaire vaudois (CHUV)
Route de Cery 60
1008 Prilly
Les auteurs n’ont pas déclaré de conflit d’intérêts en rapport avec cet article.
1. Académie Suisse des Sciences Médicales. Prise en charge et traitement des personnes atteintes de démence. 2017. Directives médico-éthiques.
2. Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, Ballard C, Banerjee S, Burns A, Cohen-Mansfield J, Cooper C, Fox N, Gitlin LN, Howard R, Kales HC, Larson EB, Ritchie K, Rockwood K, Sampson EL, Samus Q, Schneider LS, Selbæk G, Teri L, Mukadam N. Dementia prevention, intervention, and care. Lancet. 2017 Dec 16;390(10113):2673-2734.
3. Schweizerische Gesellschaft für Alterspsychiatrie und Psychotherapie (SGAP). Empfehlungen zur Diagnostik und Therapie der Behavioralen und Psychologischen Symptome der Demenz (BPSD). 2014.
4. Michalon J. Panser avec les animaux. Sociologie du soin par le contact animalier. 2014. Paris : Presse de l’Ecole des Mines.
5. Batubara SO, Tonapa SI, Saragih ID, Mulyadi M, Lee BO. Effects of animal-assisted interventions for people with dementia: A systematic review and meta-analysis. Geriatr Nurs. 2022 Jan-Feb;43:26-37.
6. Orr N, Abbott R, Bethel A, Paviour S, Whear R, Garside R, Coon JT. What are the effects of animals on the health and wellbeing of residents in care homes? A systematic review of the qualitative and quantitative evidence. BMC Geriatr. 2023 Mar 25;23(1):170.
Recevoir un patient avec des troubles anxieux est une situation courante pour les médecins généralistes, et la gestion de l’anxiété ne nécessite pas toujours une consultation spécialisée. L’anxiété en elle-même signale un déséquilibre souvent de manière non spécifique, donc son accueil dans la consultation est crucial. Il est important pour le médecin de reconnaître et d’interpréter les signaux d’alarme de l’anxiété, plutôt que de simplement la traiter avec des médicaments anxiolytiques. Une approche d’écoute active, relayée par celle d’un groupe Balint, peut aider à comprendre et à répondre aux besoins du patient anxieux. L’anxiété, souvent liée à la peur de la mort, peut influencer la relation médecin-patient et nécessite une approche empathique et réfléchie pour être gérée efficacement.
Receiving a patient with anxiety disorders is a common situation for GPs, and managing anxiety does not always require a specialist consultation. Anxiety in itself often signals an imbalance in a non-specific way, so its reception in the consultation is crucial. It is important for the doctor to recognise and interpret the warning signs of anxiety, rather than simply treating it with anxiolytic drugs. An active listening approach, supported by that of a Balint group, can help to understand and respond to the needs of the anxious patient. Anxiety, often linked to the fear of death, can influence the doctor-patient relationship and requires an empathetic and thoughtful approach if it is to be managed effectively.
Keywords: doctor-patient relationship, anxiety disorders, anxiolytic drugs, Balint group
Recevoir un patient avec des troubles anxieux relève du quotidien du médecin généraliste, et cette pratique de l’anxiété ne demande pas de consultation spécialisée. Nous allons discuter de son accueil dans le cabinet du praticien et de l’intérêt d’un regard Balint sur cet accueil et sur les interactions médecin-malade, médecin-soignant-malade.
Accueil de l’anxiété dans la consultation
L’anxiété est un signe vague, peu spécifique, analogue à la douleur et dans ce sens, elle signale un déséquilibre sans plus de précision. Sigmund Freud décrit déjà en 1895 la multiplicité des symptômes somatiques de l’angoisse, des vertiges à la pollakiurie, des spasmes intestinaux aux maux de tête (1). C’est un signal d’alarme dit Freud, au médecin et au soignant de l’entendre et de le lire pour y voir clair.
Le médecin peut en quelques questions en savoir plus. Comme la douleur, l’anxiété peut se palper pour révéler son origine. Une question sur un deuil récent, sur l’annonce d’une maladie somatique et le médecin perçoit l’anxiété augmenter, le débit verbal s’accélérer, et les anticipations négatives arriver. Si le médecin palpe juste, il va en effet voir l’anxiété varier et pourra donc l’utiliser directement dans sa consultation. Évitons donc de l’éteindre rapidement avec un anxiolytique, car on n’y verra plus rien. Nous pouvons ici nous fier à Michael Balint qui propose dans « Comment débuter » (2) une écoute active du médecin, ouverte dans un entretien qui ne s’enferme pas dans une seule collecte anamnestique. Étonnamment simple, une pratique de l’écoute offre aussi un temps de réflexion utile pour formuler la réponse « rassurante » demandée par l’anxiété. Cette réponse vise à définir les enjeux soignants à venir et d’habitude en nommant un diagnostic, elle constitue la première action psychosomatique soignante. En effet, décrire mais surtout nommer la maladie dont souffre le patient donne une prise cognitive rassurante: connaitre le «mal» relève des mêmes mécanismes qui font le bienfait des contes qui rassurent les angoisses infantiles. Le flou symptomatique est circonscrit, ainsi désigné il fait moins peur.
Lors de la consultation, l’anxiété accompagne chaque maladie et chaque symptôme, elle est l’affect qui est forcément présent, parfois de manière cachée, refoulée, transformée par les mécanismes de défense. Cette anxiété primaire, c’est l’angoisse de mort. Avec l’angoisse issue de la libido, ce sont nos deux angoisses primaires vie-mort. L’ angoisse de mort court avec la maladie, anticipe notre fin, elle est plus ou moins contenue, elle peut aussi déborder les mécanismes de défense et déferler comme une vague, une digue est rompue.
Argan, le malade imaginaire de Molière est immergé dans l’angoisse de mort. Dans sa relation avec les médecins, il est crédule, méfiant et avide, tout en même temps, comme ceux qui savent que la mort vainc la médecine. Le médecin doit répondre à l’angoisse de mort sous-jacente, sinon les traitements s’enchainent comme chez Molière. La phase d’écoute active permet d’accueillir l’angoisse de mort qui doit être contenue comme une hémorragie par la relation soignante. Parfois, l’anxiété déborde la relation et déstabilise le système soignant. Elle ne trouve plus ni son objet ni son pourquoi. Et surtout, elle ne trouve pas ou plus une partie du corps qui la contient comme dans le trouble psychosomatique.
L’anxiété débordante
Mon patient a 88 ans. Il me dit souffrir, depuis qu’il est jeune adulte, de troubles digestifs et de céphalées d’allure tensionnelles. Il me parle de ses nuits qui sont très courtes, entrecoupées par de rares plages de sommeil s’il ne prend pas de benzodiazépines. Il en a pris jusqu’à 5 cp de Temesta par nuit. Il souffre d’une polyarthrose, ses douleurs limitent sa mobilité. Il se déplace en rollator et en chaise roulante. Il ne peut faire que quelques pas et les transferts. Il a pris beaucoup de poids et sa dyspnée s’est donc exacerbée. Il reconnaît que ses problèmes de santé, ces dernières années, ont augmenté son anxiété. Des évènements contextuels ont aggravé sa situation. Son épouse est décédée récemment. Sa fille aînée a souffert d’un burn-out; raison pour laquelle elle habite chez son père depuis un an. L’ensemble de ces facteurs de stress ont dépassé les capacités d’adaptation du patient qui a présenté des attaques de panique quasi-quotidiennes. Elles se manifestent par une dyspnée aiguë et une sensation de mort imminente ou de devenir fou. Mes consultations à domicile sont soutenues par ergothérapeute, aide à domicile, physiothérapeute, diététicienne, psychiatre mais le trouble anxieux persiste.
L’approche Balint
Issus des travaux de Michael Balint, les groupes Balint (3) sont organisés pour échanger sur des situations soignant-soigné, animés par un superviseur, habituellement un psychiatre/psychanalyste. On y présente 1 ou 2 cas sans notes ni dossier et une question est posée au groupe par le présentateur. Les échanges se concentrent sur la relation thérapeutique elle-même: son début et éventuel héritage de relation soignante passée, l’évolution, la situation actuelle. La richesse des associations et des questions des pairs ouvre le champ. Le superviseur veille à ce que le groupe reste sur la thématique relationnelle sans se perdre dans l’analyse du patient ou du médecin. Si la potentialité positive placebo de la relation est bien connue, le pouvoir nocebo d’une relation soignant/soigné est à notre sens insuffisamment mise en avant. Nous assistons pourtant souvent à des symétries poussant d’un côté à l’augmentation des symptômes et de l’autre à une accumulation d’examens.
