Die pulmonale Hypertonie (PH) ist häufig und betrifft rund 1 % der Weltbevölkerung. Sie darf nicht mit der seltenen pulmonal-arteriellen Hypertonie (PAH, «Orphan disease») verwechselt werden. Die PAH stellt eine spezifisch therapierbare Untergruppe der PH dar. Die Prävalenz der PAH wird in westlichen Ländern auf rund 50 Fälle pro Million Einwohner geschätzt. Klinisch präsentiert sich die PH anfangs meist unspezifisch mit Belastungsdyspnoe und Müdigkeit, was oft zu einer verzögerten Diagnosestellung führt. Für Hausärztinnen und Hausärzte ist es daher entscheidend, bei unklarer Dyspnoe frühzeitig an eine PH zu denken. Eine strukturierte Abklärung ermöglicht die Einordnung in die verschiedenen PH-Gruppen und identifiziert diejenigen Patientinnen und Patienten, die von einer gezielten Therapie profitieren. Die frühzeitige Überweisung in ein spezialisiertes Zentrum, wie auch die hausärztliche Betreuung sind zentral für Prognose und Versorgungsqualität.
Pulmonary hypertension (PH) is common and affects approximately 1 % of the global population. It should not be confused with the rare pulmonary arterial hypertension (PAH), an orphan disease that represents a specifically treatable subgroup of PH. The prevalence of PAH in Western countries is estimated at around 50 cases per million inhabitants. Clinically, PH typically presents with non-specific symptoms such as exertional dyspnea and fatigue, often leading to delayed diagnosis at more advanced stages of the disease. For primary care physicians, it is therefore crucial to consider PH early in the evaluation of unexplained dyspnea. A structured diagnostic approach allows classification into the different PH groups and helps identify those patients who may benefit from targeted therapy. Early referral to a specialized center, as well as continued primary care management, are essential for improving prognosis and quality of care.
Keywords: Pulmonale Hypertonie; pulmonal-arterielle Hypertonie; Dyspnoe; Grundversorgung
Unspezifische Symptome
Die Symptome der pulmonalen Hypertonie beginnen oft schleichend mit Anstrengungsdyspnoe, allgemeiner Leistungsminderung, Müdigkeit, Palpitationen und Dyspnoe beim Vornüberbeugen (Bendopnoe). In fortgeschrittenen Stadien treten Zeichen der Rechtsherzinsuffizienz und Synkopen auf, selten kommt es zur Hämoptyse. Die unspezifischen Beschwerden und häufig vorhandenen Komorbiditäten führen dazu, dass von Symptombeginn bis zur Diagnosestellung trotz zunehmender Sensibilisierung für das Krankheitsbild weiterhin 1–2 Jahre vergehen. Bei Diagnosestellung ist die Symptomlast erheblich, häufig liegt eine Dyspnoe NYHA III–IV vor (1). Unabhängig von der Grunderkrankung ist die Entwicklung einer PH mit einer verminderten Lebensqualität und ungünstiger Prognose verbunden.
Diagnostisches Vorgehen bei Verdacht auf eine pulmonale Hypertonie
Bei unklarer Dyspnoe sollte initial eine Basisdiagnostik (Labor inkl. NT-proBNP, EKG, Thoraxröntgen, Lungenfunktion) durchgeführt werden. Bleibt die Ursache unklar oder besteht ein klinischer Verdacht auf eine PH, sollte eine Echokardiographie zur Abschätzung der PH-Wahrscheinlichkeit erfolgen. Bleibt der Verdacht einer PH, folgt eine strukturierte Abklärung zur Abschätzung der Wahrscheinlichkeit und zur Zuordnung in eine der fünf PH-Gruppen.
