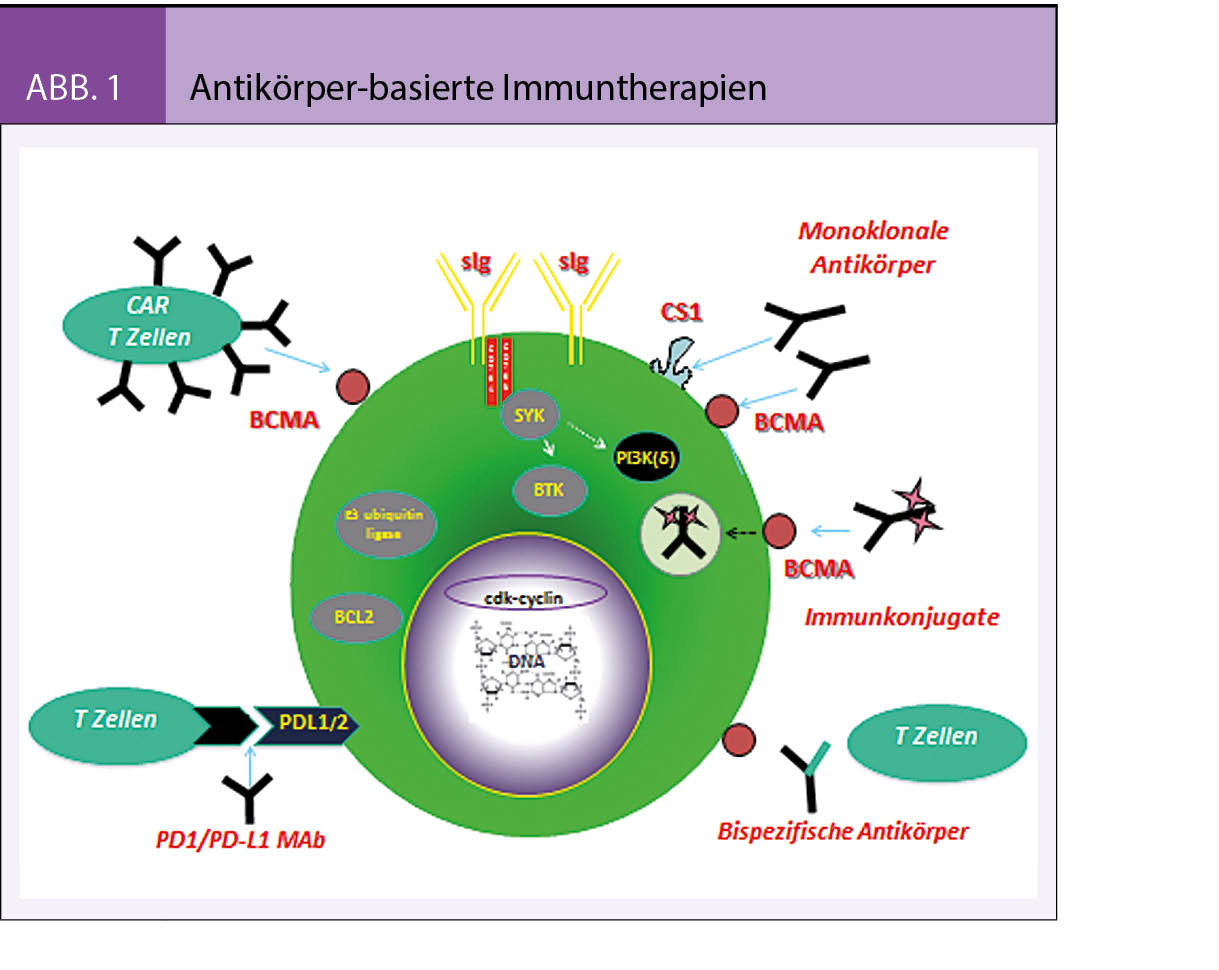
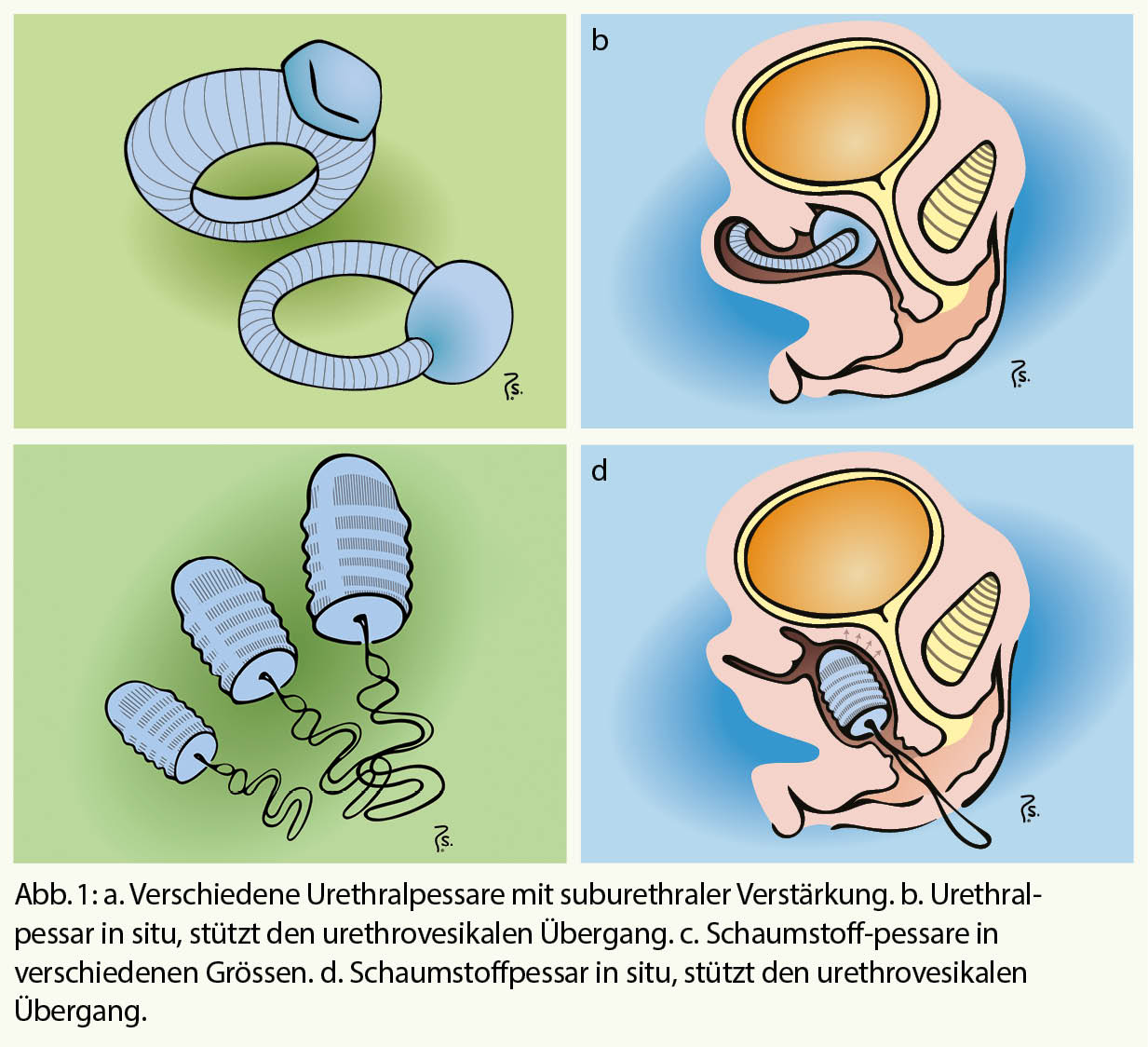
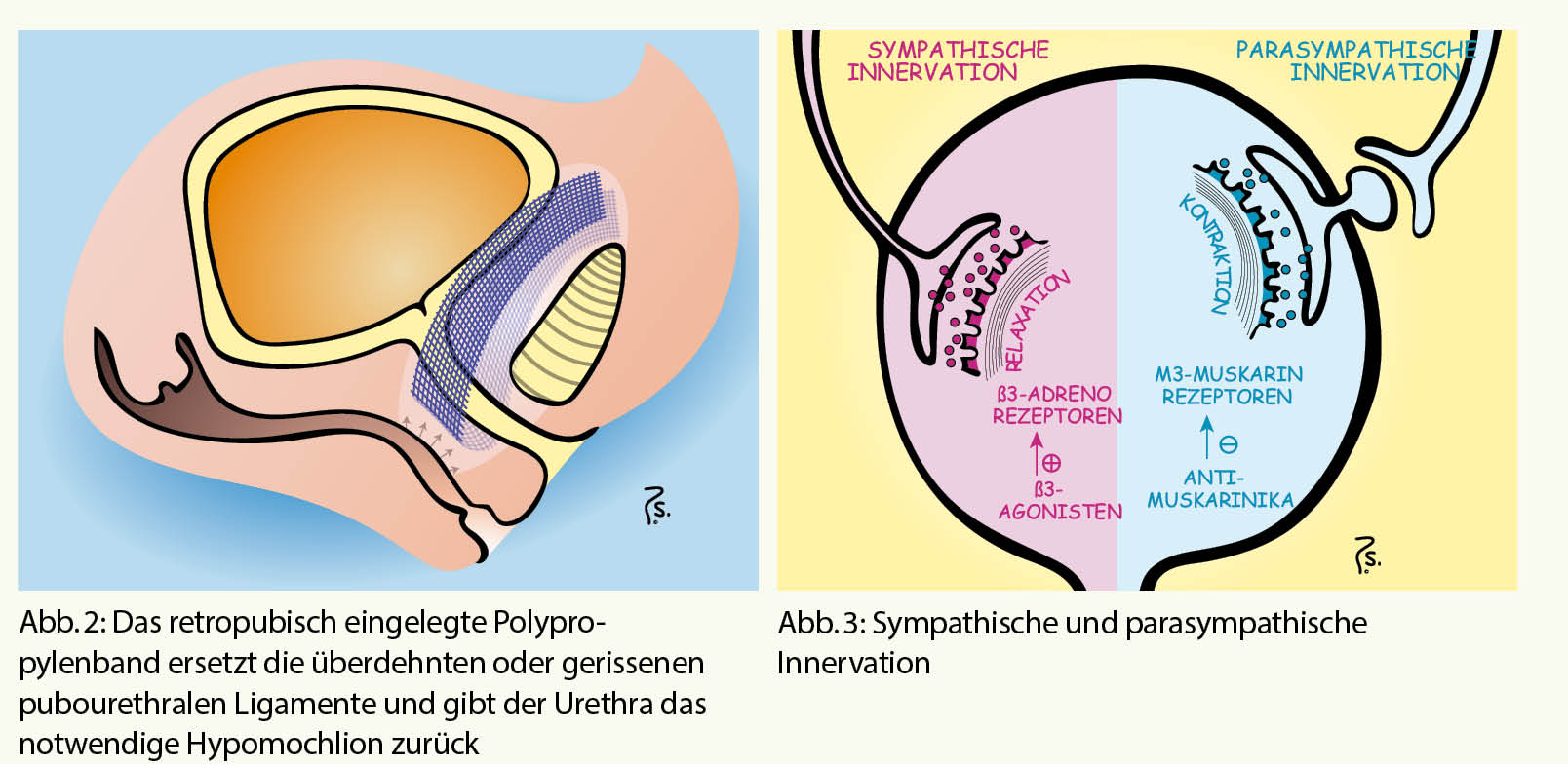
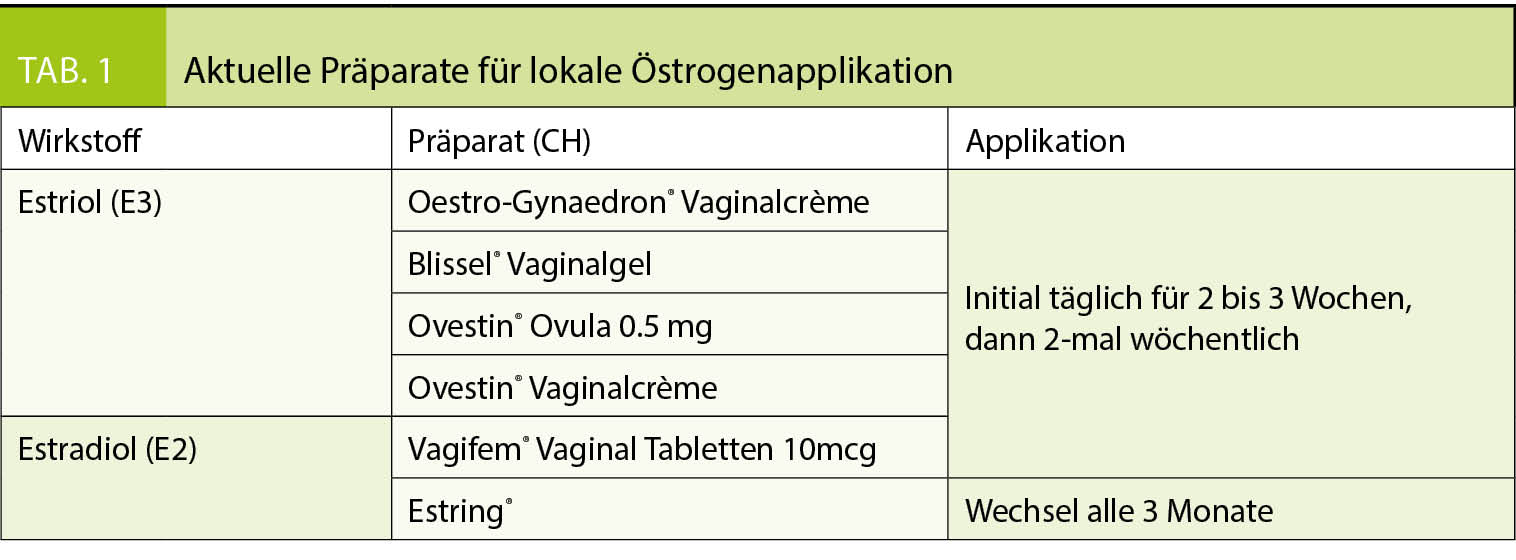
Die natürliche Hämostase befindet sich in einem Gleichgewicht der Wirkung von prokoagulatorischen und antikoagulatorischen Faktoren. Eine Entgleisung endet oft entweder in Blutung oder in Thrombose. Der operative Eingriff ist eine Intervention, welche trotz kontrollierter blutstillender Verhältnisse eine prothrombotische Wirkung induziert. Bestimmende Faktoren dafür sind die Lokalisation des chirurgischen Traumas, die Dauer des Eingriffs, die Lädierung grosser Gefässe.