En effet, l’anxiété se propage comme le feu qui prend possession du patient puis des proches et des soignants. Le médecin est désigné comme l’un des acteurs qui doit éteindre le feu. Il s’arme des défenses qu’il connait, il nomme, distingue, classifie et souvent propose une médication. Mais, s’il est contaminé par l’angoisse, il peut se sentir obligé de prescrire des traitements inappropriés parfois iatrogènes. Appliquée aux troubles anxieux, la méthode Balint produit du nouveau dans la prise en charge. Le médecin pris dans le brouillard anxieux reçoit des avis, des conseils et des validations de son travail et de ses attitudes. Exposer la relation amène une clarification des rôles et de la dynamique que l’anxiété avait brouillé entre les acteurs de la situation. Dans son chapitre dédié (4), Balint parle des rapports de spécialistes, y compris ceux des psychiatres, qui tendent à dessaisir le médecin omnipraticien de la dimension existentielle de la demande du patient, à savoir gérer l’angoisse de mort. Il peut s’ensuivre une multiplication de réponses «physiques» qui en fait laissent libre cours à la propagation anxieuse parfois même en l’amplifiant.
Un regard Balint
En soumettant l’exemple décrit précédemment à un regard Balint, on voit apparaitre de nouveaux choix d’intervention:
• Rencontrer le corps dans la relation, tenir une main, le toucher pour apaiser l’anxiété, réexaminer son patient.
• Trouver une manière de finir les consultations qui inclut l’anxiété soulevée par la séparation: savoir prévoir le vide et penser dès le début au message de la fin: vous serez peut-être plus angoissé quand je partirai, voici ce que je vous propose de faire (écrire qq mots, dessiner, tél à qqn, …)
• Être avec, marcher avec: être disponible pour que le moment de la consultation soit agréable, pouvoir se réjouir de voir le patient angoissé car je vais l’aider par ma présence.
• Organiser, structurer la journée, les rendez-vous, moments de rencontre. Pour chaque moment de la journée, trouver une réponse aux questions angoissées du patient. Que va-t-il se passer? Où vais-je me retrouver?
• Faire un réseau pour soutenir le tissu relationnel soignant et contenir l’angoisse diffuse. Ce tissu entre thérapeutes et famille forme comme un filet de sécurité qui a été désorganisé par l’angoisse.
Ces propositions visent à reprendre la main sur l’angoisse de mort circulante dans la situation. Parler de l’angoisse de mort, trouver un bon lieu (cela peut être une promenade avec le patient) et le bon moment pour l’aborder est essentiel. Alors, nous nous sommes déplacés de la position de guérir le symptôme à tout prix à la gestion commune de l’angoisse.
Actions psychosomatiques soignantes
S’il existe des maladies psychosomatiques, il existe aussi des actions psychosomatiques soignantes qui fonctionnent comme des moments psychothérapeutiques. La première d’entre elles consiste en nommant l’émotion, ici l’angoisse, et la légitimer. Il faut trouver les mots pour expliquer la force de cette émotion partagée et ces mots diffèrent pour chaque relation soignante. On peut parler de feu, d’inondation ou de tempête de l’angoisse, en tous les cas nommer ce qui emporte la relation et ses acteurs sans qu’ils puissent y échapper.
Dans la vignette, l’action psychosomatique sera d’arrêter la propagation du feu anxieux pour éviter le risque de la spirale iatrogénique. Pour réinstaller un cadre de traitement, on s’appuie sur le contexte soignant tout entier en organisant la temporalité des soins et le message délivré par les soignants. Dans la relation, on peut utiliser des médiations différentes de la parole comme le dessin (dessiner l’angoisse dans le corps), parfois une musique partagée ou un jeu. L’être avec peut devenir marcher avec, et le médecin choisit sa technique de relaxation (hypnose, méditation).
Beaucoup de médecins sont encore trop inhibés devant l’utilisation de ces actions soignantes et empêchent eux-mêmes leur créativité alors qu’elle est nécessaire devant le risque de chronicisation et de fatigue compassionnelle. Les groupes Balint et la supervision individuelle Balint (5) ainsi que les formations continues (SoPSOc) sont là pour lutter contre les résistances à cette approche.
Conclusion
L’incipit de la citation de Molière reflète sa position naturaliste, l’homme doit accepter son impuissance devant la maladie et la mort. On peut l’entendre aussi comme un message de fraternité Balintien. Ce qui compte c’est l’autre le frère, la personne du médecin et non ce qu’il prescrit; comme le dit Balint le médecin doit se prescrire lui-même. L’anxiété est une hémorragie, et une défaite pour le Moi dont le rôle est de réguler les stimuli et les instances, il n’en peut plus et donc se manifeste par l’anxiété. La nommer, la faire parler, par exemple des crocodiles sous le lit, cette mise en mot rassure. Comment l’accueillir sans agir, lui donner sa place et suivre son cours et éventuellement remonter à la source et la transformer en discours sur la séparation, l’impuissance, le deuil, la peur de la mort, le désir. L’anxiété se met alors à parler au travers de la relation au médecin, et la peur trouve son objet.
Copyright: Aerzteverlag medinfo AG
Dr Matthias Vannotti
Médecin interne Générale FMH
rue Verdaine 8/a
1095 Lutry
mvannotti@hotmail.com
Dr Dag Söderström
FMH psychiatrie et psychothérapie, psychanalyste SSPsa
Av de la Gare 16
1800 Vevey
dsoderstrom@bluewin.ch
Les auteurs n’ont pas déclaré de conflit d’intérêts en rapport avec cet article.
1. Freud S. Qu’il est justifié de séparer de la neurasthénie un certain complexe symptomatique sous le nom de « névrose d’angoisse », Névrose psychose et perversion, Parid, PUF, 1973, pp15–38
2. Balint M, comment débuter, in Le médecin, son malade et la maladie, 1966 Payot, pp128–146
3. Voir Balint.ch
4. Balint M, La survivance de la relation maître-élève, in Le médecin, son malade et la maladie 1966, Payot, pp103–117
5. Herzig L, Söderström D: Supervision individuelle: une aide face aux patients difficiles. Primary care 2013;13:5, 86–87
Les dépressions sont, avec les maladies démentielles et les maladies anxieuses, les maladies psychiatriques de la vieillesse les plus fréquentes; les troubles du sommeil sont également un symptôme fréquent chez les personnes âgées. Chez les personnes âgées, les dépressions peuvent se manifester par des symptômes atypiques, de sorte que les dépressions liées à l’ âge ne sont pas reconnues immédiatement. Souvent, des troubles physiques, des douleurs, un malaise général et des troubles cognitifs sont au premier plan et non les symptômes habituels de la dépression. En particulier, il est généralement difficile , chez les ainés, de distinguer une dépression d’ une symptomatologie dépressive dans le cadre d’ une démence débutante en raison des troubles cognitifs. Les troubles du sommeil peuvent être un symptôme de la dépression de la personne âgée. Mais d’ autres causes peuvent être à l’ origine de troubles du sommeil chez les personnes âgées, indépendantes d’ une dépression et qui, non diagnostiquées et non traitées, compliquent considérablement le traitement de la dépression.
Depression is the most common psychiatric disorders in old age, alongside dementia and anxiety; sleep disorders are also a common symptom in old age. Depression can manifest in older people through atypical symptoms, which means that old-age depression is not immediately recognized. Physical complaints, pain, general malaise and cognitive disorders are often the main cognitive disorders and not the main symptom of depression. In particular, the differentiation of depression in old age from depressive symptoms in the context of incipient dementia is usually difficult due to the cognitive disorders in old-age depression. Key Words: Old age, depression, symptoms, cognitive disorders
La dépression chez le sujet âgé
Les causes
Des facteurs somatiques et psychiques/psychosociaux peuvent être impliqués dans l’ apparition d’ une dépression chez les personnes âgées.
En font partie la diminution des capacités physiques et mentales, l’ apparition de maladies physiques avec la peur de perdre son autonomie, le changement de rôle (profession, retraite, famille – départ des enfants) associé à une réorientation personnelle pour la période de la retraite, l’ expérience de pertes, les changements/réductions du réseau social, le départ/décès d’ amis proches et de membres de la famille, mais aussi la perte du réseau social au travail.
Afin de planifier une thérapie spécifique et individuelle efficace, il est tout d’ abord important de reconnaître la dépression dans sa manifestation et son intensité et de saisir en outre les éventuels facteurs impliqués dans son apparition et son maintien (facteurs physiques et psychosociaux).
Diagnostic de la dépression du sujet âgé
Le diagnostic de la dépression du sujet âgé nécessite donc un examen étendu psychopathologique, psychosocial et physique (y compris des examens somatiques et la détermination de paramètres de laboratoire et une imagerie (IRM)). La Société Suisse de Psychiatrie de la personne âgée (SGAP/SPPA) a présenté (1) les recommandations actuelles pour le traitement d’ une dépression à l’ âge avancé. Celles-ci sont également déterminantes pour le diagnostic différentiel d’ une démence débutante. En complément, des échelles de «rating» telles que l’ échelle de dépression gériatrique (GDS) peuvent être utilisées pour déterminer l’ intensité de la dépression.
La clarification et l’ évaluation de la suicidalité, qui est nettement plus élevée aussi bien chez les personnes âgées qu’ en cas de dépression, font partie intégrante du diagnostic. La suicidalité doit être abordée et son intensité évaluée. L’ évaluation et la discussion de la suicidalité nécessitent une certaine expérience clinique. Des échelles d’ évaluation sont à disposition également dans ce domaine (1, 2).