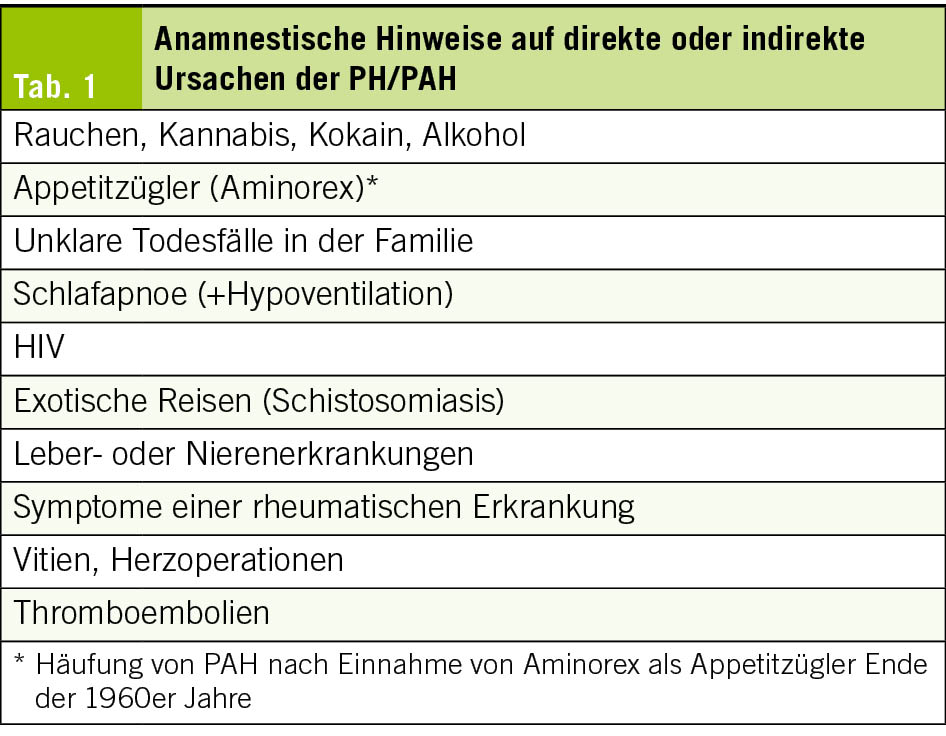
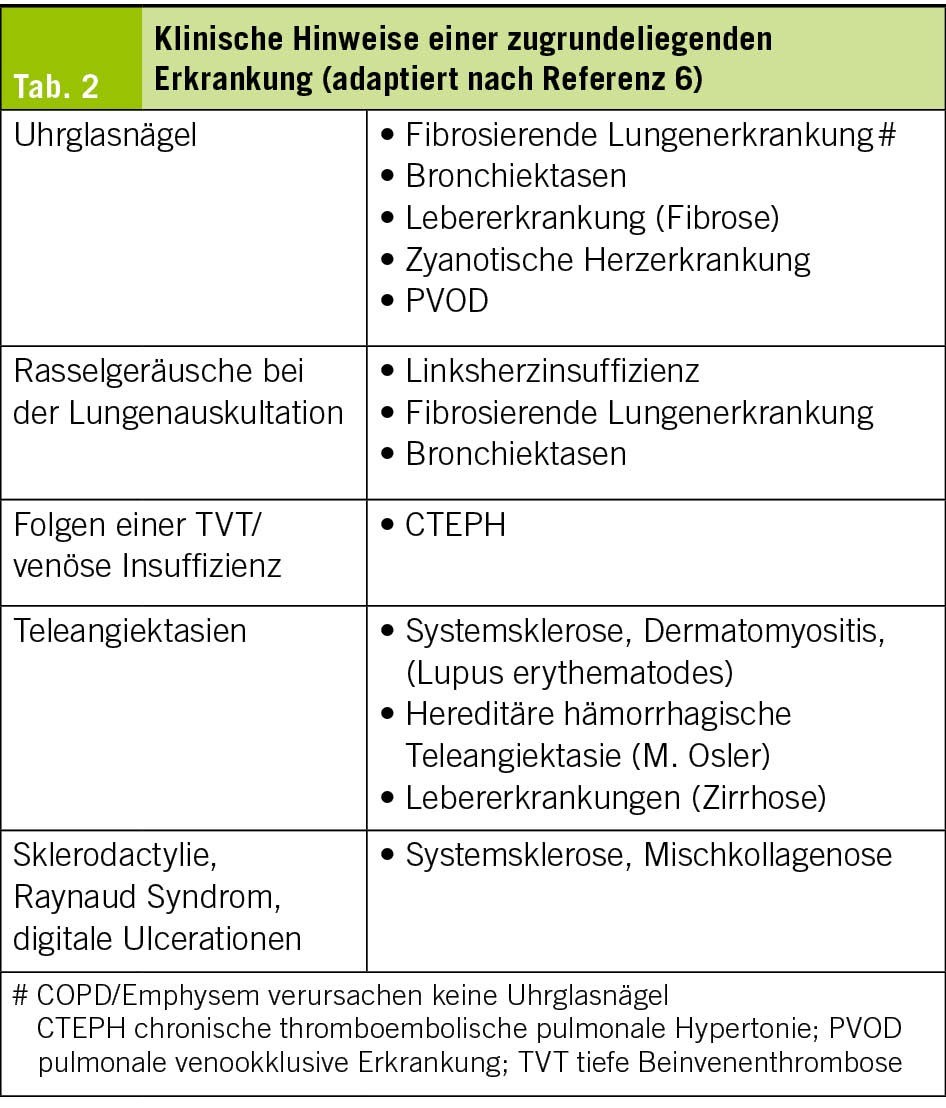
Bereits in der Anamnese können Hinweise auf die Ursache der PH oder eine zugrunde liegende Erkrankung gefunden werden (Tab. 1). In der körperlichen Untersuchung findet sich je nach Krankheitsstadium ein prominenter 2. Herzton, Zeichen der Rechtsherzinsuffizienz wie Unterschenkelödeme, positiver hepato-jugulärer Reflux (HJR), eine Hepatomegalie und ggf. Ascites oder eine Zyanose. Es ist aber möglich, dass die körperliche Untersuchung fast normal ausfällt. Klinische Hinweise auf eine Ursache der PH sind in Tab. 2 zusammengestellt.
Apparativ folgen Lungenfunktion inklusive Diffusionsmessung, arterielle Blutgasanalyse, 6-Minuten-Gehtest sowie CT-Thorax. Zur Diagnostik der chronischen thromboembolischen pulmonalen Hypertonie (CTEPH) sind Perfusionsdefizite entscheidend. Hierzu kann ein CT-Thorax mit Kontrastmittel mit der sogenannten «dual energy»-Technik oder alternativ eine Perfusionsszintigraphie durchgeführt werden. Die Echokardiographie dient zudem dem Ausschluss einer Linksherzerkrankung.
In unklaren Fällen kann eine Spiroergometrie zusätzliche Informationen liefern. Die diagnostische Aussagekraft ist hoch und auch zur Risikobeurteilung gewinnt die Spiroergometrie an Bedeutung (2, 3). Das NT-proBNP ist ein wichtiger Marker für Verlauf und Prognose.
Definition
Die definitive Diagnose erfolgt mittels Rechtsherzkatheter. Eine pulmonale Hypertonie liegt bei einem mittleren pulmonalarteriellen Druck (mPAP) > 20 mmHg vor. Die Differenzierung in prä- und postkapilläre PH erfolgt anhand des pulmonalkapillären Verschlussdrucks (PCWP) und des pulmonalen Gefässwiderstands (PVR). Kombinationen aus prä- und postkapillärer PH sind möglich.
Die Relevanz der klinischen Klassifikation
Die umfangreiche Diagnostik dient der Zuordnung der PH in eine der 5 Gruppen, da hiervon die Therapie abhängt. Eine Übersicht dieser Einteilung bietet Abb. 1.