L’ hémostase naturelle se situe dans un équilibre de facteurs procoagulatoires et anticoagulatoires. Le déraillement se termine souvent par un saignement ou une thrombose. L’ intervention chirurgicale est une intervention qui induit un effet prothrombotique malgré une hémostase contrôlée. Les facteurs décisifs à cet égard sont l’ emplacement du traumatisme chirurgical, la durée de l’ opération et la lésion des gros vaisseaux.
Die postoperative Thromboseneigung wurde bereits in den 70er Jahren erkannt und systematisch mit Heparinen behandelt. Die niedermolekularen Heparine (LMWH) waren Produkte, welche an der Handhabung, Effizienz und Sicherheit dem älteren unfraktionierten Heparin überlegen waren und sich deswegen schnell als indizierte Mittel zur postoperativen Thromboseprophylaxe durchgesetzt haben. Mittlerweile haben die Vielfalt der Operationen, die Verfeinerung der operativen Techniken und die Entwicklung neuer Antikoagulantien die Planung der Thromboseprophylaxe, sowohl für eine bestehende Antikoagulation wie auch für den rein postoperativen Thromboseschutz zur Herausforderung gemacht.
Perioperative Überbrückung einer vorbestehenden Antikoagulation («Bridging»)
Vorhofflimmern und venöse thromboembolische Ereignisse treten mit zunehmendem Lebensalter häufiger auf. Auf Grund der Alterung der Gesellschaft ist es somit wahrscheinlich, dass immer mehr Menschen mit einer längerfristigen Antikoagulation behandelt werden. Wenn bei antikoagulierten Patienten ein invasiver Eingriff ansteht, stellen sich 3 Schlüsselfragen:
Wie hoch ist das Risiko für den Patienten, wenn die Antikoagulation nicht unterbrochen wird?
Wie hoch ist das Risiko für den Patienten, wenn die Antikoagulation unterbrochen wird?
Wie hoch ist die Sicherheit und Wirksamkeit, wenn ein alternatives Antikoagulans zur Überbrückung («Bridging») eingesetzt wird?
Das klassische Konzept der Bridging-Therapie war die Überbrückung einer Antikoagulatin mit einem Vitamin-K Antagonisten (VKA; z.B. Phenprocoumon) mit einem unfraktionierten Heparin mit Dosisanpassung anhand der aktivierten Thromboplastinzeit (aPTT). In den letzten 20 Jahren wurde jedoch das unfraktionierte Heparin (LMWH) ersetzt. Diese können bei normaler Nierenfunktion in therapeutischer Dosierung subkutan und ohne Laborkontrolle in einem ambulanten Setting verabreicht werden und beinhalten zudem ein eindeutig kleineres Risiko für die Entwicklung einer Heparin-induzierten Thrombopenie. Mit der Einführung der noch neueren direkten oralen Antikoagulanzien (DOACs; z.B. Dabigatran, Apixaban, Edoxaban, Rivaroxaban) hat sich das Management der perioperativen Antikoagulation nochmals deutlich verändert.
Blutungsrisiko
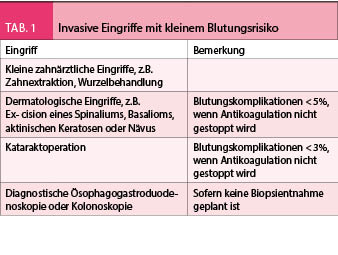
Nicht jeder operative Eingriff geht mit dem gleich grossen Blutungsrisiko einher. Bei einigen Eingriffen ist das Blutungsrisiko so klein, dass eine orale Antikoagulation gar nicht unterbrochen werden muss. Daher handelt es sich vor allem um kleine zahnärztliche, dermatologische oder ophthalmologische Eingriffe (Tab 1). Bei diesen Eingriffen lassen sich Blutungen in den meisten Fällen auch bei antikoagulierten Patienten mit lokalen Massnahmen bestens beherrschen. Das Blutungsrisiko muss jedoch selbstverständlich für jeden Patienten individuell abgeschätzt werden.
Thromboembolierisiko
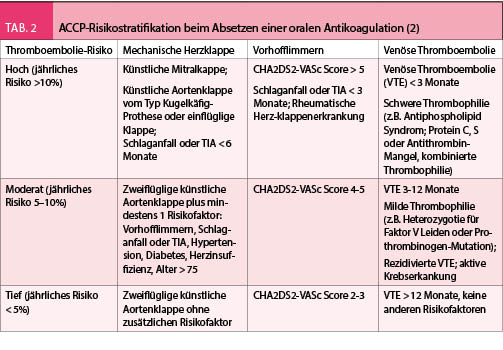
Die häufigsten Gründe für eine zeitliche unlimitierte Antikoagulation sind die Schlaganfallprophylaxe beim Vorhofflimmern, die unprovozierte und/oder rezidivierte venöse Thromboembolie und der mechanische Herzklappenersatz. Wird die Antikoagulation mit einem VKA durchgeführt, so ist bei einem präoperativen Absetzen des VKA eine überbrückende Antikoagulation nur bei einem relevanten Thromboembolierisiko notwendig. Dieses Risiko ist sowohl von der Indikation der Antikoagulation als auch vom Patienten selber abhängig. Gemäss den ACCP Guidelines von 2012 werden die Patienten bezüglich ihres jährlichen Thromboembolierisikos in der 3 Risikokategorien stratifiziert (Tab. 2). Während die Guidelines für Patienten mit einem hohen Risiko grundsätzlich ein Bridging empfehlen, kann bei Patienten mit einem niedrigen Risiko die Antikoagulation in den meisten Fällen ohne Bridging mit einer alternativen Substanz unterbrochen werden. Bei Patienten mit intermediärem Risiko müssen sowohl die Risiken des jeweiligen Eingriffs als auch die individuellen Risiken des Patienten zur Festlegung der Strategie einbezogen werden.
Praktisches Vorgehen bei Marcoumar und anderen VKA
Grundsätzlich ist das periprozedurale Blutungs- und Thromboembolie-Risiko eines invasiven Eingriffs bei allen Patienten mit dauerhafter Antikoagulation höher als bei Patienten ohne Antikoagulation. Diese Tatsache ist unabhängig von den Modalitäten der überbrückenden Antikoagulation und muss mit den Patienten in einem Aufklärungsgespräch besprochen werden. Die publizierten Leitlinien zur Überbrückung einer Antikoagulation beziehen sich meistens auf Warfarin, welches eine deutlich kürzere Halbwertszeit als das Pheprocoumon (Marcoumar®) hat. Beispielsweise wird in den ACCP Guidelines empfohlen, eine VKA-Therapie etwas 5 Tage vor einem invasiven Eingriff mit relevantem Blutungsrisiko zu stoppen und dann falls notwendig auf eine Bridging-Substanz umzusteigen. In der Praxis bewährt es sich jedoch, Phenprocoumon schon mindestens 7 Tage vor einem geplanten Eingriff zu pausieren. Sobald die INR unter 2.0 abgefallen ist, muss in Abhängigkeit des Risikos (Tab. 2) die Bridging-Therpie gestartet werden. Die aktuellen Guidelines sehen UFH und LMWH als gleichwertig an, enthalten jedoch keine genauen Angaben über die zu verwendende LMWH-Dosierung. In der Praxis werden heutzutage vorwiegend LMWH zum Bridging verwendet und es hat sich bewährt, die Dosierung Risiko-adaptiert zu wählen: bei hohem Thromboembolierisiko LMWH in therapeutischer Dosis (200 E/kg Körpergewicht/Tag); bei moderatem Thromboembolierisiko LMWH in (hoch-) prophylaktischer Dosis (100 E/kg Körpergewicht/Tag). Gemäss den ACCP Guidelines soll das UFH 6h vor einem invasiven Eingriff gestoppt werden, die letzte Dosis LMWH soll nicht später als 24h vorher appliziert werden. Es muss unbedingt sichergestellt werden, dass zum Zeitpunkt des Eingriffs keine relevante VKA-Wirkung mehr vorliegt. Dehalb empfiehlt es sich, die INR 24h vor dem Eingriff nochmals zu kontrollieren. Sollte sie zu diesem Zeitpunkt noch nicht unter 1.5 abgefallen sein, kann eine Dosis Vitamin K (z.B. 2-10 mg Konakion® p.o. oder i.v.) appliziert werden, um eine Korrektur bis zum Zeitpunkt des Eingriffs herbeizuführen (INR am Morgen des Eingriffs nochmals messen!). Nach erfolgtem Eingriff soll 48-72h bis zur Wiederaufnahme einer therapeutischen Antikoagulation in jedem Fall erst dann erfolgen, wenn klinisch kein relevanted Blutungsrisiko mehr besteht. Bei Patienten mit hohem Thromboembolierisiko kann postoperativ die Zeit bis zum Einsetzen einer therapeutischen Antokoagulation mit LMWH in prophylaktischer Dosierung überbrückt werden. Sobald postoperativ die Hämostase gesichert ist, darf auch der VKA wieder begonnen werden. DIe Bridging-Substanz sollte so lange gegeben werden, bis ein therapeutischer INR an zwei aufeinander folgenden Tagen dokumentiert ist.
Praktisches Vorgehen bei DOAC
Das perioperative Procedere bei Patienten, die mit einem DOAC antikoaguliert sind, ist auf Grund der relativ kurzen Halbwertszeit dieser Substanz denkbar einfach. In der Regel sollen DOAC 24h vor einem invasiven Eingriff mit Blutungsrisiko gestoppt werden, bei hohem Blutdruckrisiko schon 48h vorher. Sobald nach erfolgtem Eingriff die lokale Hämostase wieder gewährleistet ist, kann dann die Antikoagulation mit dem DOAC wieder aufgenommen werden, oft schon ab dem ersten postoperativen Tag. Bei hohem Blutungsrisiko ist es auch möglich, kurzzeitig prophylaktische Dosen zu verwenden. Bezüglich der Details für die einzelnen Substanzen ist auf www.swissmedicinfo.ch verwiesen. Ein eigentliches Bridging mit LMWH entfällt also bei den DOACs, weil die Pharmakokinetik dieser Medikamentenklassen identisch ist. Gelegentlich kann es aber notwendig sein, postoperativ beispielsweise bei nicht möglicher peroraler Medikamenteneinnahme vorübergehend auf eine parenterale Substanz auszuweichen.
Postoperative Thromboseprophylaxe ohne vorbestehende Antikoagulation
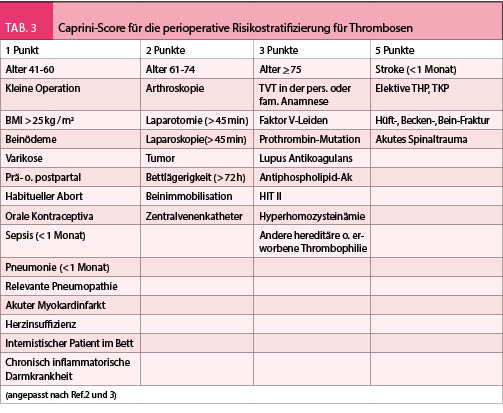
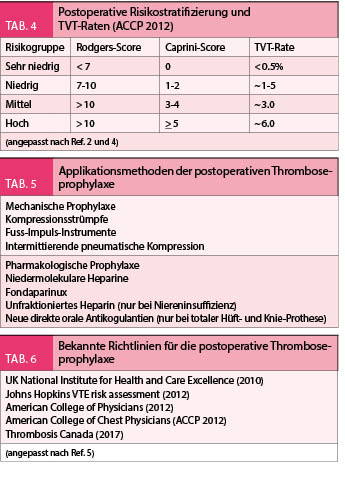
Das Risiko für postoperative Thrombosen wurde in verschiedenen Studien bereits früher vor 15 Jahren abgeschätzt, z.B. bei der totalen Hüft- oder Knieprothese lag die Rate der postoperativen Thrombosen zwischen 25-40%. Die prophylakrische postoperative Behandlung mit LMWH konnte diese Rate auf < 20% reduzieren. Neuere Operationstechniken und Materialien haben aktuell dieses Risiko auf 4-5% reduziert. Dem entsprechend besteht eine Tendenz das postoperative Risiko genauer zu gewichten und die Patienten demnach in Risikogruppen für Thrombosebildung zu stratifizieren. Dafür werden detaillierte Score-Systeme verwendet (z.B. Caprini-Score oder Rodgers-Score, Tab. 5), welche verschiedene klinische, anamnestische und medikamentöse Parameter, die mit Thrombosen assoziiert sind, festhalten. Die Stratifizierung kategorisiert die Patienten in 4 Stufen (sehr niedrig, niedrig, mittel, hoch). Die geschätzte Thromboserate je nach Stufe wird in Tabelle 4 angegeben. Je nach Risikokategorie wird ebenfalls die Auswahl der Form der Prophylaxe empfohlen, mechanische oder pharmakologische (Tab. 5). Je nach Operationstyp kann diese Empfehlung auch variieren.