Traitement de la dépression du sujet âgé
Le traitement de la dépression du sujet âgé devrait, tout comme le diagnostic, s’ orienter aux recommandations de traitement de la SGAP/SPPA) (1). En principe, en cas de dépression secondaire survenant dans le cadre d’ une autre maladie de base (p. ex. trouble de la fonction thyroïdienne), il s’ agit d’ abord de traiter la maladie de base; le cas échéant, il faut cependant – même en présence d’ une autre maladie – traiter en parallèle l’ état dépressif ou les symptômes particuliers (p. ex. troubles du sommeil, agitation). En cas de dépression légère, une psychothérapie seule est préférable; en cas de dépression modérée, le traitement peut être soit médicamenteux, soit psychothérapeutique, soit combiné; en cas de dépression grave, il faut toujours associer un traitement antidépresseur médicamenteux et une psychothérapie. A cela s’ ajoutent d’ autres thérapies adjuvantes non médicamenteuses comme la luminothérapie, l’ activité physique et la thérapie sportive ainsi que l’ ergothérapie et la thérapie par l’ art.
Le traitement de la dépression devrait être effectué par un spécialiste en psychiatrie et psychothérapie, surtout chez les personnes âgées, après une tentative de thérapie infructueuse (au plus tard après deux), car une «vision psychosomatique» globale avec l’ application de stratégies psychothérapeutiques spécifiques est généralement nécessaire pour le traitement.
La psychothérapie
La psychothérapie des personnes âgées doit s’ orienter aux contenus et les thèmes décrits comme facteurs de stress pour l’ apparition de dépressions chez les personnes âgées (voir ci-dessus). Un aspect important à relever est qu’ une perspective d’ avenir doit être développée pour les patients âgés, voire parfois très âgés, même si le temps pour cet avenir est beaucoup plus court chez eux que chez les plus jeunes. Un autre aspect important qui devrait être abordé en psychothérapie est celui de «l’ acceptation». Il s’ agit ici d’ accepter la situation actuelle, par exemple la présence d’ une maladie physique (généralement chronique) qui réduit la mobilité et l’ autonomie, ou encore les erreurs déplorées lors de phases antérieures de la vie, qui ne se laissent plus corriger. Ce n’ est qu’ alors que l’ on peut travailler sur les possibilités d’ organiser l’ avenir et d’ améliorer la qualité de vie.
La plupart des résultats positifs sont disponibles pour la thérapie cognitivo-comportementale (TCC), pour la thérapie interpersonnelle (TIP) et pour la thérapie focale psychodynamique. Pour les méthodes «de troisième vague», soit ACT = Acceptance and Commitment Therapy, MBCT = Mindfulness-Based Cognitive Therapy et CBASP = Cognitive Behavioral Analysis System of Psychotherapy, les données relatives à l’ utilisation chez les personnes âgées sont encore peu nombreuses. Cela ne signifie pas pour autant que ces méthodes thérapeutiques – appliquées au cas par cas – ne sont pas efficaces (1).
Le traitement médicamenteux
Si un traitement médicamenteux est nécessaire, tous les antidépresseurs autorisés sont en principe efficaces. Les antidépresseurs peuvent déjà être utilisés en cas de dépression légère à modérée avec des troubles du sommeil prononcés, en particulier lorsqu’ une psychothérapie n’ est pas possible (par ex. pas de disponibilité, troubles cognitifs prononcés). Le choix des antidépresseurs se fait alors en premier lieu en fonction du profil des effets secondaires et, le cas échéant, des co-morbidités présentes simultanément. La règle de base pour le traitement médicamenteux des personnes âgées est «start low, go slow». Il convient néanmoins de viser et d’ atteindre un dosage suffisant du médicament, qui peut être évalué à l’ aide de contrôles des taux sanguins. Les médicaments ayant des effets secondaires anticholinergiques doivent être évités.
Résistance au traitement
En cas de résistance au traitement, les étapes mentionnées dans les recommandations suisses de traitement de la dépression chez le sujet âgé ou unipolaire (changement, combinaison de médicaments, augmentation, procédés biologiques supplémentaires) devraient être appliquées (1, 3).
Il convient de noter ici que si une augmentation de la dose d’ une substance n’ est pas possible en raison de l’ apparition d’ effets secondaires, une combinaison avec un deuxième antidépresseur à une dose également faible à moyenne peut être utile. pour minimiser des effets secondaires. La prise en charge d’ une dépression du sujet âgé résistante au traitement doit être effectuée par un spécialiste en psychiatrie et psychothérapie. Des troubles du sommeil persistants peuvent être une raison pour l’ absence de réponse à un traitement antidépresseur.
Les troubles du sommeil chez les personnes âgées
Comme nous l’ avons déjà mentionné, les troubles du sommeil peuvent être un symptôme de la dépression. Mais il existe de nombreuses autres raisons qui peuvent être à l’ origine de troubles du sommeil, en particulier chez les personnes âgées.
La régulation du sommeil
Pour comprendre les troubles du sommeil, il est nécessaire de connaître la régulation du sommeil.
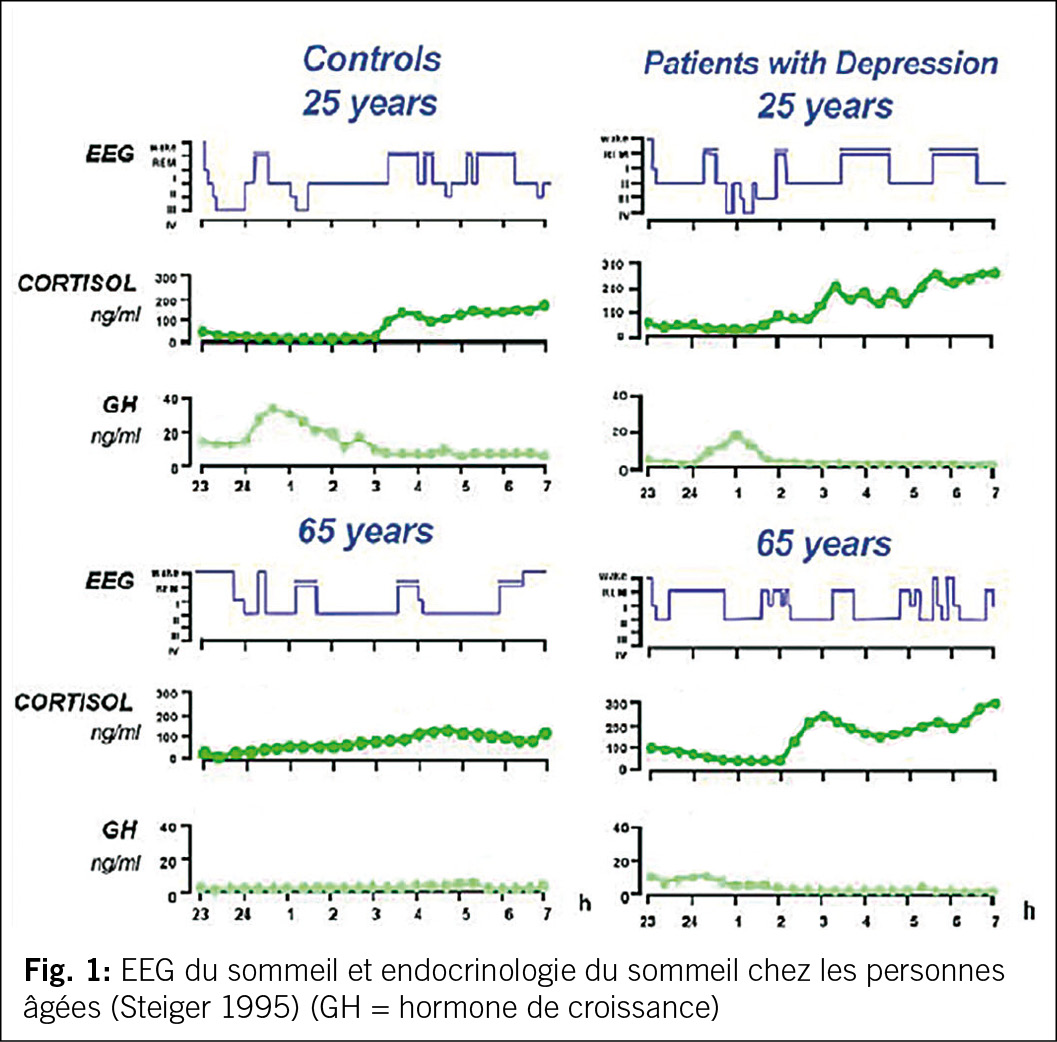
Les enregistrements EEG effectués pendant le sommeil nous donnent une indication de l’ activité électrophysiologique pendant la nuit. L’ analyse de ces enregistrements montre une alternance de sommeil non paradoxal et de sommeil paradoxal (cycle de sommeil) avec un sommeil non paradoxal profond en début de nuit et un sommeil non paradoxal léger en fin de nuit (dans les cycles de sommeil ultérieurs). Ces phases de sommeil sont également associées à une libération régulière de différentes hormones, qui présentent chacune un schéma caractéristique (4, 5, cf. fig. 1).