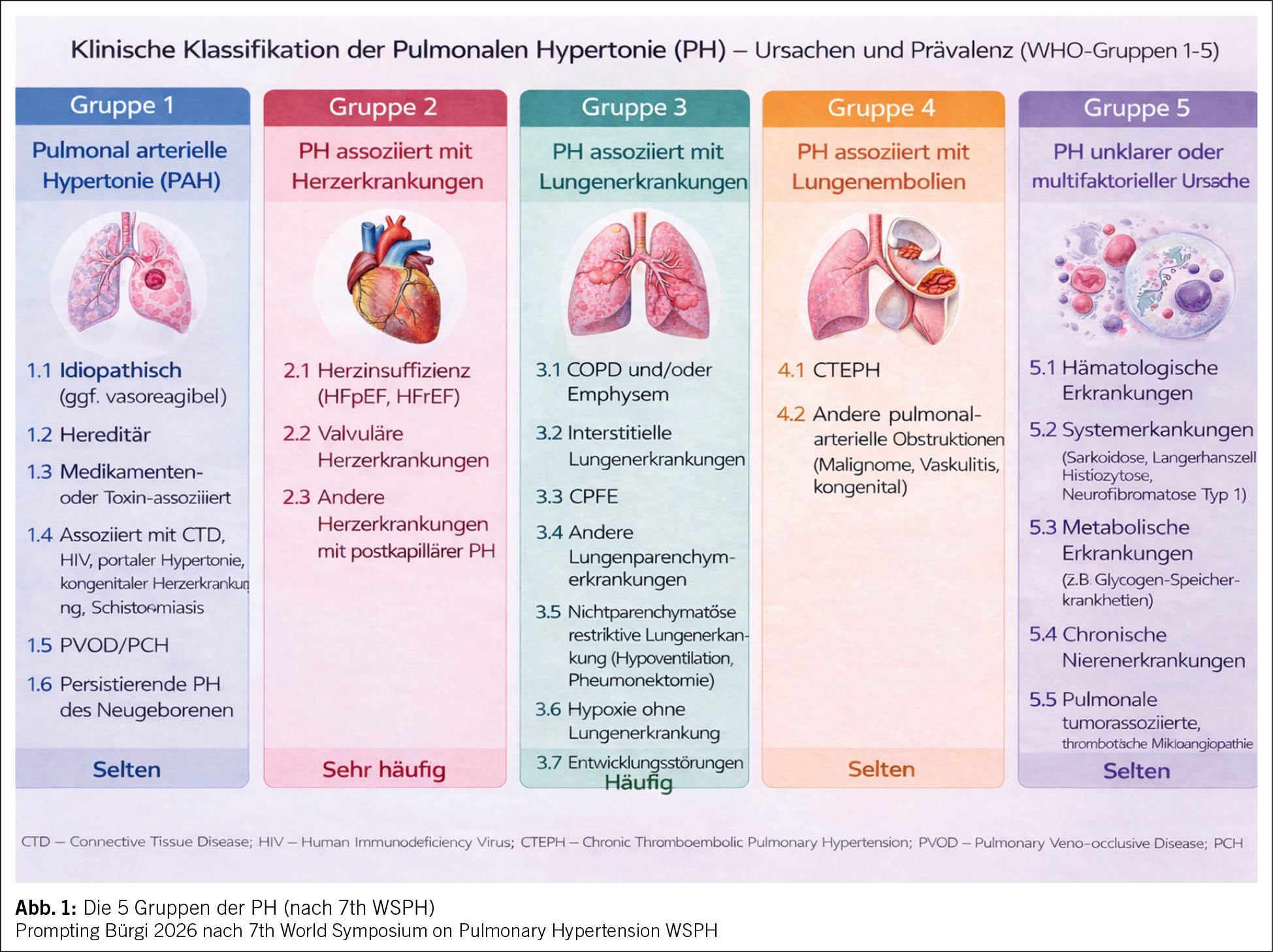
Die häufigsten Ursachen sind Linksherz- und Lungenerkrankungen (ca. 95 % der Fälle) (4). Nur Patienten mit einer PAH oder einer CTEPH qualifizieren für eine PH-spezifische medikamentöse Therapie, bei den anderen Gruppen steht die Therapie der Grunderkrankung im Vordergrund. Bei der CTEPH gibt es je nach Lokalisation der Gefässobstruktion in den Lungenarterien chirurgische (Thrombendarterektomie) und interventionelle (Ballonangioplastie) Therapieoptionen in Referenzzentren.
Therapie: supportive Massnahmen
Unabhängig von der zugrundeliegenden PH-Form sind supportive Massnahmen ein zentraler Bestandteil der Behandlung. Dazu gehören insbesondere eine sorgfältige Volämiekontrolle sowie eine Sauerstoffsupplementierung bei relevanter Hypoxämie (pO₂ < 8 kPa). Regelmässige, angepasste körperliche Aktivität – idealerweise im Rahmen eines strukturierten Rehabilitationsprogramms – wird empfohlen.