In der Literatur sind verschiedene Richtlinien für die Handhabung der perioperativen Thromboseprophylaxe bekannt (Tab. 6). Alle münden in die gleiche Richtung ein, eine Risikostratifizierung z.B. nach Caprini-Score wird präoperativ verlangt, je nach Risikokategorie wird die Art der Prophylaxe ausgewählt. Bei den nicht-orthopädischen Eingriffen mit sehr niedrigem oder niedrigem Thromboserisiko werden konservative oder mechanische Massnahmen empfohlen, für mittleres oder hohes Thromboserisiko ist die pharmakologische Prophylaxe die bessere Auswahl. In der Schweiz sind die Empfehlungen der ACCP 2012 breiter bekannt. Die pharmakologische Prophylaxe wird mit niedermolekularen Heparinen appliziert (Dalteparin, Enoxaparin, Nadroparin), die Dosierung bleibt bei den allermeisten Fällen fixiert auf ca. 75 E/ Kg KG/ Tag als subkutane Gabe einmal pro Tag. Einige Einschränkungen sind hier zu berücksichtigen (kleines Gewicht < 50 Kg, Niereninsuffizienz GFR < 30 ml / min), welche zur Dosisadaptation führen. Die Herstellerangaben der Produkte sind hier zu befolgen.
Je nach Operation und Gewichtung der Blutungskonsequenzen kann diese Dosierung reduziert werden (z.B. ZNS-Operationen initial mit halbierter Dosis der LMWH).
Dauer der Thromboseprophylaxe
Lange sahen die Empfehlungen vor, die Thromboseprophylaxe nur während der Hospitalisation zu verabreichen. Neuere Studien insbesondere bei den orthopädischen Eingriffen empfehlen eine Dauer von 5 Wochen postoperativ, bzw. bei persistierenden Risikofaktoren für Thrombosen (Beinimmobilisation, Bettlägerigkeit) auch länger.
Start der Prophylaxe präoperativ oderpostoperativ
Die offizielle Zulassung der LMWH in Europa schreibt vor, dass die erste Gabe der Prophylaxe 12h präoperativ verabreicht werden soll. Die Rationale dafür liegt Jahrzehnte zurück, als man festgestellt hatte, dass die meisten postoperativen Thrombosen bereits intraoperativ initiiert wurden. Mit der präoperativen Gabe wollte man allfällige thrombogene Herde im Gefässnetz Stunden vor der Provokation unterdrücken, sodass intraoperativ keine Thrombusbildung aktiviert wird. Mittlerweile haben Vergleichsstudien aus den USA mit Enoxaparin, wie auch die Vergleichsstudie mit den neuen direkten Antikoagulantien in der orthopädischen Chirurgie gezeigt, dass die präoperative Gabe eines LMWH keine Vorteile bedeutet. Demzufolge erlaubt die Empfehlung der ACCP 2012 die erste Gabe sowohl 12h präoperativ wie 12h postoperativ zu planen. Kürzeres postoperatives Fenster war mit mehr Blutungskomplikationen assoziiert.
Aspirin zur postoperativen Thromboseprophylaxe
Bereits seit den 90er Jahren war es bekannt, dass Aspirin einen Schutz gegen Thrombosen ausüben könnte, allerdings mit einer Effizienz von nur 50-60%. Im Vergleich zu den LMWH war die eindeutig ungenügend und daher war Aspirin nie für diese Indikation empfohlen. Neuere Daten signalisieren hier eine Wende. Eine kürzlich publizierte Vergleichsstudie zwischen Aspirin 100 mg/Tag und Rivaroxaban 10mg/Tag in einer selektionierten Patientenkohorte mit totaler Hüft- oder Knieprothese hat gezeigt, dass Aspirin gleich effizient und sicher war wie Rivaroxaban. Die Europäische Gesellschaft für Anästhesie hat dies bereits in den offiziellen Empfehlungen aufgenommen, allerdings für Patienten mit niedrigem Thromboserisiko oder für Patienten mit hohem Blutungsrisiko (1). Diese Empfehlung kann leider nicht auf Patienten mit nicht-orthopädischen Eingriffen übertragen werden.
Dr. med. Lukas Graf
Zentrum für Labormedizin, Hämostase- und Hämophiliezentrum St. Gallen
Frohbergstrasse 3
9001 St. Gallen
lukas.graf@zlmsg.ch
Prof. Dr. med. Dimitrios Tsakiris
Klinik für Hämatologie
Hämatologische Diagnostik Labormedizin
Universitätsspital Basel und Blutspendezentrum beider Basel SRK
Petersgraben 4
4031 Basel
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Beitrag deklariert.
Ein Bridging der therapeutischen Antikoagulation mit Vitamin K-Antagonisten kann mit einem LMWH je nach Thromboserisiko in therapeutischer (200 E/Kg KG / d) oder subtherapeutischer (100 E / Kg KG / d) Dosierung erfolgen.
Ein eigentliches Bridging der DOAC mit LMWH entfällt aufgrund der identischen Pharmakokinetik.
Postoperativ sind 2018 niedrigere Thromboserisiken zu beobachten als vor 15 Jahren.
Die Risikostratifizierung für Thrombosen (Caprini- o. Rodgers-Score) postoperativ definiert die Art (mechanisch oder pharmakologisch) und die Dauer der Thromboseprophylaxe.
Aspirin kann neu bei orthopädischen Eingriffen (Total-Knie- oder Hüftprothese) bei Patienten mit niedrigem Thromboserisiko postoperativ zur Thromboseprophylaxe eingesetzt werden (100 mg/d p.o.)
Messages à retenir
Le pontage de l’ anticoagulation thérapeutique avec des antagonistes de la vitamine K peut être effectué avec un LMWH à des doses thérapeutiques (200 E / Kg KG / j) ou sous-thérapeutiques (100 E / Kg KG / j), selon le risque de thrombose.
En raison de la pharmacocinétique identique, il n’ est pas nécessaire d’établir un pontage entre le DOAC et le LMWH
En postopératoire, le risque de thrombose est plus faible en 2018 qu’ il y a 15 ans.
La stratification du risque de thrombose (score Caprini ou Rodgers) post-opératoire définit le type (mécanique ou pharmacologique) et la durée de la prophylaxie thrombotique.
L’aspirine peut maintenant être utilisée en postopératoire pour prévenir la thrombose (100 mg / j p.o.) dans les procédures orthopédiques (prothèse totale du genou ou de la hanche) chez les patients présentant un faible risque de thrombose.
Literatur
1. Jenny J-Y, Pabinger I, Samama CM. European guidelines on perioperative venous thromboembolism prophylaxis: Aspirin. Eur J Anaesthesiol. England; 2018 Feb;35(2):123–9.
2. Gould MK, Garcia DA, Wren SM, Karanicolas PJ, Arcelus JI, Heit JA, et al. Prevention of VTE in nonorthopedic surgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. United States; 2012 Feb;141(2 Suppl):e227S–e277S.
3. Caprini JA. Risk assessment as a guide for the prevention of the many faces of venous thromboembolism. Am J Surg. United States; 2010 Jan;199(1 Suppl):S3-10.
4. Falck-Ytter Y, Francis CW, Johanson NA, Curley C, Dahl OE, Schulman S, et al. Prevention of VTE in orthopedic surgery patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. United States; 2012 Feb;141(2 Suppl):e278S–e325S.
5. Murphy PB, Vogt KN, Lau BD, Aboagye J, Parry NG, Streiff MB, et al. Venous Thromboembolism Prevention in Emergency General Surgery: A Review. JAMA Surg. United States; 2018 May;153(5):479–86.
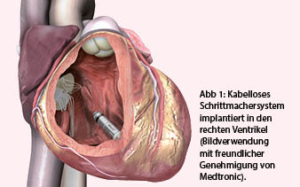
Konventionelle Herzschrittmacher besitzen Elektroden, die über lange Kabel mit dem Schrittmacheraggregat verbunden sind. Diese Kabel stellen die Achillesferse herkömmlicher Systeme dar und können Komplikationen verursachen. Die Implantationsrate von kabellosen Schrittmachern nimmt daher rasch zu. Der folgende Beitrag gibt eine Übersicht über Vor- und Nachteile kabelloser Systeme, Indikationen, Implantationsablauf, Patientennachsorge und mögliche Zukunftsperspektiven.
Les stimulateurs cardiaques conventionnels ont des électrodes qui sont reliées à l’unité du stimulateur cardiaque par de longs câbles. Ces câbles sont le talon d’Achille des systèmes conventionnels et peuvent causer des complications. Le taux d’implantation des stimulateurs cardiaques sans fil augmente donc rapidement. L’article suivant donne un aperçu des avantages et des inconvénients des systèmes sans fil, des indications, de la procédure d’implantation, du suivi des patients et des perspectives d’avenir possibles.
Konventionelle Schrittmacher – sind wir am Ende einer Erfolgsgeschichte?
Bereits wenige Jahre nach der Erstimplantation eines voll implantierbaren Herzschrittmachers 1958 hatte sich die Schrittmachertherapie als Methode der Wahl zur Behandlung bradykarder Rhythmusstörungen etabliert. Technische Neuerungen in den kommenden Jahren verbesserten die therapeutischen Möglichkeiten rasch. Die aufgrund der beschränkten Energiespeicherkapazität initial noch kurze Lebensdauer der Geräte vergrösserte sich mit Lithium-basierter Batteriechemie. Mikroprozessor-basierte elektronische Schaltungen und drahtlose Programmierbarkeit erlaubten patientenspezifische Optimierungen. Beschleunigungssensoren und andere Sensoren ermöglichten mit der «rate-response» eine physiologischere Stimulation des Herzens. Die Implementierung eines zusätzlichen Kabels im Rahmen der Resynchronisationstherapie gestattete auch die Stimulation des linken Ventrikels in Patienten mit Herzinsuffizienz. Moderne Aggregate verfügen über vielfältigste Funktionen. Automatische Optimierungen von Stimulationsoutput (zum Energiesparen), Herzinsuffizienz- und Schlafapnoemonitoring oder Fernüberwachung der Gerätefunktion zuhause repräsentieren nur einen kleinen Teil der heute verfügbaren Möglichkeiten.
Trotzdem besitzen heutige konventionelle Systeme gewichtige Nachteile. Nebst der endlichen Lebensdauer der Geräte aufgrund limitierter Batteriereserven sind insbesondere die Schrittmacherkabel eine Achillesferse. Die Komplikationsrate nach Schrittmacherimplantation ist erheblich. Bereits zwei Monate nach Implantation erleiden mehr als 10% aller Patienten eine Komplikation, am häufigsten bedingt durch die Schrittmacherkabel (1). Initial handelt es sich oft um Kabeldislokationen, im Laufe der Zeit mehren sich Isolationsdefekte und Kabelbrüche (vgl. auch Fallbeschreibung zu Abb. 2). Es liegt daher auf der Hand, durch Vermeidung von Kabeln und Entwicklung von kabellosen Schrittmachern diese Komplikationen zu umgehen. Der Grundgedanke besteht darin, dass das ganze Schrittmachersystem in den rechten Ventrikel eingeführt wird und somit keine Kabel mehr notwendig sind (Abb. 1).
Kabellose Schrittmacher – die aktuelle Studienlage
Die ersten Patienten, bei denen kabellos der Herzmuskel stimuliert wurde, waren Empfänger eines Gerätes zur kardialen Resynchronisation (CRT). Es handelte sich bei den implantierten Geräten nicht um eigentliche Schrittmacher, sondern lediglich um einen kleinen piezoelektrischen Wandler. Dieser konvertierte im linken Ventrikel einen von aussen zugeführten Ultraschallpuls in einen elektrischen Stimulationspuls (2). Der Ultraschallpulsgenerator wird dabei zusätzlich zum CRT-Generator subkutan implantiert. Im Rahmen der WiSE-CRT-Studie wurde dieses Gerät 13 von 17 Patienten erfolgreich implantiert. Es kam jedoch zu drei Perikard-
ergüssen, einer der betroffenen Patienten verstarb (3). Die Studie wurde daher vorzeitig gestoppt. Auch die später publizierte SELECT-LV-Studie zeigte mit 8.6% eine hohe Anzahl an Akutkomplikationen nach Implantation des Gerätes (4). Der Hersteller des WiSE-CRT®-Systems (EBR Systems) modifizierte daraufhin das Implantat – konklusive Resultate zu diesem Device stehen aus.