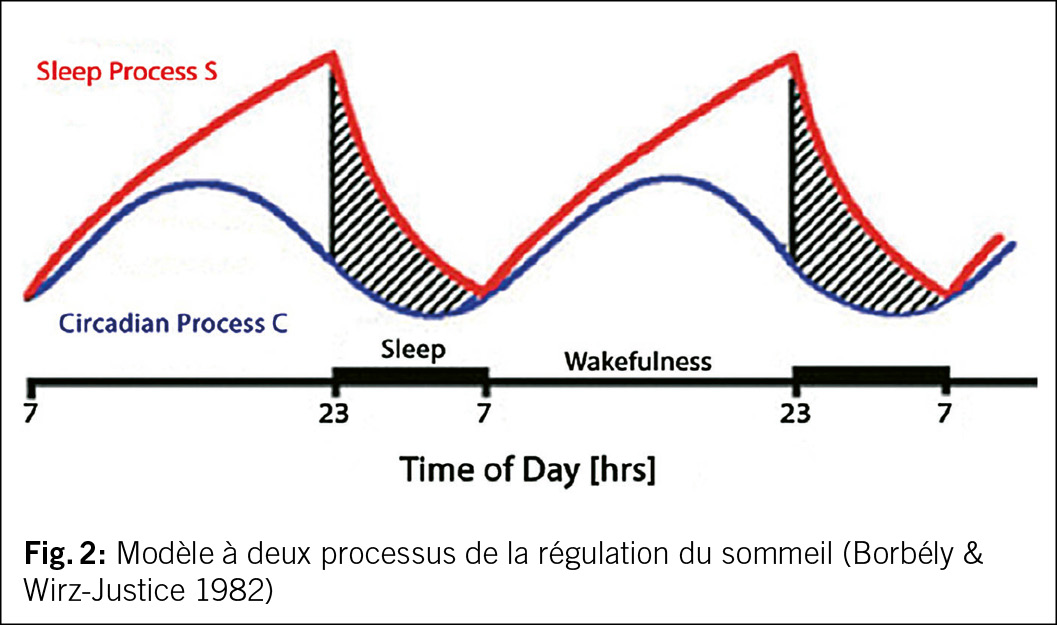
Le modèle à deux processus (6) fournit une explication pour la régulation du sommeil. Le sommeil est lié au rythme clair-obscur de la journée de 24 heures imposé par le soleil et est donc soumis à un rythme circadien. Celui-ci est contrôlé par un pacemaker endogène dans le diencéphale (nucleus suprachiasmaticus) (= processus C – circadian). Parallèlement, indépendamment du rythme lumière/obscurité du soleil, la durée de l’ éveil précédent influence notre sommeil en augmentant la pression de sommeil (processus S – pression de sommeil), (cf. fig. 2). Plus on reste éveillé longtemps, plus le sommeil de la nuit suivante est profond (processus de pression de sommeil S). On attribue au sommeil profond non REM une fonction de récupération physique et psychique ainsi qu’ une fonction de stimulation de la mémoire, notamment en raison de la réduction de l’ activité neuronale corticale et de la modification de l’ activité de certaines hormones et du système immunitaire. Le sommeil paradoxal est moins influencé par la variation de la durée d’ éveil précédente, mais plutôt lié au rythme circadien (5).
D’ après les connaissances des dernières décennies, il importe de viser avec le traitement un sommeil sain, naturel, avec des phases suffisantes de sommeil profond et de phases de sommeil non paroxysmale (sommeil REM) stables. De manière physiologique, chez la personne âgée, le sommeil se raccourcit et devient globalement plus léger. De plus, le processus C de la régulation du sommeil (composante circadiane) subit des changements, en particulier avec une montée plus précoce de la cortisole dans la deuxième partie de la nuit (réf 4, 5, 7, cf. aussi fig. 1).
Raisons des troubles du sommeil chez les personnes âgées
Outre ces modifications physiologiques qui entraînent un sommeil plus léger avec un rythme circadien affaibli pour la température corporelle centrale, la mélatonine et le cortisol avec une avance de phase (phase advance) d’ environ une heure, on trouve chez les personnes âgées comme conséquence de ces modifications du sommeil liées à l’ âge et de la sécrétion hormonale associée au sommeil (en particulier aussi le cortisol) une sensibilité accrue aux troubles du sommeil dus à des facteurs exogènes (facteurs de stress). Les facteurs de stress qui peuvent influencer le sommeil chez les personnes âgées sont analogues à ceux mentionnés pour la dépression liée à l’ âge (voir ci-dessus). En tant que facteurs de stress spécifiques à l’ âge ils peuvent conduire plus rapidement et plus intensément à une insomnie prononcée en raison du sommeil plus léger des personnes âgées (5).
Les maladies physiques jouent un rôle important dans le développement de l’ insomnie, surtout chez les personnes âgées. Il s’ agit notamment des troubles du sommeil spécifiques du «Restless Legs Syndrome» (syndrome des jambes sans repos) et des troubles respiratoires liés au sommeil (arrêts respiratoires, apnée du sommeil), qui sont tous deux plus fréquents avec l’ âge avançant et qui restent généralement non diagnostiqués pendant longtemps, malgré les troubles du sommeil déjà existants.
Parmi les troubles spécifiques du sommeil, il convient également de mentionner le trouble du comportement du sommeil paradoxal, qui s’ accompagne de mouvements moteurs pendant le sommeil paradoxal, le plus souvent en deuxième partie de nuit, et qui peut être associé à une maladie neurodégénérative du groupe des synnucléinopathies (maladie de Parkinson, démence de corps de Lewy) et qui apparaît plus fréquemment avec l’ âge.
A ces troubles primaires du sommeil s’ ajoutent une multitude de maladies physiques qui peuvent avoir un effet négatif sur le sommeil. Il s’ agit en premier lieu de différents syndromes douloureux, mais aussi de maladies cardiovasculaires, pulmonaires et urogénitales, ainsi que de leur traitement médicamenteux avec des substances qui perturbent parfois le sommeil (p. ex. des préparations à base de théophylline le soir pour traiter l’ asthme) (9). En cas de co-morbidités somatiques (mais aussi psychiques) existantes, une étroite collaboration interdisciplinaire entre les disciplines médicales (médecin du sommeil, interniste, neurologiste, autres) est indiquée afin d’ obtenir un résultat optimal. Le traitement des troubles du sommeil secondaires nécessite un traitement aussi optimal que possible de la maladie de base, ainsi qu’ un ajustement médicamenteux des patients en tenant compte des propriétés perturbatrices du sommeil de certains médicaments somatiques.
Diagnostic des troubles du sommeil
En premier lieu, il convient de déterminer si les troubles du sommeil dont le patient fait état ont une valeur pathologique ou s’ il s’ agit uniquement d’ un comportement de sommeil erroné ou d’ un trouble de la perception du sommeil. Pour ce faire, il est possible d’ utiliser l’ agenda du sommeil et l’ actigraphie, en plus d’ une anamnèse personnelle et externe approfondie. Si au niveau phénoménologique un trouble du sommeil est constaté, il s’ agit alors de rechercher les causes. Il faut tout d’ abord découvrir ou exclure les causes somatiques décrites ci-dessus (co-morbidités somatiques, médicaments potentiellement perturbateurs du sommeil) et les causes psychiques (facteurs de stress, maladies psychiques).
Comme ce processus diagnostique complet peut durer longtemps selon les cas avant d’ atteindre une certaine clarté, il est souvent nécessaire de traiter l’ insomnie de manière symptomatique en parallèle.
Cela se justifie également par le fait qu’ un traitement spécifique supplémentaire de l’ insomnie est souvent nécessaire même si on trouve et traite une cause.
Il existe des options médicamenteuses et non médicamenteuses pour le traitement de l’ insomnie (9).