Ebenfalls wichtig sind präventive Massnahmen wie konsequente Impfungen. Bei Frauen im gebärfähigen Alter sollte eine zuverlässige Kontrazeption besprochen werden, da Schwangerschaften bei PH mit einem hohen Risiko für Mutter und Kind verbunden sind und daher davon abgeraten wird. Tritt dennoch eine Schwangerschaft ein, ist eine engmaschige Betreuung in einem spezialisierten Zentrum erforderlich. Psychosoziale Unterstützung ist ein weiterer wesentlicher Bestandteil der Betreuung.
Insgesamt kommt der hausärztlichen Mitbetreuung eine entscheidende Rolle in der langfristigen Versorgung dieser Patientinnen und Patienten zu.
PH-spezifische Therapien nur bei ausgewählten Formen
Seit den 1990er-Jahren stehen uns spezifische Medikamente für die PAH (Gruppe 1) zur Verfügung. Am Anfang war es lediglich ein intravenöses Prostacyclin-Analogon, welches mittels Pumpe kontinuierlich verabreicht werden musste. Unterdessen stehen uns 4 Medikamentenklassen zur Verfügung. Diese Therapien sind zwar sehr teuer, aber auch effektiv; die Sterblichkeitsrate ist immer noch deutlich erhöht, sie hat sich über die vergangenen 30 Jahre aber fast halbiert (5). Eine Übersicht über die Therapien und typische Nebenwirkungen bietet Tab. 3. Die Therapieempfehlungen basieren auf einer Risikostratifizierung.
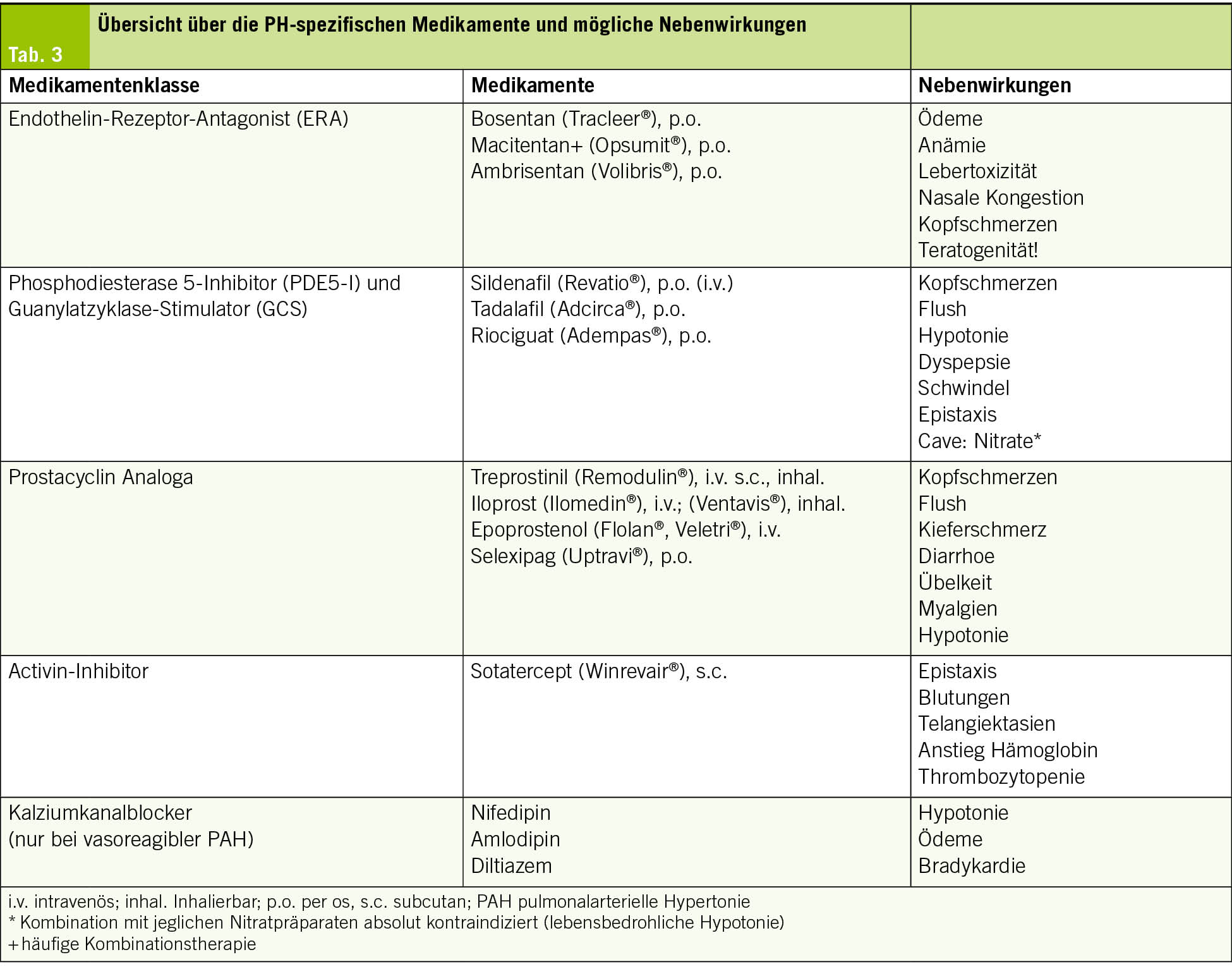
Eine Antikoagulation wird ausser bei der CTEPH nicht mehr generell empfohlen. In ausgewählten Fällen sollte frühzeitig die Möglichkeit einer Lungentransplantation evaluiert und mit den Betroffenen besprochen werden.
Komorbiditäten
In frühen Studien waren jüngere Patientinnen ohne relevante Komorbiditäten übervertreten. In der Sprechstunde sehen wir aber häufig ältere und polymorbide Patientinnen und Patienten. Dies spiegelt sich auch in Registerdaten, wo unterdessen bis zu 85 % der Patientinnen und Patienten mit PAH kardiopulmonale Komorbiditäten aufweisen und das Alter bei Diagnosestellung > 60 Jahre beträgt. Die Abgrenzung zwischen den Gruppen 1, 2 und 3 ist deshalb nicht einfach. Dieser Tatsache wurde in den Leitlinien Rechnung getragen, es gibt unterdessen einen Therapiealgorithmus für PAH-Patienten mit kardiopulmonalen Komorbiditäten (6) und es steht die Frage im Raum, ob PAH-Patienten mit Komorbiditäten in künftigen Leitlinien nicht eine eigene PH-Gruppe darstellen sollten.