Die erste Implantation eines kompletten kabellosen Schrittmachers im Menschen wurde 2012 durchgeführt. 2014 wurden im Rahmen der LEADLESS-Studie Resultate zu 33 Patienten publiziert (5). Sicherheit und Implantationserfolg des ersten kabellosen Schrittmachers (Nanostim®, Abbott) wurden dabei untersucht. In 32 von 33 Patienten war die Implantation erfolgreich, bei einem Patienten kam es zu einer Myokardperforation, an deren Komplikationen der Patient verstarb. In der nachfolgenden multizentrischen LEADLESS II-Studie wurden 526 Patienten nach Nanostim®-Implantation untersucht. Nach sechs Monaten konnten bei 6.7% der Patienten schwerwiegende Komplikationen beobachtet werden (darunter u. a. 1.6% kardiale Perforationen, 1.1% Dislokationen, 1.2% vaskuläre Komplikationen, und 0.8% interventionsbedürftige Reizschwellenerhöhungen) (6). Nach mehreren Todesfällen wurde die weitere Implantation des Gerätes zunächst kurzzeitig gestoppt. Im weiteren Verlauf wurden dann Batterieprobleme mit komplettem Deviceversagen beobachtet, was im Herbst 2016 zu einem weltweiten Implantationsstopp dieser Geräte führte.
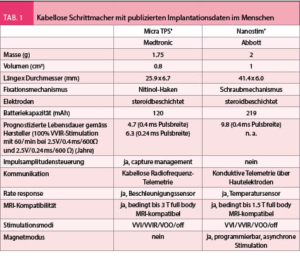
LEADLESS I und II hatten gezeigt, dass kabellose Schrittmacher funktionieren und die Implantation einfach möglich ist, das Gerät hatte aber ernsthafte Probleme verursacht. Eine grössere Studie zu einem kabellosen Schrittmacher eines anderen Herstellers wurde 2016 publiziert. Es handelte sich dabei um eine prospektive multizentrische Studie, welche die Sicherheit des Micra TPS® (Medtronic) in 725 Patienten untersuchte (7). Auch dieses Gerät schien einfach implantierbar zu sein und gute Stimulationsparameter zu erzielen. 25 Patienten (3.4%) entwickelten akute Komplikationen (ein Todesfall, 1.6% kardiale Perforationen, keine Dislokationen). Verglichen mit einer historischen Kontrollgruppe mit konventionellen Schrittmachern war die Komplikationsrate der kabellosen Geräte signifikant geringer. Die Resultate dieser Studie konnten im Rahmen einer weltweiten Registerstudie bestätigt werden (8). Das Micra TPS® konnte in 99.6% der Patienten erfolgreich implantiert werden. Im ersten Monat nach Implantation wurden in 1.51% der Patienten ernste Komplikationen beschrieben (darunter 0.13% Perforationen, 0.13% Dislokationen). Die Rate an schwerwiegenden Komplikationen lag damit tendenziell sogar leicht tiefer als in den initialen Studien zum Micra TPS®. Dieser kabellose Schrittmacher ist derzeit als einziger frei erhältlich und in Europa zugelassen. Weltweit haben mittlerweile gegen 20 000 Patienten diesen Schrittmacher erhalten. Einen Überblick über die technischen Spezifikationen des Micra TPS® und Nanostim® liefert (Tab. 1).
Kabellose Schrittmacher – Implantationsablauf und Besonderheiten in der Nachsorge
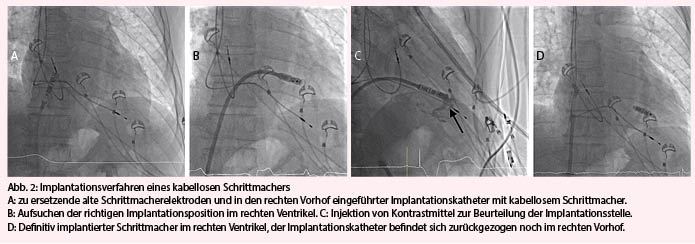
Im Gegensatz zu konventionellen Schrittmachern werden kabellose Aggregate ganz in den rechten Ventrikel eingeführt. Dazu wird in Lokalanästhesie (ggf. Sedation) die V. femoralis punktiert, nach einer kleinen Hautinzision der Zugangsweg schrittweise aufgedehnt und schliesslich eine 27-F-Schleuse (Aussendurchmesser) in der Vene platziert. Dadurch wird anschliessend unter Durchleuchtung das ganze Schrittmachersystem mittels eines steuerbaren Katheters via V. cava inferior in die rechte Herzkammer eingeführt (Abb. 2A und 2B). Nach präferenziell septaler Platzierung – allenfalls unter Zuhilfenahme von Kontrastmittel zur RV-Ventrikulografie (Abb. 2C) – wird der Schrittmacher durch ein Fixationssystem (Haken oder Schraubmechanismus) am Myokard fixiert. Die mechanische Fixierung wird mittels sanftem Zug überprüft. Nach Kontrolle der üblichen Stimulationsparameter kann der immer noch am Katheter fixierte Schrittmacher definitiv freigelassen oder umplatziert werden (Abb. 2D). Die femoral eingeführte Schleuse wird schliesslich entfernt und die Hautinzision mit einer Z-Naht verschlossen.
Nach Implantation kann der Patient auf einer intermediate care unit oder einer Normalstation mit geschultem Personal weiter überwacht werden. Einer ambulanten Implantation stehen wir sehr zurückhaltend gegenüber, da es auch Stunden später noch zu einer lebensbedrohlichen Perikardtamponade kommen kann. Einige Stunden nach Implantation kann der Patient mobilisiert werden. Üblicherweise führen wir am Folgetag eine Optimierung der Frequenzadaptierung durch Anpassung des Beschleunigungssensor-Vektors durch. Auch die Z-Naht kann meist bereits entfernt und durch einen Verband ersetzt werden. Auf das Heben schwerer Lasten und Benetzen der Wunde sollte in den Folgetagen verzichtet werden, ansonsten sind keine spezifischen Vorsichtsmassnahmen er-forderlich. Wie üblich erfolgt nach Einheilung des Schrittmachers nach ca.
2 Monaten die erste ambulante Kontrolle, die später in jährlichem Abstand wiederholt wird. Patienten mit kabellosem Schrittmacher lassen sich nach Abheilung der inguinalen Punktionsstelle nicht mehr klinisch identifizieren. Das Gerät kann im Thoraxröntgenbild jedoch noch ausgemacht werden (Abb. 2d). Sämtliche bislang in den Menschen implantierbaren Schrittmacher sind bedingt MRI-kompatibel. Sie müssen aber wie konventionelle Schrittmacher vor und nach der Bildgebung umprogrammiert werden. Auch ist ein MRI erst 6 Wochen nach Implantation zulässig.
Kabellose Schrittmacher – für welchen Patienten?
Bei welchen Patienten sollte nun – bei bestehender Schrittmacherindikation – die Implantation eines kabellosen Schrittmachers erwogen werden? Die 2013 publizierten Guidelines der Europäischen Gesellschaft für Kardiologie machen noch keine Aussagen dazu (9). Werden sie sinngemäss angewendet, stellen kabellose Schrittmacher in erster Linie eine Option dar bei Patienten mit AV-Block und Vorhofflimmern. Gemäss einer multinationalen Umfrage ist dies nebst venösen Zugangsproblemen (z.B. Verschluss der Vv. subcaviae, Dialysekatheter) und Komplikationen nach konventioneller Schrittmachereinlage tatsächlich die häufigste Implantationsindikation (10). Eine mechanische Trikuspidalklappe, ein V. cava-Filter und morbide Adipositas mit einem erwarteten Abstand des Gerätes zum Programmierkopf > 12.5 cm stellen Kontraindikationen zur Implantation dar.
Kabellose Herzschrittmacher – Limitationen und Zukunftsperspektiven
Bei den derzeitig verfügbaren kabellosen Schrittmachern handelt es sich nur um 1-Kammerschrittmacher zur Implantation in den rechten Ventrikel. Die Indikationen zur Implantation solcher Systeme sind limitiert (9), die überwiegende Zahl der heute implantierten konventionellen Schrittmacher sind 2-Kammerschrittmacher oder Resynchronisationsgeräte. Entsprechend werden grosse Anstrengungen unternommen, kabellose Geräte mit VDDR- oder DDDR-Modi zu entwickeln. Diese Programmiermodi erlauben die Detektion der Vorhofaktion und eine entsprechend darauf abgestimmte Schrittmacherstimulation des Ventrikels. Im DDDR-Modus würde sogar eine Vorhofstimulation ermöglicht. Zur Entwicklung genannter Modi werden verschiedene Ansätze verfolgt. Eine Wahrnehmung der aktiven mechanischen Vorhofaktion (A-Welle) scheint theoretisch mit einem Beschleunigungssensor des im rechten Ventrikel sitzenden kabellosen Geräts möglich. Dieser Ansatz wurde im Rahmen der MASS- und MARVEL-Studien untersucht. Es zeigt sich, dass das Konzept grundsätzlich funktioniert, sich damit aber derzeit während lediglich 87% aller Herzschläge eine gewisse VDD-Stimulation erzielen lässt (11). Sollte zusätzlich auch eine Vorhofstimulation gewünscht sein, müsste auch dort noch ein Gerät implantiert werden. Aufgrund der teilweise äusserst geringen Wanddicke des rechen Vorhofs stellt die sichere und komplikationsarme Verankerung eines Gerätes ebendort eine grosse Herausforderung dar. Schliesslich müssten zwei (oder sogar mehrere) kabellose Geräte im Herzen drahtlos miteinander kommunizieren können. Konventionelle Radiofrequenz-Telemetrie scheint dazu aufgrund des relativ hohen Energieverbrauchs nicht geeignet. Eine mögliche Alternative stellt die «intra-body-communication» dar, die die elektrische Leitfähigkeit von Gewebe und Blut nutzt. Dabei werden kurzzeitige hochfrequente Wechselstromimpulse an Myokard und Blut abgegeben, was eine sehr energiesparende, schnelle und bidirektionale Kommunikation erlaubt (12). Kürzlich konnten in Tierversuchen mit dieser Technologie erstmals erfolgreich kabellose Zweikammerschrittmacher implantiert werden.
Dr. med. Dr. phil. Andreas Häberlin
Universitätsklinik für Kardiologie
Inselspital
3010 Bern
Universität Bern
andres.haeberlin@artorg.unibe.ch
MScLukas Bereuter
Universitätsklinik für Kardiologie
Inselspital
3010 Bern
Prof. Dr. med. Hildegard Tanner
Leitende Ärztin Rhythmologie und Elektrophysiologie
Universitätsklinik für Kardiologie
Inselspital
Freiburgstrasse
3010 Bern
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel.
Kabellose Schrittmacher stellen bei Patienten mit Schrittmacher-Indikation eine potentielle Alternative zu herkömmlichen Systemen dar.
Die kabelbedingten Komplikationen konventioneller Schrittmacher können durch diese Geräte vermieden werden. Es besteht schwache Evidenz, dass die Overall-Komplikationsrate kabelloser Geräte geringer als diejenige konventioneller Systeme sein könnte.
Speziell geeignet zur Implantation eines kabellosen Schrittmachers scheinen Patienten mit venösen Zugangsproblemen oder AV-Block mit Vorhofflimmern.
Kabellose 2-Kammerschrittmacher sind derzeit noch nicht kommerziell erhältlich, hierzu müssen zunächst technische Herausforderungen bewältigt werden (z.B. energiesparende Kommunikation, Verankerung eines Gerätes im rechten Vorhof).
Messages à retenir
Les stimulateurs cardiaques sans fil sont une alternative potentielle aux systèmes conventionnels pour les patients ayant des indications de stimulateur cardiaque.
Les complications liées aux câbles des stimulateurs cardiaques conventionnels peuvent être évitées grâce à ces dispositifs. Il y a peu de preuves que le taux de complication global des dispositifs sans fil pourrait être inférieur à celui des systèmes conventionnels.
Les patients présentant des problèmes d’accès veineux ou un bloc AV avec fibrillation auriculaire semblent particulièrement appropriés à l’implantation d’un stimulateur cardiaque sans fil.
Les stimulateurs cardiaques sans fil à 2 chambres ne sont pas encore disponibles dans le commerce, pour cela, il faut d’abord maîtriser les défis techniques (p. ex. communication économe en énergie, ancrage d’un appareil dans l’oreillette droite)
Literatur
1. Udo, E.O., et al., Incidence and predictors of short- and long-term complications in pacemaker therapy: the FOLLOWPACE study. Heart Rhythm, 2012. 9(5): p. 728-35.
2. Auricchio, A., et al., First-in-man implantation of leadless ultrasound-based cardiac stimulation pacing system: novel endocardial left ventricular resynchronization therapy in heart failure patients. Europace, 2013. 15(8): p. 1191-7.
3. Auricchio, A., et al., Feasibility, safety, and short-term outcome of leadless ultrasound-based endocardial left ventricular resynchronization in heart failure patients: results of the wireless stimulation endocardially for CRT (WiSE-CRT) study. Europace, 2014. 16(5): p. 681-8.
4. Reddy, V.Y., et al., Cardiac Resynchronization Therapy With Wireless Left Ventricular Endocardial Pacing: The SELECT-LV Study. J Am Coll Cardiol, 2017. 69(17): p. 2119-2129.
5. Reddy, V.Y., et al., Permanent leadless cardiac pacing: results of the LEADLESS trial. Circulation, 2014. 129(14): p. 1466-71.
6. Reddy, V.Y., et al., Percutaneous Implantation of an Entirely Intracardiac Leadless Pacemaker. N Engl J Med, 2015. 373(12): p. 1125-35.
7. Reynolds, D., et al., A Leadless Intracardiac Transcatheter Pacing System. N Engl J Med, 2016. 374(6): p. 533-41.
8. Roberts, P.R., et al., A leadless pacemaker in the real-world setting: The Micra Transcatheter Pacing System Post-Approval Registry. Heart Rhythm, 2017. 14(9): p. 1375-1379.