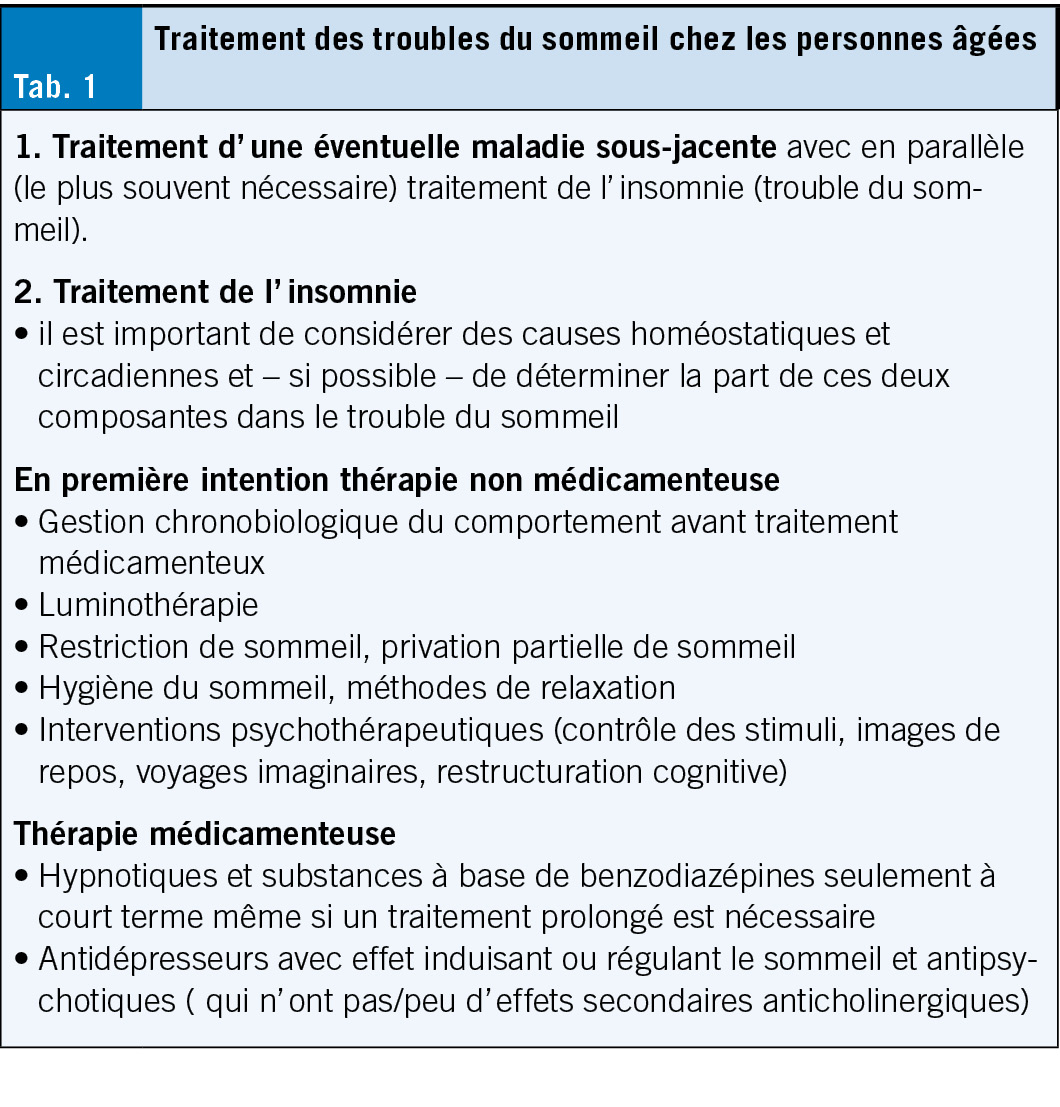
Traitement de l’ insomnie chez les personnes âgées (Tab. 1)
Mesures non médicamenteuses
Parmi les mesures non médicamenteuses, à relever surtout des heures de sommeil et d’ éveil constantes (renforcement du processus C), éventuellement combinées avec une restriction de sommeil et une sieste limitée (renforcement du processus S).
L’ utilisation individuellement adaptée de la luminothérapie (agit sur le processus C) ainsi que toutes les mesures d’ hygiène du sommeil (les deux processus sont concernés) sont d’ autres possibilités thérapeutiques non médicamenteuses qui agissent directement sur ces deux processus de régulation du sommeil. L’ activité physique au sens d’ une activité sportive d’ intensité moyenne a également un effet positif de soutien, tout comme les thérapies actives créatives et stimulantes sur le plan intellectuel (ergothérapie, art-thérapie). Ces activités peuvent également servir pour stabiliser le rythme circadien en les pratiquant toujours à des heures fixes de la journée. Récemment, des études contrôlées ont mis en évidence l’ effet positif de l’ activité physique et du sport sur l’ amélioration des troubles du sommeil et de la dépression. Les programmes d’ intensité moyenne avec une fréquence de trois séances d’ entraînement par semaine pendant trois à six mois étaient particulièrement efficaces. Plus de 50 % des études examinées ont montré des effets positifs sur les troubles du sommeil ainsi que sur l’ amélioration de la dépression en général (10).
Chez les personnes âgées, les activités et programmes sportifs devraient être utilisés en cas de dépression ou/et d’ insomnie, en concertation avec les médecins traitants et sous leur supervision.
Traitement médicamenteux
En cas de troubles du sommeil, il faut toujours commencer par utiliser des méthodes non médicamenteuses, mais il est parfois indispensable de recourir à des médicaments favorisant le sommeil. Les hypnotiques benzodiazépines et les analogues des benzodiazépines (les substances Z comme le zolpidem) sont les plus utilisés, mais uniquement pour une utilisation à court terme. Ils sont autorisés pour le traitement de l’ insomnie, mais doivent être administrés avec beaucoup de retenue, surtout chez les personnes âgées, en raison de leur profil d’ effets secondaires (surtout le risque de chute, les troubles cognitifs, la tolérance et les problèmes de dépendance).
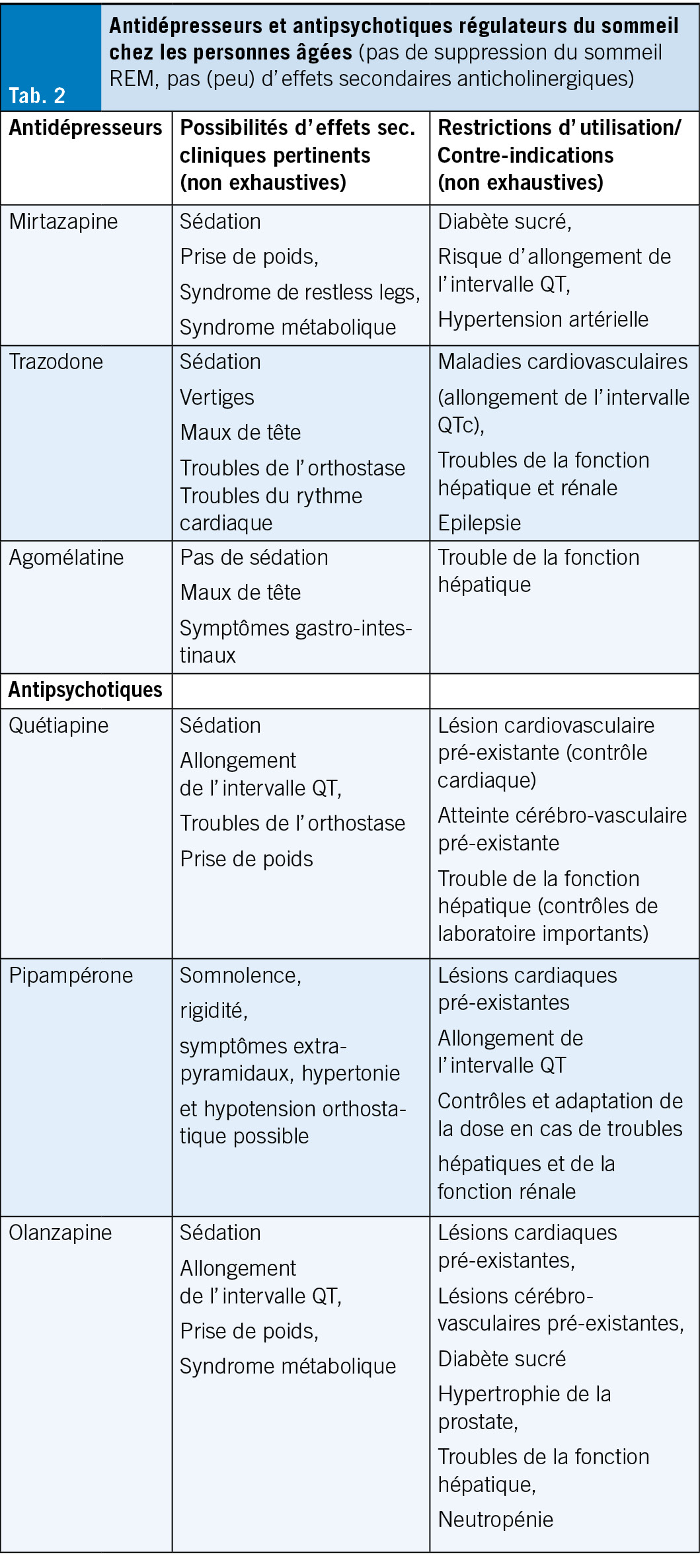
Si un traitement médicamenteux prolongé de l’ insomnie est nécessaire (en cas d’ insomnie primaire ou secondaire), des substances de la classe des antidépresseurs ou des antipsychotiques peuvent être administrées (cf. tableau 2).