Der PAH-Patient in der Hausarztpraxis
In der hausärztlichen Praxis ist zu beachten, dass die überwiegende Mehrzahl der PH-Fälle durch Linksherzerkrankungen oder chronische Lungenerkrankungen verursacht wird, während die PAH eine seltene Differenzialdiagnose darstellt.
Patientinnen und Patienten mit einer PAH sind heute älter und weisen relevante Komorbiditäten sowie eine komplexe Medikation auf. Die hausärztliche Betreuung ist deshalb für eine ganzheitliche Versorgung entscheidend. Neben der Behandlung der Begleiterkrankungen und den oben erwähnten supportiven Massnahmen sind Kenntnisse der PH-spezifischen Medikamente inklusive deren Nebenwirkungen und potenziellen Interaktionen wichtig (Tab. 3).
Warnzeichen einer klinischen Verschlechterung müssen frühzeitig erkannt werden. Dazu gehören eine zunehmende Belastungsdyspnoe, Leistungsknick, Zeichen der Rechtsherzinsuffizienz (periphere Ödeme, Gewichtszunahme) oder Synkopen. Bei entsprechenden Veränderungen sollte zeitnah Rücksprache mit dem betreuenden PH-Zentrum erfolgen oder eine erneute spezialisierte Beurteilung initiiert werden. Ein frühzeitiges Reagieren ist entscheidend, da eine rasche Eskalation der Therapie einen Einfluss auf die Prognose hat.
Welche Patienten sollen in eine Spezialsprechstunde für PH überwiesen werden?
Eine Überweisung zur Abklärung einer PH sollte bei klinischem Verdacht frühzeitig erfolgen, insbesondere wenn die Symptomatik nicht durch häufige kardiale oder pulmonale Erkrankungen erklärt werden kann. Eine unbehandelte PH verläuft progredient und ist mit einer hohen Morbidität und Mortalität verbunden. Leitsymptom ist dabei meist eine progrediente Belastungsdyspnoe unklarer Genese oder eine Diskrepanz zwischen subjektiver Symptomatik und objektivierbaren Befunden.