9. Brignole, M., et al., 2013 ESC Guidelines on cardiac pacing and cardiac resynchronization therapy: the Task Force on cardiac pacing and resynchronization therapy of the European Society of Cardiology (ESC). Developed in collaboration with the European Heart Rhythm Association (EHRA). Eur Heart J, 2013. 34(29): p. 2281-329.
10. Boveda, S., et al., Use of leadless pacemakers in Europe: results of the European Heart Rhythm Association survey. Europace, 2018. 20(3): p. 555-559.
11. Chinitz, L., et al., Accelerometer-based atrioventricular synchronous pacing with a ventricular leadless pacemaker: Results from the Micra atrioventricular feasibility studies. Heart Rhythm, 2018.
12. Bereuter L., et al., Leadless dual-chamber pacing – a novel communication
method for wireless pacemaker synchronization. JACC Basic to Translational
Science, accepted.
Die pädiatrische AML ist seltener aber aggressiver als die ALL. Heutzutage können ca. 60% der Patienten geheilt werden. Dieser Erfolg konnte erreicht werden, einerseits durch die Intensivierung der konventionellen Chemotherapie, eine umfassende supportive Therapie und eine sorgfältige Behandlung von Komplikationen und Rezidiven, andererseits durch eine zunehmend individualisierte prognostische Einordnung zum optimalen Einsatz verfügbarer Therapieoptionen.
La leucémie myéloïde aiguë pédiatrique est plus rare mais plus agressive que la leucémie lymphoblastique aigüe (LLA). Aujourd’ hui, environ 60 % des patients peuvent être guéris. Ce succès a été obtenu d’ une part par l’ intensification de la chimiothérapie conventionnelle, une thérapie de soutien complète et un traitement soigneux des complications et des récidives et, d’ autre part, par une classification pronostique de plus en plus individualisée pour une utilisation optimale des options thérapeutiques disponibles.
Epidemiologie
Die akute myeloische Leukämie (AML) gilt allgemein als Erkrankung des älteren Menschen. Bei jungen Leuten unter 20 Jahren hat sie eine Inzidenz von etwa 0,7 / 100.000 / Jahr (1). Wie bei den Erwachsenen wird zwischen primären und sekundären AML unterschieden. Die primären (oder «de novo») AML sind beim Kind deutlich häufiger und zeigen sich als akutes Krankheitsbild. Sekundäre AML entwickeln sich auf der Basis eines myelodysplastischen Syndroms (MDS) oder im Verlauf einer angeborenen (Fanconi Anämie, schwere kongenitale Neutropenie, Shwachman-Diamond Syndrom, Dyskeratosis congenita, u.a.) oder erworbenen aplastischen Anämie. Kinder mit Trisomie 21 haben ein 15-fach erhöhtes Risiko an Leukämie zu erkranken und entwickeln typischerweise ein transientes myeloproliferatives Syndrom als Neugeborene oder später das Vollbild einer AML, i.d.R. einer akuten megakaryozytischen Leukämie (AMKL-DS) (2).
Therapieinduzierte AML (t-AML) oder MDS (t-MDS) entstehen klassischerweise als Zweitmalignome einige Jahre nach Einsatz gewisser Zytostatika, die bekanntermassen kanzerogen wirken, insbesondere Anthrazykline und Epipodophyllotoxine (FAB M4 oder M5 Morphologie, typischerweise mit MLL-Rearrangement 11q23), Alkylantien (häufig mit Monosomie 7 oder Deletion 5q-) oder nach Radiotherapie.
Pathologie
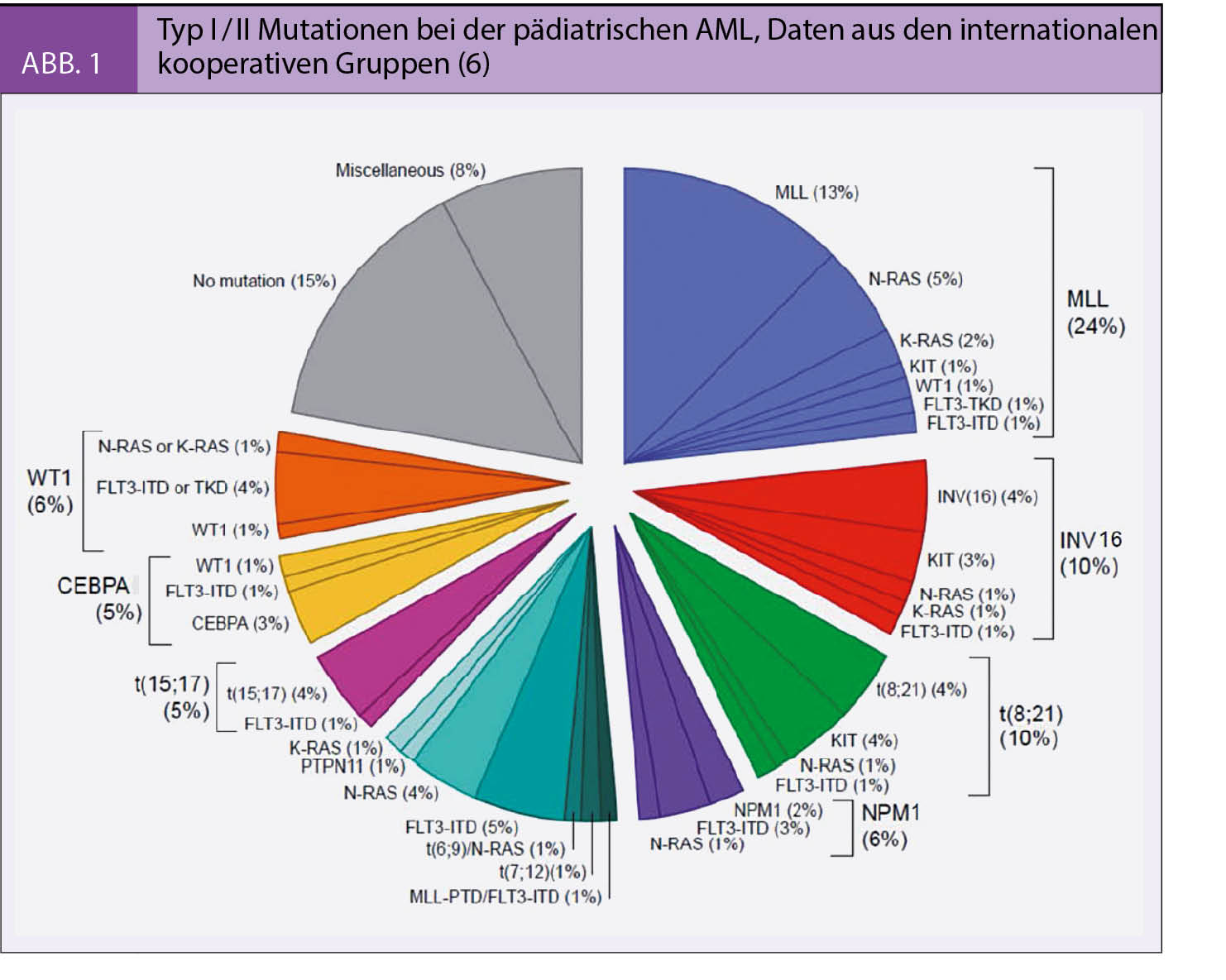
Bei der AML kommt es in frühen Progenitoren der Myelopoese oder in hämatopoetischen Stammzellen zur malignen Entartung. Zum Auftreten der offenen Leukämie sind wahrscheinlich mindestens zwei genetische Ereignisse in einem Zellklon erforderlich, die auch in grösserem zeitlichem Abstand auftreten können (3). Zudem spielen weitere Faktoren wie vulnerable Phasen der Immunentwicklung oder Veränderungen des Knochenmark-Mikroenvironments eine Rolle (4). Es gilt als gesichert, dass eine Differenzierungshierachie vorliegt, das heisst, dass sich der Grossteil der leukämischen Blasten aus der originären leukämischen Stammzelle (Stammzellklon) entwickelt (5). Bei der Mehrzahl der AML können leukämieassozierte zyto- und molekulargenetische Veränderungen nachgewiesen und entsprechend der Einteilung von Gilliland et al. als Typ I oder Typ II Mutationen (Abb. 1) eingeordnet werden, die entweder isoliert oder kombiniert nachweisbar sind (6). In einer umfassenden europäischen Kollaboration zur AML bei Kindern und Jugendlichen konnten die Häufigkeit und prognostische Relevanz dieser Mutationen belegt werden (7, 8, 9). Heutzutage sind diese Erkenntnisse ans Licht der Entwicklung molekular wirkender Substanzen, die als individualisierte, rationale Therapieoptionen infrage kommen, von besonderer Bedeutung.
Therapie
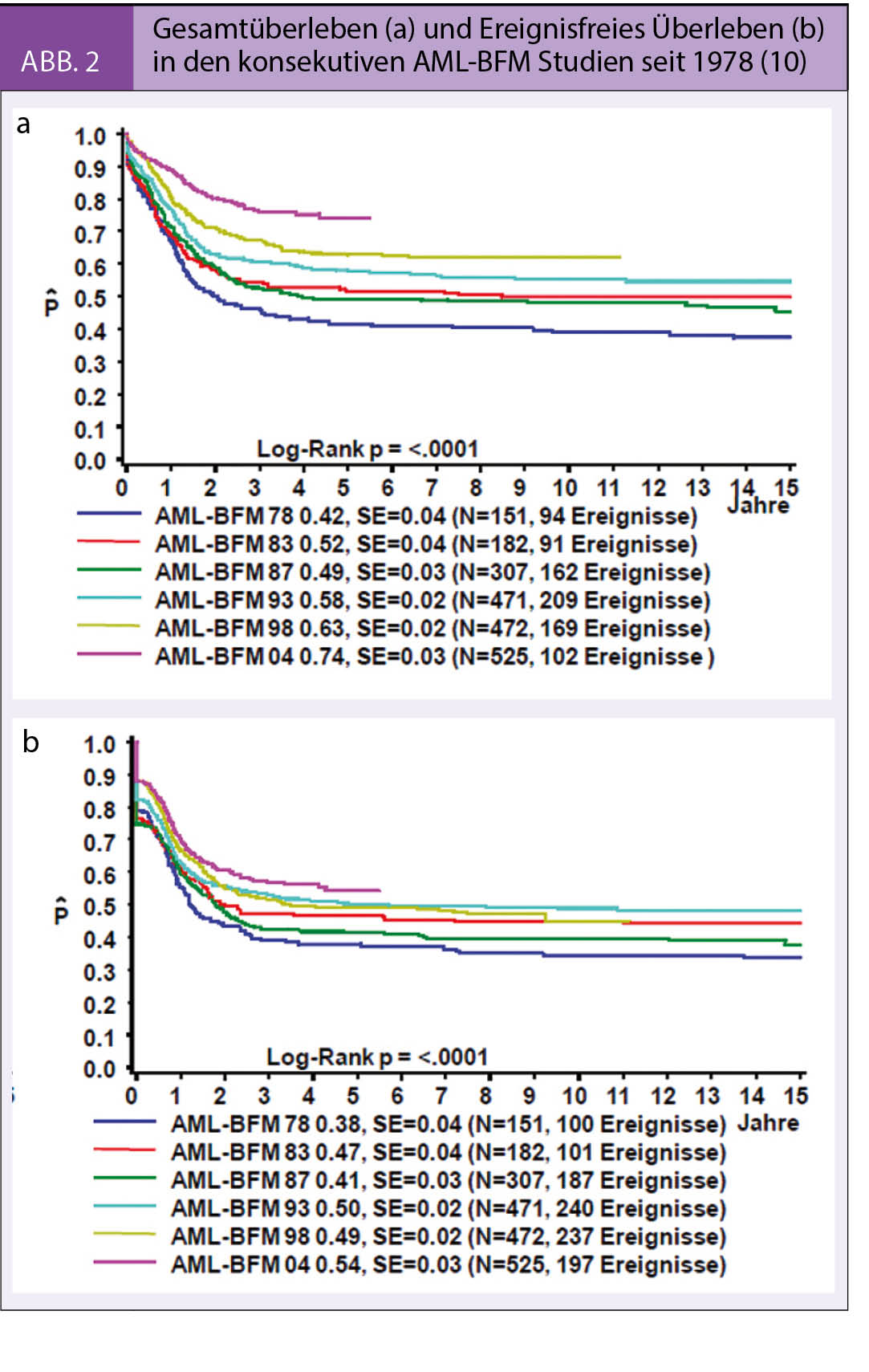
Die Prognose der pädiatrischen AML hat sich im Laufe der letzten 40 Jahre dank der Entwicklung und Umsetzung internationaler Therapieoptimierungsstudien (u. a. AML-BFM Gruppe in Europa, POG / COG in Amerika) kontinuierlich verbessert. Die 10-Jahres-Überlebensrate lag Ende der 70-er Jahre bei 42% und konnte bis heute auf 74% verbessert werden (Abb. 2) (10). Dieser Fortschritt wurde einerseits durch die Verbesserung der Kenntnisse der Molekularbiologie der AML mit anschliessender Verfeinerung der Therapiestratifizierung der Patienten, andererseits durch eine deutliche Intensivierung der Primärtherapie –insbesondere der Induktionstherapie – sowie durch verbesserte prophylaktische und supportive Massnahmen erreicht. Hinzu kommt eine konsequentere und verstärkte Behandlung der Rezidive (11). Weltweit wird aktuell die pädiatrische AML mit kurzen und intensiven Therapieblöcken behandelt, welche hauptsächlich aus Cytosinarabinosid (Ara-C) in verschiedenen Dosenintensitäten, Anthrazyklinen (Daunorubicin, liposomales Daunorubicin, Idarubicin und Mitoxantron) sowie Etoposid bestehen.