Lors de l’ utilisation de ces substances chez les personnes âgées, c’ est le profil d’ effets secondaires de chaque substance qui détermine le choix. En principe, aucune substance présentant des effets secondaires anticholinergiques (notamment sécheresse buccale, rétention urinaire, constipation, troubles de l’ accommodation et cognitifs) ne devrait être utilisée chez les personnes âgées; en présence de co-morbidités, il faut surtout faire attention au potentiel de déclenchement de troubles moteurs extrapyramidaux, à l’ allongement de l’ intervalle QT et à l’ induction d’ un état métabolique diabétogène ou d’ un syndrome métabolique.
Après clarification du profil d’ effets secondaires et d’ éventuelles co-morbidités, il convient d’ utiliser chez les personnes âgées avant tout des substances qui n’ induisent pas de grandes modifications de l’ architecture du sommeil et qui provoquent de préférence une augmentation du sommeil profond sans suppression du sommeil REM (comme p. ex. la trazodone, l’ agomélatine, la mirtazapine ou, dans la classe des antipsychotiques, surtout la quétiapine et le pipampérone) (cf. tab. 2). L’ utilisation de ces substances s’ oriente – comme mentionné ci-dessus au profil des effets secondaires et se voit limitée par la présence d’ éventuelles co-morbidités (5).
On oublie souvent l’ agoniste de la mélatonine Circadine, qui est autorisé pour les troubles du sommeil chez les personnes âgées et qui contrecarre la diminution de la force du processus C avec l’ âge.
Une nouvelle option de traitement est disponible depuis la fin de l’ année dernière: le daridorexant, un antagoniste des récepteurs de l’ orexine. Il entraîne une amélioration de l’ endormissement et du maintien du sommeil, une augmentation du sommeil profond et du sommeil paradoxal et se caractérise surtout par une bonne tolérance, sans dépendance physique, ce qui en fait une option de traitement sérieuse, notamment pour les patients âgés (11). Le traitement des troubles du sommeil chez les personnes âgées ne se limite toutefois jamais au seul traitement médicamenteux, il faut toujours inclure des éléments de traitement non médicamenteux. Après un diagnostic minutieux et une anamnèse du sommeil, le traitement doit être planifié et mis en œuvre de manière personnalisée en fonction de la constellation présente chez chaque patient, avec comme objectif le traitement complet du trouble du sommeil et la restauration des capacités physiques et mentales.
Nécessité de la prévention
Une insomnie non traitée entraîne un risque accru d’ apparition de maladies physiques, notamment cérébrovasculaires, cardiovasculaires et métaboliques (diabète de type II, syndrome métabolique), mais aussi de maladies psychiques, notamment des troubles anxieux, cognitifs et de dépression.
Il est donc impératif de traiter rapidement et systématiquement les troubles du sommeil ayant valeur de maladie.
Le risque d’ apparition de troubles du sommeil chez les personnes âgées peut être réduit en respectant un rythme jour-nuit régulier avec des heures de sommeil fixes ( le coucher en règle générale pas avant 22h30) et des horaires réguliers de prise de nourriture. Une sieste peut être autorisée à midi, mais au maximum une demi-heure et pas après 15 heures.
S’ y ajoutent des mesures visant à maintenir la forme physique et mentale et le respect des mesures d’ hygiène du sommeil. Toutes ces mesures comportementales jouent un grand rôle dans la prévention des troubles du sommeil et de la dépression et peuvent être appliquées par les personnes âgées elles-mêmes.
Cet article est une traduction de «der informierte arzt – die informierte ärztin 03_2024»
Copyright Aerzteverlag medinfo AG
PD Dr. med. Dr. phil. Ulrich Michael Hemmeter
Chefarzt Psychiatrie St. Gallen Nord
Zürcherstrasse 30
9500 Wil
L‘ auteur n’ a pas déclaré de conflit d’ intérêts en rapport avec cet article.
1. Hatzinger M, Hemmeter U, Hirsbrunner T, Holsboer-Trachsler E, Leyhe T, Mall JF, Mosimann U, Rach N, Trächsel N, Savaskan E. Empfehlungen für Diagnostik und Therapie der. Depression im Alter. Praxis 2019; 107(3): 127–144.
2. Hemmeter U., Suidizalität erkennen und einschätzen, der informierte Arzt, 2023, 10, 13–16
3. Holsboer-Trachlser et al, Die somatische Behandlung der unipolaren depressiven Störungen: Update 2016, Teil 1, Die Akutbehandlung depressiver Episoden, SWISS MEDICAL FORUM – SCHWEIZERISCHES MEDIZIN-FORUM 2016;16(35):716–72
4. Steiger A: Schlafendokrinologie. Nervenarzt. 1995;66:15–27.
5. Hemmeter U, Ngamsri Th. Gestörter Schlaf: Ursachen und Behandlungsmöglichkeiten. Psychiatrie + Neurologie, 2023, 2, 16–23.
6. Borbély AA et al.: Sleep, sleep deprivation and depression. A hypothesis derived from a model of sleep regulation. Hum Neurobiol. 1982;1(3):205–210.
7. Lorette A et al.: Sleep in the elderly. In:, 2nd edition, Editors: Claudio Bassetti, Walter McNicholas, Tiina Paunio, Philippe Peigneux, Publisher: European Sleep Research Society (ESRS) in Regensburg, Germany. Sleep Medicine Textbook. 2021;2.
8. Patel D et al.: Insomnia in the Elderly: A Review. J Clin Sleep Med. 2018;14(6):1017–1024.
9. Riemann D et al.: S3-Leitlinie Nicht erholsamer Schlaf/Schlafstörungen, Kapitel «Insomnie bei Erwachsenen» (AWMFRegisternummer063–003), Update 2016. Somnologie. 2017;21:2–44
10. Hemmeter UM, Ngamsri T. [Physical Activity and Mental Health in the Elderly]. Praxis (Bern 1994). 2022;110(4):193–8.
11. Park J, Render KP, Cates DW., Daridorexant: Comprehensive Review of A New Oral Agent for the Treatment of Insomnia. Ann Pharmacother. 2023 Sep;57(9):1076–1087.
12. Hertenstein E et al.: Insomnia as a predictor of mental disorders: A systematic review and meta-analysis. Sleep Med Rev. 2019;43:96–105.
Un nombre considérable de personnes souffrent de maladies dépressives, d’ angoisses pathologiques, présentent des symptômes psychosomatiques, ont des problèmes de sommeil chroniques ou sont épuisées sur le plan psychophysique. Elles cherchent souvent d’ abord une aide médicale dans le cabinet du médecin de famille. Les médecins de famille sont donc confrontés chaque jour à des patients atteints de troubles psychiques et doivent alors agir. Il est donc d’ autant plus important d’ avoir des compétences de base concernant les différents psychotropes et leur utilisation ciblée, mais aussi de connaître leurs limites thérapeutiques. En ces temps de prétendu engouement pour la neurobiologie les psychotropes ne peuvent toutefois jamais remplacer une bonne relation de confiance entre le médecin et le patient. Ils doivent être considérés comme un complément dans le traitement exigeant des patients souffrant de troubles psychiques.
A considerable number of people suffer from depressive disorders, pathological anxiety, show psychosomatic symptoms, have chronic sleep problems or are psycho-physically exhausted and often first seek medical help in the GP’ s practice. GPs are therefore confronted with psychologically impaired patients every day and must then act. This makes it all the more important for them to have a basic knowledge of the various psychotropic drugs and their targeted use, but also to know about their therapeutic limitations. Even in times of supposed neurobiology hype, psychopharmaceuticals can never replace a good, trusting relationship between doctor and patient. They are to be seen as a supplement in the demanding treatment of mentally ill patients. Key words: depressive disorders, anxiety, insomnia, psychotropic drugs, neurobiology, psychopharmaceuticals
Aspects généraux du traitement par psychotropes
Les maladies psychiatriques de nature plus grave entraînent des déficiences permanentes et une espérance de vie plus courte que de nombreuses maladies somatiques. Ils imposent donc des exigences élevées au praticien. De nombreux patients atteints de maladies mentales consultent d’ abord leur médecin de famille. La plupart du temps, ce sont des personnes présentant des symptômes dépressifs où se plaignant d’ anxiété pathologique, puisque ces deux maladies mentales continuent d’ être en tête du classement des maladies mentales. Le diagnostic est parfois difficile, surtout dans le cas des troubles dépressifs, car les patients ne s’ adressent pas immédiatement à leur psychisme altéré. Ou ils ne peuvent pas relier les symptômes à des processus psychologiques et signaler seuls les problèmes physiques. Le médecin de famille a souvent l’ avantage de connaître le patient depuis longtemps, de s’ attaquer aux changements de comportement et de réactions et de rendre les symptômes explicables. Si aucune référence n’ est faite au psychiatre, la responsabilité de la thérapie lui incombe uniquement. Il joue ainsi un rôle important dans la détection initiale, le triage et le traitement des troubles mentaux, ainsi que dans la prescription et la gestion des médicaments psychotropes.