Ein weiterer Zuweisungsgrund können auffällige bildgebende Befunde sein. Hierzu zählt zum Beispiel ein dilatierter Truncus pulmonalis (ab 30 mm) im CT-Thorax (6), vor allem wenn gleichzeitig eine ungeklärte Dyspnoe besteht. Ebenso sollte bei echokardiographischem Verdacht auf eine PH eine weiterführende Abklärung erfolgen, sofern keine hinreichende Erklärung durch eine linksventrikuläre Erkrankung oder eine Lungenerkrankung vorliegt.
Besondere Aufmerksamkeit erfordern Patientinnen und Patienten mit Grunderkrankungen, die mit einer erhöhten Prävalenz einer P(A)H einhergehen, wie z. B. bei der Systemsklerose oder den Mischkollagenosen. In diesen Risikogruppen ist ein regelmässiges Screening empfohlen.
Schliesslich sollte bei Patientinnen und Patienten nach Lungenembolien an eine CTEPH gedacht werden, wenn die Belastungsdyspnoe trotz adäquater Antikoagulation nach drei bis sechs Monaten persistiert.
Fazit
Die pulmonale Hypertonie (PH) ist häufig und stellt ein facettenreiches Syndrom dar. Demgegenüber stellt die pulmonal-arterielle Hypertonie (PAH) eine definierte Patientengruppe dar, welche mit PH-spezifischen Medikamenten behandelt werden kann.
Das Ziel einer Spezialsprechstunde ist es, mittels einer strukturierten Abklärung herauszufinden, bei welchen Patienten diese Therapien eingesetzt werden können. Unterdessen gibt es 4 potente Medikamentenklassen. Die Mehrheit der Patienten mit PAH sind > 60 Jahre alt und polymorbide. Eine abgestimmte Zusammenarbeit zwischen Hausärzt/-innen und spezialisierten Zentren ermöglicht ein frühzeitiges Erkennen von Komplikationen und verbessert die Prognose und Versorgungsqualität.
Abkürzungen
ABGA Arterielle Blutgasanalyse
CTEPH Chronisch thromboembolische pulmonale Hypertonie
mPAP Mittlerer pulmonalarterieller Druck
NYHA New York Heart Association (Funktionsklassifikation)
PAH Pulmonal-arterielle Hypertonie
PCWP Pulmonalkapillärer Verschlussdruck
PH Pulmonale Hypertonie
PVR Pulmonalvaskulärer Widerstand
TTE Transthorakale Echokardiographie
Copyright
Aerzteverlag medinfo AG
Pneumologie und Schlafmedizin
Stadtspital Triemli
Birmensdorferstrasse 497
8063 Zürich
Der Autor hat keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Khou V, McGoon MD, Strange G, et al. Diagnostic delay in pulmonary arterial hypertension: Insights from the Australian and New Zealand pulmonary hypertension registry. Respirology. 2020;25(8):863–871.
2. Zhao L, et al. Cardiopulmonary exercise testing improves diagnostic specificity in patients with echocardiography-suspected pulmonary hypertension. Clin Cardiol. 2017;40(2):95–101.
3. Baccelli A, et al. Prognostic value of cardiopulmonary exercise testing in pulmonary arterial hypertension. Eur Respir J. 2025;66(2):2402026.
4. Hoeper MM, Humbert M, Souza R, Idrees M, Kawut SM, Sliwa-Hahnle K, et al. A global view of pulmonary hypertension. Lancet Respir Med. 2016;4:306–322.
5. Boucly A, Weatherald J, Savale L, Jaïs X, Cottin V, Prevot G, Picard F, de Groote P, Jevnikar M, Bergot E, Chaouat A, Chabanne C, Bourdin A, Parent F, Montani D, Simonneau G, Humbert M, Sitbon O. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J. 2017;50(2):1700889.
6. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS, Escribano-Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke-Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Vonk Noordegraaf A, Delcroix M, Rosenkranz S; ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023;61(1):2200879.