Wie in der ALL hat auch in der AML, neben den genetischen und biologischen Eigenschaften der Krankheit, das frühe Therapieansprechen einen sehr hohen prognostischen Stellenwert. Die Entwicklung hochsensitiver Methoden zur Erfassung der sogenannten minimal residuellen Erkrankung (minimal residual disease, MRD) und die entsprechende Anpassung der Therapieintensität, insbesondere früh in der Behandlung, haben somit eine zentrale Rolle in der Besserung der Resultate gespielt. Zur MRD-Bestimmung kommen prinzipiell methodisch die Morphologie, Immunphänotypisierung, Monitoring von genetischen Aberrationen (FISH) oder Fusionsgenen (qPCR), Monitoring ausgewählter Genexpressionen und die Nachverfolgung von klonspezifischen Mutationen (qPCR) infrage.
Die verschiedenen diagnostischen Methoden unterscheiden sich insbesondere hinsichtlich der Sensitivität (Morphologie <FISH < Immunphänotypisierung < qPCR) und Spezifität (Mutationen > Fusionsgene > FISH > Immunphänotypisierung > Morphologie) (12). Wichtige Aspekte der Anwendung dieser Technologien sind darüber hinaus die Zielsetzung und der Zeitpunkt innerhalb der einzelnen Therapiephasen. Für die Bestimmung des Therapieansprechens können prinzipiell alle Methoden herangezogen werden, da es in erster Linie auf die Kinetik ankommt. Für das spätere Monitoring, mit der Zielsetzung ein drohendes Rezidiv (molekulares Rezidiv) frühzeitig zu erkennen, kommen nur sehr spezifische und sensitive Methoden infrage.
Die zunehmenden Verbesserungen der diagnostischen Methoden (multi-color-Flowcytometry) und die Identifikation neuer Mutationen (FLT3-ITD/TDK, NPM1, c-kit, ras, CEBPΑ etc.) könnten die Aussagefähigkeit der MRD-Diagnostik entscheidend verbessern und eine noch geeignetere Therapiestratifizierung sowohl im Sinne der Therapiereduktion bei gutem Ansprechen als auch der Intensivierung bei ungünstigem Ansprechen ermöglichen (13).
Dieses bekommt einen umso höheren Stellenwert, als dass durch die Einführung einer Vielzahl neuer Inhibitoren/molekular wirksamer Substanzen, zusätzliche Werkzeuge zur Bestimmung des Therapieansprechens erforderlich werden.
Rolle der ZNS-Bestrahlung
Ein initialer ZNS-Befall der AML ist in ca. 5-15% der Kinder vorhanden. Verschiedene Studien haben gezeigt, dass eine intensive, wöchentliche intrathekale Therapie bis zur Klärung des Liquors genau so wirksam ist wie die ZNS-Bestrahlung. Dies gilt auch bei der Behandlung der meisten Chlorome. Auch die prophylaktische ZNS-Behandlung mit systemisch und intrathekal applizierter Chemotherapie hat sich gegen die Bestrahlung durchgesetzt. In der aktuellen AML-BFM 2012 Studie wird auf die prophylaktische ZNS-Bestrahlung verzichtet. Es erfolgen je nach Therapiearm 9 bzw. 11 intrathekale Tripletherapien (Prednison, Methotrexat und Cytarabin), ausser bei gleichzeitiger Applikation von intravenösem hochdosiertem Cytarabin, um unnötige Neurotoxizität zu vermeiden. Hier besteht die intrathekale Therapie aus einer Monotherapie mit Cytarabin.
Stellenwert der Erhaltungstherapie
International ist die Erhaltungstherapie umstritten. Aus Studien mit Erwachsenen ist bekannt, dass die Ergebnisse bei weniger intensiver Therapie mit Erhaltungstherapie für einen Teil der Patienten von Vorteil sind (14), jedoch bei Einsatz einer intensiven Anfangstherapie keinen Vorteil bringen (15).
Bei Kindern wurde in der französischen LAME-Studie 89/91 gezeigt, dass die Ergebnisse mit und ohne Erhaltungstherapie für das erkrankungsfreie Überleben (DFS) im gleichen Bereich liegen, während die Wahrscheinlichkeit des Gesamtüberlebens (OS) sogar besser war, wenn keine Erhaltungstherapie durchgeführt wurde – begründet durch eine höhere Salvage-Rate nach Rezidiven ohne vorherige Erhaltungstherapie (16). Ebenso zeigte die amerikanische CCG-Studie 213, dass die Erhaltungstherapie nach einer Intensivierung mit Hochdosis-Cytarabin nicht notwendig war. Andererseits spielte sie eine Rolle im Zweig mit Standardintensität in der Induktion (17). Aufgrund der Heterogenität der AML und der unterschiedlichen Proliferationskinetik ist es auch denkbar, dass nur für bestimmte Subtypen der AML eine Erhaltungstherapie von Vorteil sein kann.
Stellenwert der allogenen Stammzelltransplantation
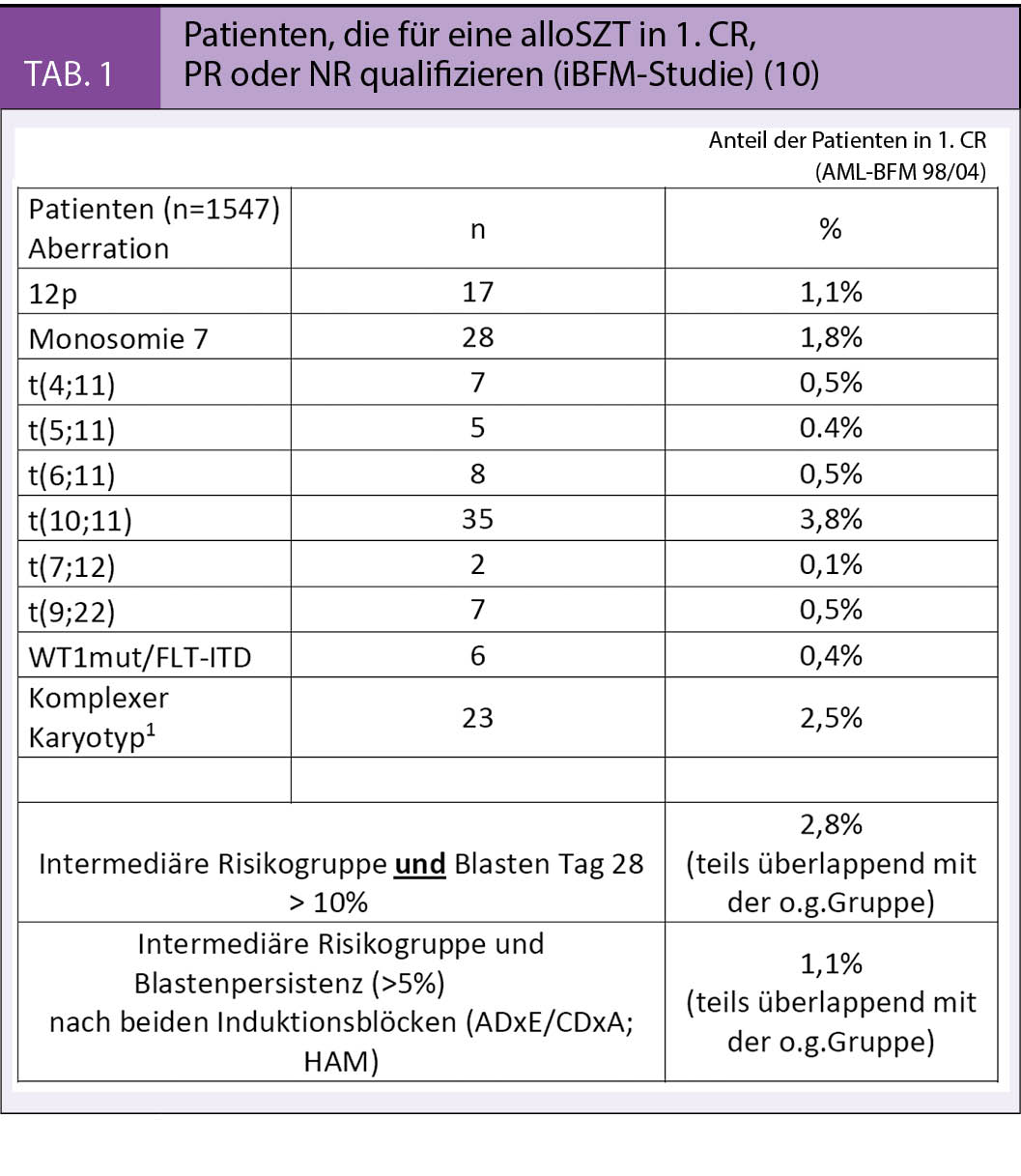
Der Stellenwert der allogenen Stammzelltransplantation (alloSZT) in erster kompletter Remission (1. CR) wird nach wie vor diskutiert. Die prospektive Analyse der alloSZT von einem Geschwisterspender für die Hochrisikogruppe der AML-BFM 98 Studie konnte keinen signifikanten Vorteil belegen. Dieses deckt sich mit den Ergebnissen der britischen MRC-Studie und der skandinavischen NOPHO Studie. Andererseits ergab eine Metaanalyse bei Kindern und Jugendlichen einen Vorteil der alloSZT in 1. CR (18). Die Aufarbeitung der verschiedenen Studien weist daraufhin, dass die Ergebnisse der alloSCT in den verschiedenen Gruppen ähnlich sind in Bezug auf das Rezidivrisiko, die Morbidität und das Überleben. Deutlichere Unterschiede treten eher beim Vergleich der jeweiligen Chemotherapiearme auf. Hinzu kommt, dass ein möglicher Vorteil der SZT auch in den unterschiedlichen Risikogruppen sehr unterschiedlich ist (19).
Die Tabelle 1 stellt die aktuellen Indikationen der iBFM AML Gruppe für eine alloSZT sowohl vom Geschwister- als auch vom passenden Fremdspender in der Frontline Therapie dar. Alle prognostisch ungünstigen molekular-biologischen Subtypen sind dabei vertreten, sowie die Patienten mit fehlendem Ansprechen auf die Induktionstherapie (sog. Non-Responders, ca. 4% der Hochrisikopatienten) für welche die alloSZT die einzige kurative Option bietet.
Unumstritten ist die SZT jedoch im Rezidiv Fall.
Neue Therapien
Trotz allen Fortschritten bleibt die Prognose der pädiatrischen AML deutlich unter jener der ALL. Patienten mit refraktärer AML (non-responders), diejenigen mit Rezidiv nach allogener Stammzelltransplantation aber auch einige Patienten mit bestimmten sehr Hoch-Risiko Mutationen haben alle noch sehr eingeschränkte Überlebenschancen. Deshalb sind neue, wenn möglich gezielte, Therapieeinsätze notwendig. Dank der oben erwähnten Fortschritte in der molekular-biologischen Charakterisierung der AML konnten eine Vielzahl neuer Mutationen (Typ I / II, Abb. 1) nachgewiesen werden, die sowohl bei der Leukä-mogenese als auch für Proliferation, Differenzierungsgrad oder Phänotyp relevant sind, aber auch therapeutisch genutzt werden können. Es wurden spezifische Substanzen (Tyrosin-Kinase Inhibitoren, «small molecules» etc.) entwickelt, die einzelne Signalkaskaden blockieren können. Trotz der meist guten Effektivität in vitro, ergaben die klinischen Studien als Monotherapien in der Regel eher begrenzte Behandlungserfolge. In Kombination mit konventionellen Chemotherapien ergeben sich allerdings zunehmend Hinweise, dass bei einem gezielten Einsatz dieser Substanzen, die Ansprechraten von AML-Patienten verbessert werden können.
Sorafenib (als Beispiel einer gezielten Therapie)
Sorafenib ist ein Arzneimittel aus der Gruppe der Tyrosin-Kinase-Inhibitoren (TKI) mit Wirkung auf multiple Tyrosinkinasen (multi-TKI) und hat somit mehrere Angriffspunkte: i) Inhibition der Rezeptor-Tyrosinkinasen (FLT-3,c-kit, VEGFR-2, VEGFR-3, PDGFR-b) mit Blockade der entsprechenden Signalkaskaden und reduzierter Tumor-Angiogenese; ii) Inhibition der Serin/Threonin-Kinasen (Raf-Kinasen: CRAF, BRAF, V600EBRAF) mit Hemmung der Raf-Signalkaskade. In beiden Fällen führt Sorafenib zu einer verminderten Zellteilung und Proliferation. Vorläufige Ergebnisse in Fallberichten und Studien bei Erwachsenen zeigen, dass eine gezielte Therapie mit Sorafenib bei AML mit FLT3-ITD eine vielversprechende Option anbieten könnte (20). Weitere experimentelle Daten zeigen, dass Inhibitoren wie Sorafenib, die die FLT3-ITD spezifischer hemmen als andere multi-TKI (Lestaurtinib[CEP701], Midostaurin [PKC412]), effektiver für FLT3-ITD/TDK positive AML sein können und weniger Nebenwirkungen zeigen (21).