En Allemagne, les médecins de famille prescrivent un tiers des psychotropes. Aux États-Unis, les trois quarts des antidépresseurs ne sont pas prescrits par des psychiatres mais par des médecins généralistes, des pédiatres ou des gynécologues. Et le nombre de ventes de psychotropes ne cesse d’ augmenter, notamment ceux des antidépresseurs, des tranquillisants, des neuroleptiques et aussi ceux du méthylphénidate. Selon les données des caisses-maladie, environ 20 % de tous les assurés en Suisse prennent des psychotropes, dont la moitié sont prescrits par les médecins de famille et 30 % par les psychiatres. La pratique croissante de la prescription de médicaments psychotropes est à plusieurs reprises évaluée de manière critique par le corps médical, les caisses d’ assurance maladie, les patients et les organisations de patients, mais cela n’ a pas entraîné de changements positifs à ce jour. Les médicaments psychotropes doivent toujours être utilisés de manière très précise et pendant une période limitée, et le patient doit être supervisé par le médecin pendant le traitement régulièrement accompagné par le médecin prescripteur. Le traitement médicamenteux des maladies mentales est inférieur à une combinaison avec la psychothérapie médicale et n’ est fondamentalement ni raisonnable ni opportun. Les médicaments psychotropes sont importants sur le plan thérapeutique en psychiatrie et en psychothérapie, mais leur seul spectre d’ effet est limité. L’ idée que leur utilisation puisse résoudre les problèmes sous-jacents d’ un trouble mental est insuffisante. Sans une clarification approfondie et compétente avec une analyse de la situation psychosociale et professionnelle très pertinente, l’ utilisation de psychotropes reste problématique. En outre, leur effet et leurs effets secondaires doivent être mis en balance les uns avec les autres et les traitements alternatifs et les mesures d’ auto-assistance doivent être pris en compte dans la stratégie thérapeutique. L’ orientation biologique de la psychiatrie et des neurosciences tente d’ établir une image des troubles mentaux selon laquelle lorsque le psychisme souffre, le cerveau est simplement censé être malade. En relation avec l’ affirmation générale d’ aujourd’ hui de la «solution rapide» pour le médecin généraliste, des questions importantes se posent sur la pratique adéquate ou critique de l’ utilisation des psycho pharmaceutiques: Alors, quand faut-il utiliser les médicaments psychotropes ? Quelle qualité des troubles mentaux justifie le traitement médicamenteux? Quels sont les bénéfices pour les patients et quels sont les effets négatifs possibles des médicaments utilisés ?
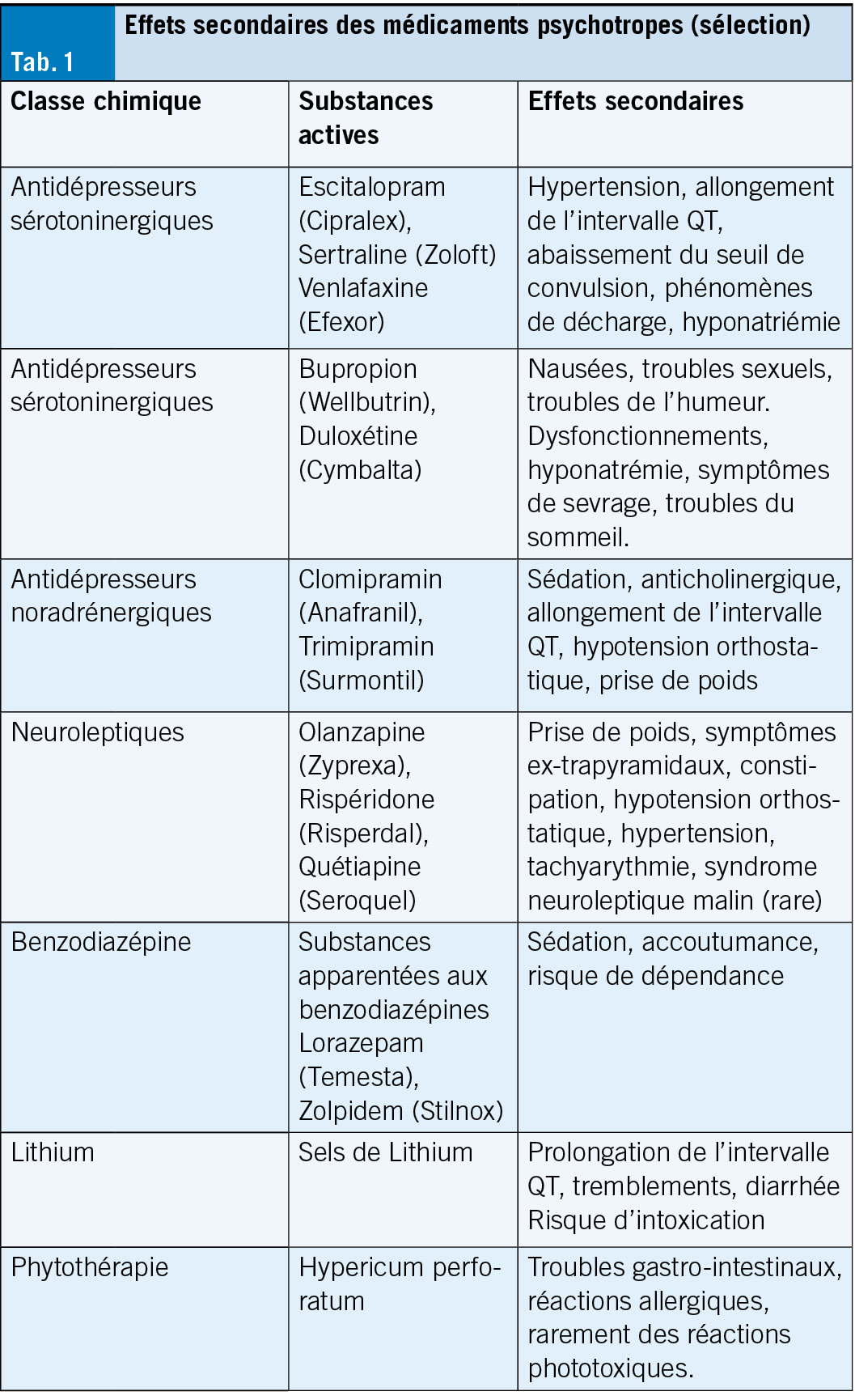
Antidépresseurs
Les données sur les bénéfices des antidépresseurs sont contradictoires. Les antidépresseurs avec un nouveau mécanisme d’ action, une efficacité rapide et moins d’ effets secondaires ne sont pas en vue. De nouvelles études montrent que si la majorité des antidépresseurs examinés diffèrent du placebo plus qu’ aléatoirement, c’ est-à-dire qu’ ils ont un effet, ils restent très faibles dans leur taille d’ effet. Les effets sont si faibles que l’ on suppose que 90 % de l’ effet des antidépresseurs est basé sur les effets placebo. Néanmoins, les patients peuvent bénéficier d’ antidépresseurs, même si nous ne savons toujours pas comment et pourquoi. L’ hypothèse de la monoamine, c’ est-à-dire qu’ une concentration réduite de sérotonine ou de noradrénaline est seule responsable de la déviation de l’ humeur de base ne peut pas être maintenue même sur la base de les dernières études. Une causalité neurobiologique linéaire entre les antidépresseurs et l’ amélioration de la santé mentale n’ a probablement jamais existé, et son effet thymoleptique partiellement obtenu reste incertain.
Pour les états dépressifs légers et modérés, les antidépresseurs ne sont plus considérés comme indiqués de toute urgence. Ils peuvent être traités avec succès par une psychothérapie médicale. Cependant, les antidépresseurs sont utilisés comme antidépresseurs activateurs (ISRS ou IRSN) ou sédatifs (par exemple trazodone (Trittico), mirtazapine (Remeron) ou tricycliques) en cas de symptômes dépressifs prononcés. Les ISRS sont également indiqués pour les troubles anxieux et avec les antidépresseurs sédatifs. Dans ce contexte, il faut tenir compte de l’ aptitude à la conduite consécutivement limitée et du risque de chute. Les ISRS sont également utilisés pour les troubles obsessionnels compulsifs et les syndromes de douleur chronique. Le syndrome sérotoninergique dans les ISRS est rare et peut survenir lorsqu’ il est associé à du triptan, à des opioïdes ou à l’ administration concomitante d’ autres antidépresseurs. En raison des incertitudes concernant le mécanisme d’ action des antidépresseurs décrites ci-dessus, il n’ existe pas d’ option médicamenteuse sur mesure pour un certain type de patient. Leur utilisation dépend de la tolérance individuelle, de la présence de comorbidités et de la propre expérience du praticien. En pratique générale, quelques préparations suffisent, mais elles doivent être bien connues. Les antidépresseurs doivent toujours être dosés (dose d’ essai) et doivent toujours être arrêtés lentement après une stabilisation soutenue de la santé mentale en raison d’ éventuels symptômes de sevrage.
Les phytothérapies (Hypericum perforatum) sont comparables aux antidépresseurs synthétiques dans les cas modérés et peuvent être proposées au patient comme premier choix car leurs effets secondaires et le risque d’ interaction sont plus faibles. La kétamine est autorisée comme antidépresseur en Suisse depuis 2020 uniquement pour la dépression résistante au traitement et est utilisée sous forme de spray nasal et de façon intraveineuse.
Ces dernières années, les neuroleptiques ont également été utilisés comme thérapie d’ augmentation pour les troubles dépressifs sans symptômes psychotiques et pour les troubles du sommeil. L’ indication semble discutable en raison des bénéfices souvent insuffisants pour les patients et surtout à cause des effets secondaires. Dans les maladies dépressives récurrentes, le lithium est incompatible avec le change le stabilisateur d’ humeur de choix, son mécanisme d’ action n’ est pas connu. Le schéma posologique doit être suivi strictement et se conformer aux tests sanguins réguliers selon le fabricant. Le domaine thérapeutique est étroit (cardiotoxicité) et le patient doit en être informé ainsi que des autres effets secondaires possibles et de l’ intervalle de temps (12 heures) entre la dernière prise de Lithium et la prise de sang.
Neuroleptiques
Il existe un grand nombre de neuroleptiques et leur sélection n’ est pas non plus facile. Ils sont antipsychotiques, sédatifs et soulagent l’ agitation psychomotrice. Ils sont utilisés p. o. ou i. m. dépôt dans les symptômes psychotiques dans le contexte de la schizophrénie ou de la dépression majeure, comme prophylaxie de phase dans les troubles bipolaires, dans les épisodes maniaques aigus et dans les états d’ éveil chez les patients schizophrènes et maniaques application i. m. aigu. Ils agissent sur différents récepteurs de la dopamine, de la sérotonine et de l’ histamine et entraîneraient une normalisation de l’ activité cérébrale, notamment via leur antagonisme D2. Dans le cas de Latuda, le mécanisme d’ action est inconnu, mais dans le cas de Reagila, l’ effet doit être obtenu via un agonisme partiel des récepteurs D3 ou D2. À ce jour, les études n’ ont pas montré que les nouvelles préparations sont significativement plus efficaces que les neuroleptiques classiques. Cependant, les antipsychotiques de la nouvelle génération (par exemple la rispéridone, l’ olanzapine, la quétiapine) sont propagés comme particulièrement efficaces contre les symptômes négatifs de la schizophrénie. La situation est similaire à celle des antidépresseurs: l’ avantage des préparations plus récentes se reflète principalement dans leurs profils d’ effets secondaires parfois plus favorables, par exemple en ce qui concerne la prise de poids, la dyskinésie tardive, l’ akathisie, la constipation, l’ hypotension orthostatique, l’ hypertension et les tachyarythmies. Dans le cas des neuroleptiques, il est également important de bien connaître quelques préparations, leurs effets secondaires et les interactions possibles et de faire attention au dosage approprié. Des études ont montré qu’ un bon effet antipsychotique est obtenu en occupant les récepteurs de la dopamine de 60 à 65 %, et à partir de 80 %, les effets secondaires extrapyramidaux augmentent fortement. Ainsi, la dose de neuroleptiques peut être maintenue à un niveau bas (par exemple, 2 à 6 mg de Risperdal, 5 à 20 mg de Zyprexa). La prudence est recommandée lors de l’ utilisation de neuroleptiques en raison d’ une agitation sévère et d’ un déficit cognitif préexistant chez les personnes âgées. Dans le cas de l’ agitation nocturne des patients âgés, les prescriptions de neuroleptiques reflètent malheureusement trop souvent le manque lamentable de personnel dans les établissements. Les gardes de siège externes sont souvent plus utiles comme alternative et doivent être discutées. Même les neuroleptiques qui ne sont utilisés que pendant une courte période peuvent également accélérer le développement de la démence.
Les benzodiazépines
Après les antidépresseurs, les benzodiazépines sont considérées comme les deuxièmes médicaments psychotropes les plus fréquemment prescrits. Ils sont utilisés dans les maladies psychiatriques comme anxiolytiques/sédatifs et hypnotiques. En raison de leur action rapide via une liaison allostérique (non compétitive) au récepteur GABA-A et de leur bonne ampleur thérapeutique, ils sont souvent prescrits trop longtemps, souvent pendant des années, ce qui entraîne des déficits cognitifs et des dépendances à faible dose. Les benzodiazépines étant des relaxants musculaires en plus de leurs effets anxiolytiques, sédatifs, sédatifs, somnifères et antispasmodiques, le risque de chutes augmente, en particulier chez les patients âgés. Les benzodiazépines peuvent être associées à des antidépresseurs et à des neuroleptiques. Cependant, il convient de souligner que l’ effet sédatif est potentialisé, surtout si de l’ alcool ou d’ autres substances psychotropes sont consommés en même temps. Il existe également la possibilité d’ une accumulation de médicaments avec certaines préparations, bien qu’ aucun effet correspondant ne puisse être détecté avec Xanax et Temesta, mais avec Valium et Rivotril, de tels effets peuvent être détectés. L’ arrêt des benzodiazépines en cas de dépendance physique entraîne des symptômes de sevrage (crise de sevrage des grottes) et doit toujours être effectué très lentement et sous surveillance médicale stricte. Des alternatives aux benzodiazépines sont disponibles pour l’ agitation et l’ augmentation des sentiments d’ anxiété, par exemple dans le cadre d’ un trouble dépressif, ainsi que pour le traitement des problèmes de sommeil. Les patients doivent se voir proposer de telles préparations principalement en raison de leur potentiel de dépendance faible ou nul. L’ huile de lavande (Laitea), la combinaison de houblon et de baldrian (Hova), la mélatonine (Circadin) ont fait leurs preuves chez de nombreux patients, et la mirtazapine (Remeron), la miansérine (Tolvon) et la tradozone (Trittico) sont également préférables aux benzodiazépines comme antidépresseurs induisant le sommeil.
Conclusion
Les troubles mentaux ne sont pas des infections et les psychotropes ne sont pas des antibiotiques. Une position médicale qui considère l’ équilibre des neurotransmetteurs comme un remède à une maladie mentale est sans aucun doute beaucoup trop myope. Elle nie le développement psychosocial pertinent en tant que facteur décisif pour le développement des troubles mentaux. La psychothérapie seule ne peut pas traiter durablement les maladies mentales, elles sont manifestement plus efficaces en combinaison avec la psychothérapie médicale et doivent donc être considérées comme un complément à une stratégie de traitement de pointe avec une psychothérapie médicale. L’ utilisation de médicaments psychotropes nécessite une compétence de base appropriée, une prise en compte précise en cas de diagnostic clarifié et d’ indication exacte, ainsi qu’ un suivi médical régulier. En outre, le patient a besoin d’ informations adéquates sur les effets secondaires possibles. Leur apparence doit toujours être prise au sérieux. Les slogans de persévérance ne sont pas utiles. Les nausées, les étourdissements, la prise de poids, l’ augmentation du pouls et de la pression artérielle ou la perte de libido représentent une réduction significative de la qualité de vie au-delà de la maladie sous-jacente. Ils perpétuent l’ affect négatif et les peurs. Chez les patients âgés, l’ altération du métabolisme doit toujours être prise en compte lors de l’ utilisation de médicaments psychotropes, notamment en ce qui concerne le risque d’ un éventuel délire en cas de polypharmacie préexistante.
L’ interface entre le médecin de famille et le psychiatre est multidimensionnelle et parfois caractérisés par des évaluations différentes et peut-être aussi par des intérêts divergents, mais l’ objectif commun est la prise en charge optimale des patients atteints de maladies mentales. Une clarification et un traitement spécialisés doivent être envisagés rapidement.
Cet article est une traduction de «der informierte arzt – die informierte ärztin» 11/2023
Copyright Aerzteverlag medinfo AG
Dr méd. Michael Sacchetto-Mussetti
Spécialiste en psychiatrie et psychothérapie FMH
Dorfstrasse 5
8700 Küsnacht
www.zentrumkuesnacht.ch
m.sacchetto@hin.ch
L’ auteur n’ a déclaré aucun conflit d’ intérêts en relation avec cet article.
Contrairement à l’ esprit du temps, le traitement des patients psychiatriques nécessite pas seulement une thérapie psychopharmaceutique, mais aussi un accompagnement permanent. Un examen et un traitement psychothérapeutique par le médecin spécialiste sont indiqués dans la plupart des cas.
Il convient de prendre peu de médicaments psychotropes, mais de bien connaître le mode d’ action, les effets secondaires et les interactions. Il est particulièrement important de discuter précisément de la médication. Leur dosage respectif doit être choisi avec retenue (effets secondaires). Les médicaments psychotropes doivent être utilisés de façon limitée dans le temps. Les neuroleptiques ne doivent être considérés que comme une solution de dernier recours.
Une bonne collaboration entre le médecin de famille et le psychiatre augmente le taux de réussite du diagnostic, surtout pour les troubles dépressifs souvent non diagnostiqués.
Les effets secondaires des médicaments psychotropes doivent toujours être pris au sérieux. Même si la prise de médicaments n’ est que de courte durée, il convient d’ adapter la dose ou un changement de médicament.