Das gegenwärtige Therapieprotokoll der iBFM Gruppe (International Relapsed AML 2010/01) wird die Wirkmechanismen dieses TKI bei Patienten mit einer FLT3-ITD berücksichtigen. Durch die sequenzielle Gabe von Sorafenib (3 Tage Intervall zwischen Sorafenib und Chemotherapiestart) wird die gleichzeitige Applikation mit der Chemotherapie vermieden und somit das Risiko einer möglichen Wirkminderung der Chemotherapie durch Änderung des Zellzyklus reduziert. Zudem werden die mehrfach in vitro nachgewiesenen schnellen aber transienten Resistenzentwicklungen vermieden, die höchstwahrscheinlich nicht auf einen resistenten Klon zurückzuführen sind.
Der Autor hat keine Interessenskonflikte im Zusammenhang mit diesem Beitrag deklariert.
Die pädiatrische AML ist eine seltene, molekularbiologisch sehr
heterogene Erkrankung
Die Behandlung beruht auf kurze, intensive und sehr myelotoxische Chemotherapieblöcke
Wie bei der ALL ist eine Stratifizierung der Patienten auf der Basis der Molekularbiologie und des frühen Ansprechens unentbehrlich
Der Stellenwert der alloSZT in der initialen Therapie ist umstritten, für bestimmte Subtypen der AML aber sicher relevant
Neue Therapien, insbesondere TKI, könnten die düstere Prognose bestimmter Patientengruppen verbessern
Messages à retenir
La leucémie myéloïde aiguë pédiatrique est une maladie rare, moléculairement très hétérogène
Le traitement repose sur des blocs de chimiothérapie courts, intensifs et très myélotoxiques
Comme pour la LLA, la stratification des patients sur la base de la biologie moléculaire et de la réponse précoce est indispensable
Le rôle de la transplantation de cellules souches allogéniques en traitement initial est controversé, mais certainement pertinent pour certains sous-types de leucémie myéloïde aiguë
De nouvelles thérapies, en particulier les ITK, pourraient améliorer le pronostic sombre de certains groupes de patients
1. Kaatsch P, Spix C. Jahresbericht / Annual Report 2005. Book. 2008.
2. Zwaan MC, Reinhardt D, Hitzler J, Vyas P. Acute leukemias in children with Down syndrome. Pediatr Clin North Am. 2008;55:53-70,x.
3. Greaves M. Childhood leukaemia. BMJ. 2002;324:283-287.
4. Rubnitz JE, Gibson B, Smith FO. Acute myeloid leukemia. Pediatr Clin North Am. 2008;55:21-51, ix.
5. Appelbaum FR, Rowe JM, Radich J, Dick JE. Acute myeloid leukemia. Hematology (Am Soc Hematol Educ Program ). 2001;:62-86.
6. Lane SW, Gilliland DG. Leukemia stem cells. Semin Cancer Biol. 2009.
7. Goemans BF, Zwaan CM, Miller M, Zimmermann M, Harlow A, Meshinchi S, Loonen AH, Hahlen K, Reinhardt D, Creutzig U, Kaspers GJ, Heinrich MC. Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia.
8. Hollink IHIM, Zwaan CM, Zimmermann M, Arentsen-Peters TCJM, Pieters R, Cloos J, Kaspers GJL, de Graaf SSN, Harbott J, Creutzig U, Reinhardt D, Heuvel-Eibrink MM, Thiede C. Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia. 2009;23:262-270.
9. Zwaan CM, Meshinchi S, Radich JP, Veerman AJ, Huismans DR, Munske L, Podleschny M, Hahlen K, Pieters R, Zimmermann M, Reinhardt D, Harbott J, Creutzig U, Kaspers GJ, Griesinger F. FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood. 2003;102:2387-2394.
10. Creutzig U, Zimmermann M, Ritter J, Reinhardt D, Hermann J, Henze G, Jurgens H, Kabisch H, Reiter A, Riehm H, Gadner H, Schellong G. Treatment strategies and long-term results in paediatric patients treated in four consecutive AML-BFM trials. Leukemia. 2005;19:2030-2042.
11. Sander A, Zimmermann M, Dworzak M, Fleischhack G, von Neuhoff C, Reinhardt D, Kaspers GJ, Creutzig U. Consequent and intensified relapse therapy improved survival in pediatric AML: results of relapse treatment in 379 patients of three consecutive AML-BFM trials. Leukemia. 2010;24:1422-1428.
12. Reinhardt D, Langebrake C, Creutzig U, Vormoor J, Brune C, Thorwesten M, Ingiliz P, Hrusak O, Dworzak M, Griesinger F. Minimal residual disease in acute myeloid leukemia in children – standardization and evaluation of immunophenotyping in the AML-BFM-98 study. Klinische Padiatrie. 2002;214:179-187.
13. Kern W, Haferlach C, Haferlach T, Schnittger S. Monitoring of minimal residual disease in acute myeloid leukemia. Cancer. 2008;112:4-16.
14. Büchner T, Hiddemann W, Löffler H, Nowrousian MR, Maschmeyer G, Aul HC, Straif K, Hossfeld D, Heinecke A, for the AMLCG. Long-term results in adult AML: maintenance versus no maintenance, and double versus standard induction. Blood. 1990;74, Suppl. 1:105.
15. Mandelli F, Vegna ML, Avvisati G, et al. A randomized study of the efficacy of postconsolidation therapy in adult acute nonlymphocytic leukemia: a report of the Italian Cooperative Group GIMEMA. Ann Hematol. 1992;64:166-172.
16. Perel Y, Auvrignon A, Leblanc T, Vannier JP, Michel G, Nelken B, Gandemer V, Schmitt C, Lamagnere JP, de Lumley L, Bader Meunier B, Couillaud G, Schaison G, Landman-Parker J, Thuret I, Dalle JH, Baruchel A, Leverer, and for the Group LAME of the French Society of Pediatric Hematology and Immunology. Impact of addition of maintenance therapy to intensive induction and consolidation chemotherapy for childhood acute myeloblastic leukemia: results of a prospective randomized trial, LAME 89/91 [abstract]. J Clin Oncol. 2002;20:2774-2782.
17. Wells RJ, Woods WG, Lampkin BC, Nesbit ME, Lee JW, Buckley JD, Versteeg C, Hammond GD. Impact of high-dose cytarabine and asparaginase intensification on childhood acute myeloid leukemia: a report from the Children’s Cancer Group. J Clin Oncol. 1993;11:538-545.
18. Bleakley M, Lau L, Shaw PJ, Kaufman A. Bone marrow transplantation for paediatric AML in first remission: a systematic review and meta-analysis. Bone Marrow Transplant. 2002;29:843-852.
19. Horan J, Korones D. Intensive chemotherapy and bone marrow transplantation for children with acute myeloid leukemia. Blood. 2001;97:3672-3673.
20. Metzelder S, Wang Y, Wollmer E, Wanzel M, Teichler S, Chaturvedi A, Eilers M, Enghofer E, Neubauer A, Burchert A. Compassionate use of sorafenib in FLT3-ITD-positive acute myeloid leukemia: sustained regression before and after allogeneic stem cell transplantation. Blood. 2009;113:6567-6571.
21. Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, Grandin W, Lebwohl D, Wang Y, Cohen P, Fox EA, Neuberg D, Clark J, Gilliland DG, Griffin JD. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54-60.
MDS sind klonale Bluterkrankungen des älteren Menschen und werden vorwiegend bei Personen im Alter über 70 Jahre diagnostiziert. In europäischen Ländern beträgt die alters-standardisierte Inzidenzrate 2-3 pro 100’000 Patientenjahre mit einer zweifach höheren Inzidenz bei Männern als bei Frauen. Die einzige Ausnahme stellt dabei das MDS mit del(5q) dar, welche eine weibliche Prädominanz hat (1-4). Aufgrund der demographischen Alterung und der zunehmenden diagnostischen Möglichkeiten muss man in Zukunft davon ausgehen, dass die Entität der MDS zu einer der häufigsten hämatologischen Neoplasien aufsteigen wird, mit relevanter Auswirkungen auf die Gesundheitsversorgung (1).
Les MDS sont des maladies clonales du sang des personnes âgées qui sont principalement diagnostiquées chez les personnes de plus de 70 ans. Dans les pays européens, le taux d’incidence normalisé selon l’âge est de 2 à 3 pour 100 000 années-de patients, l’incidence étant deux fois plus élevée chez les hommes que chez les femmes. La seule exception est le MDS avec del (5q), qui a une prédominance féminine (1-4). En raison du vieillissement démographique et de l’augmentation des possibilités de diagnostic, le MDS devrait devenir l’une des néoplasies hématologiques les plus courantes à l’avenir, avec un impact important sur les soins de santé (1).
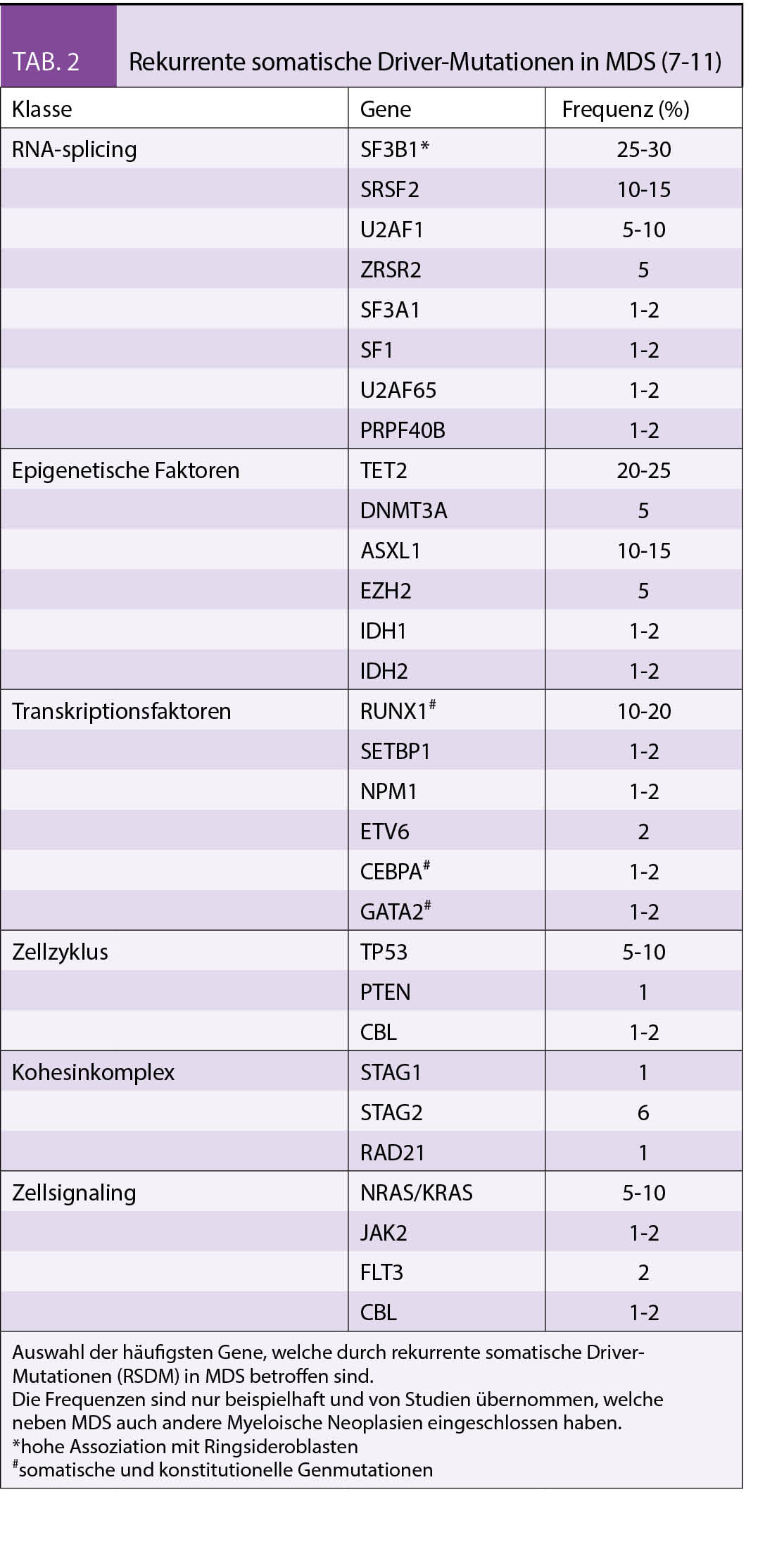
MDS sind heterogene Erkrankungen, die durch sequenzielle Ansammlung von genetischen Läsionen in den hämatopoetischen Stammzellen (HSC) verursacht werden (5). Die genetischen Läsionen, die bisher bei MDS identifiziert werden konnten, waren strukturelle Chromosomenaberrationen, die sich durch eine konventionelle Metaphasen-Zytogenetik oder Fluoreszenz-In-Situ Hybridisierung (FISH) nachweisen lassen. Die Analyseverfahren und dadurch auch das Verständnis genetischer Veränderungen in MDS und anderen myeloischen Neoplasien haben sich jedoch in den letzten Jahren rasant weiterentwickelt (6). Dank der Next Generation Sequencing (NGS) ist es nun möglich, rekurrente somatische Driver-Mutationen (RSDM) nachzuweisen. Diese RSDM treten in Genen mit folgenden Funktionen auf: RNA-Splicing, epigenetische Regulation, Transkriptionsfaktoren, Zellzyklus, Kohesinkomplex und Zellsignalling (7 - 11).
Diagnostik und Klassifizierung
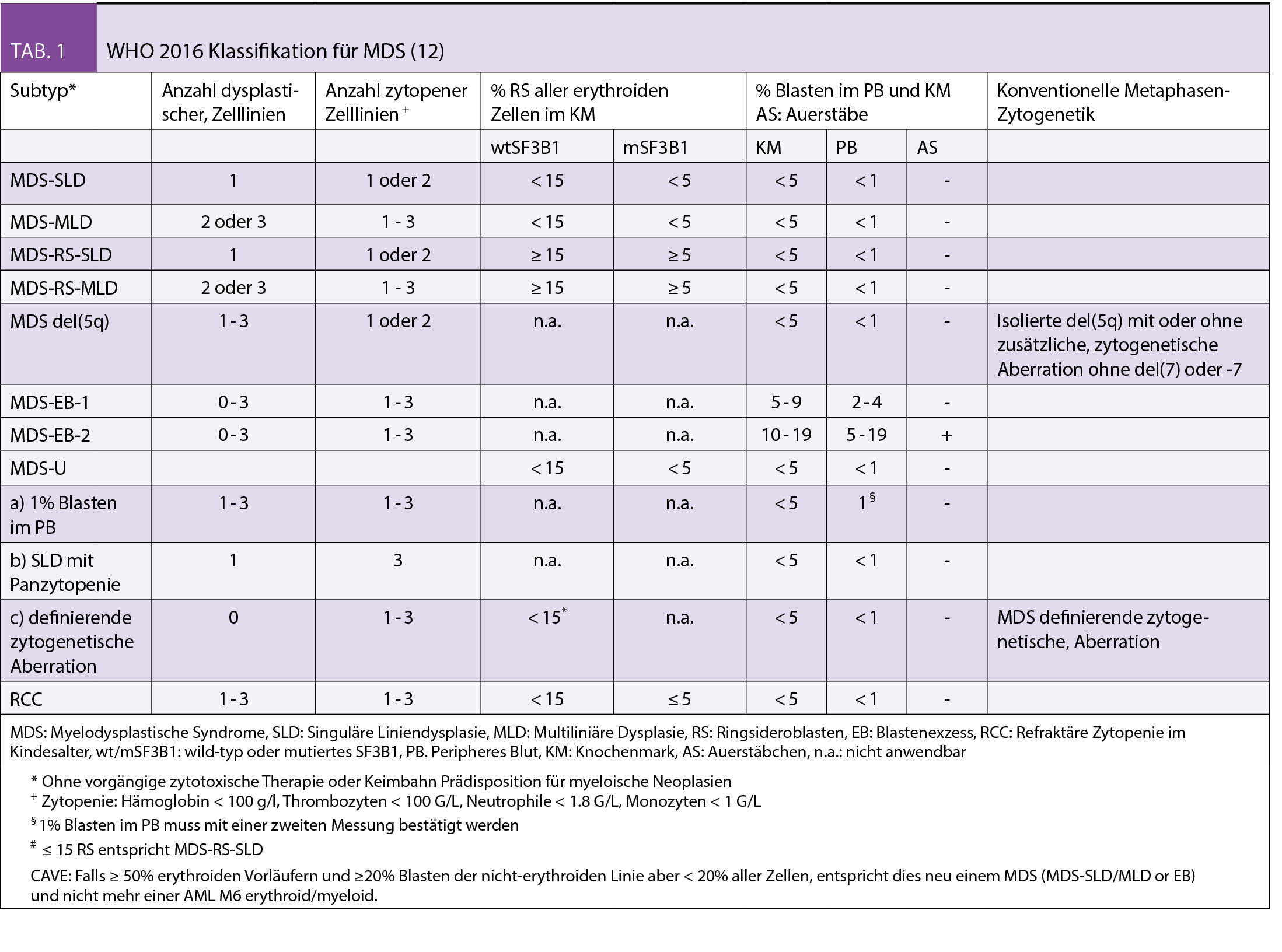
Die diagnostischen Empfehlungen bei erwachsenen Patienten mit vermutetem MDS wurden in den ELN-Empfehlungen 2013 zusammengefasst (7). Eine gründliche persönliche Anamnese (symptomatische Anämie, Infekte, Blutungen und insbesondere Expositionen gegenüber Chemo-/Radiotherapie, Pestizide, Insektizide und Lösungsmittel), Familienanamnese (Hinweise für hereditäre Prädisposition bei jüngeren MDS Patienten) sowie eine körperliche Untersuchung (Blutungszeichen, Organomegalien, Lymphadenopathien) stellen eine wichtige Grundlage dar. Zwingende Laboranalysen umfassen die morphologische Beurteilung des peripheren Blutes, des Knochenmarkaspirates/der Biopsie und eine zytogenetische Analyse. Zusätzlich empfohlene Untersuchungen umfassen die FISH (falls Zytogenetik nicht aussagekräftig) sowie die fluoreszenzaktivierte Zellsortierung (FACS). In speziellen Situationen werden zusätzlich auch molekulare Analysen (array-CGH, PCR und NGS) zum Nachweis von kryptischen Gendefekten und RSDMs empfohlen. Für die Klassifizierung der MDS werden die Anzahl von Zell-Linien, die von Zytopenie und Dysplasie betroffen sind, das Vorhandensein von Ringsideroblasten (RS) oder der Mutationen in SF3B1 (das mit RS assoziiert ist), Anzahl Blasten im peripheren Blut oder Knochenmark und das Vorliegen von speziellen MDS-definierenden zytogenetischen Anomalien (zB del 5q) berücksichtigt (Tab. 1) (12).
Zytopenien im Alter
Eine ungeklärte Anämie findet sich bei etwa 10 - 15% der Patienten im Alter von > 65 Jahren und es ist nicht immer einfach, reaktive von klonalen Zuständen abzugrenzen. Patienten mit einer Zytopenie, jedoch ohne ausreichende dysplastische Veränderungen oder MDS-definierende zytogenetische Veränderungen, werden als idiopathische Zytopenie unklarer Signifikanz (ICUS) bezeichnet (13). Bei diesen Patienten wird eine Verlaufskontrolle nach 3 - 6 Monaten und ggf. eine Wiederholung der Knochenmarksuntersuchung empfohlen (7). RSDM können mit einer altersabhängigen, erhöhten Häufigkeit bei älteren Patienten (10-20%) nachgewiesen werden. Diese Personen haben normale periphere Blutwerte oder nur eine leichte Zytopenie, welche aber die diagnostischen Kriterien für MDS nicht erfüllen. Diese Zustände werden als klonale Hämatopoiese mit indeterminiertem Potential (CHIP: normale periphere Blutwerte ohne RSDM) (14, 15) oder klonale Zytopenie unklarer Signifikanz (CCUS: Zytopenie mit RSDM) (16) bezeichnet. Die Anwendung der NGS ermöglicht neuerdings Patienten mit reaktiven und klonalen Zuständen zu unterschieden. Auf diese Weise lassen sich Patienten in frühen Stadien einer klonalen Hämatopoiese identifizieren, welche ein erhöhtes Risiko für die Entwicklung einer overten hämatologischen Neoplasie haben.
Allgemeine Überlegungen zum MDS Patientenmanagement
MDS sind sehr heterogene Erkrankungen mit einem sehr variablen natürlichen Verlauf von chronischen, asymptomatischen Zytopenien bis zu einem schnellen Fortschreiten in eine sekundäre AML. Zwei Drittel der Patienten mit MDS sterben an zytopenieassoziierten Komplikationen und ein Drittel erliegt der AML-Progression (17). Das Management basiert auf einer krankheits- und patientenassoziierten Risikostratifizierung. Eine exakte Diagnose und Risikostratifizierung sind für eine korrekte Behandlung daher entscheidend. Patienten mit niedrigem Risiko haben ein medianes Überleben von 3 bis 8 Jahren auf und sterben vorwiegend an zytopenieassoziierte Komplikationen (kardiovaskuläre Ereignisse, Infektionen und Blutungen). Die Ziele einer Behandlung von Patienten mit niedrigem Risiko liegen daher vor allem in der Verbesserung der Lebensqualität, Verringerung von zytopenieassoziierten Komplikationen und einem Hinauszögern eines Progresses in ein höhergradiges MDS (18, 19). Bei MDS Patienten mit hohem Risiko liegt hingegen die mediane Überlebenszeit nur bei 1 bis 3 Jahre und die AML-assoziierte Mortalität steht im Vordergrund. Bei diesen Patienten sollte die Behandlung darauf ausgerichtet sein, die Progression in eine AML hinauszuzögern und das Gesamtüberleben zu verbessern (20, 21).
Krankheitsassoziierte Risikofaktoren
Das Risiko für eine Progression in eine sekundäre AML und das Gesamtüberleben kann durch den International Prognostic Scoring System (IPSS) und den revidierten IPSS (IPSS-R) abgeschätzt werden) (22 - 24). Die Anzahl der Zelllinien, die von Zytopenien betroffen sind, wird in allen Prognosescoring-Systemen verwendet. Zudem sind Blastenzahl und Art der zytogenetischen Veränderungen relevant. Weniger als 5% der Blasten im Knochenmark, keine zirkulierenden Blasten im peripheren Blut, isolierte Anämie, Transfusionsunabhängigkeit und normaler Karyotyp oder günstige zytogenetische Veränderungen charakterisieren weniger fortgeschrittene MDS Formen. Im Gegensatz dazu wird ein fortgeschrittenes MDS durch > 5% Blasten im Knochenmark, Zytopenien mehrerer Zelllinien, komplexe zytogenetische oder andere ungünstige Veränderungen definiert.
Patientenassoziierte Risikofaktoren
Bei onkologischen Behandlungen älterer Patienten ist es stets wichtig, die Wirksamkeit und die Verträglichkeit einander gegenüber zu stellen. Karnofsky und Eastern Cooperative Oncology Group (ECOG) Scoring können verwendet werden, um die Performance zu bewerten. Diese sind jedoch altersbedingt und daher nicht ausreichend, um Komorbidität und Gebrechlichkeit zu beurteilen. Der Charlson Komorbiditätsindex wurde von Sorror für Patienten, die sich einer allogenen, hämatopoietischen Stammzelltransplantation (allo-HSCT) unterziehen, angepasst (25, 26) und wurde auch für MDS-Patienten validiert (HCT-CI) (27). Gebrechlichkeit und Funktionalität im täglichen Leben kann mit allgemeinen geriatrischen Bewertungsinstrumenten bewertet werden. Basierend auf einer steigenden Krebsinzidenz bei älteren Patienten sowie einer steigenden Anzahl von zielgerichteten Therapien, ist die Beurteilung patientenbezogener Faktoren ein zunehmendes Erfordernis für eine geeignete Behandlungszuteilung.
MDS-Patienten mit niedrigem Risiko
Watch-and-wait
Die Lebenserwartung von Patienten > 70 Jahre mit MDS-SLD oder MDS mit del(5q) ist nicht signifikant kürzer, als diejenige einer altersangepassten, älteren Bevölkerung (24). Patienten mit niedrig/intermediär-1 IPSS und asymptomatischen Zytopenien sollten daher nur regelmässig kontrolliert werden ohne Behandlung (7). Diese Empfehlung könnte sich in Zukunft jedoch gegebenenfalls ändern, da mit NGS in dieser Patientengruppe auch ungünstige Mutationen nachgewiesen werden können, welche von einer früheren Behandlung profitieren.
Supportive Massnahmen
Die supportiven Massnahmen umfassen Transfusionen, Infektionsprophylaxe, Antiemetika, Analgetika, Eisenchelation und Wachstumsfaktoren. Eine Transfusionsabhängigkeit ist im Allgemeinen mit einem fortgeschrittenen Krankheitsstadium und einer schlechteren Prognose assoziiert (24). Erythrozytenkonzentrate (EKs) werden in der Regel ab einem Hämoglobinspiegel < 80 g / l (oder auf höherem Niveau, falls symptomatisch) transfundiert. Thrombozytenkonzentrate sollten nur zurückhaltend zur Prophylaxe von Blutungen eingesetzt werden. Üblicherweise steigt das Risiko spontaner Blutungen bei Thrombozyten < 5 - 10 G / L oder < 20 G / L mit zusätzlichen Risikofaktoren wie Fieber oder Mukositis. Transfusionen sind in der Regel mit einem höheren Risiko für unerwünschte Ereignisse wie Alloimmunisierung und transfusionsassoziierte Komplikationen assoziiert (28). EKs müssen bei Patienten, die potentielle Kandidaten für eine allo-HSCT sind, bestrahlt werden, um das Risiko einer HLA-Alloimmunisierung und transfusionsassoziierten Graft-versus-Host-Erkrankung zu reduzieren.
Bei stark transfundierten Patienten (> 20 - 25 EKs) und/oder Ferritinwerten > 1000 mg / l kann eine Chelation eine Eisenüberladung vorbeugen, Zytopenien in ca. einem Drittel der Patienten verbessern und möglicherweise auch das Gesamtüberleben günstig beeinflussen (29 - 31). Weiterhin bleibt aber die Frage nicht sicher geklärt, welche Patienten cheliert werden sollen, da bei MDS Patienten (noch) keine randomisierten Studien vorliegen (32). Eine Eisenchelation wird im Allgemeinen mit Deferasirox Patienten angeboten, welche Kandidaten für eine allo-HSCT sind oder eine Lebenserwartung von > 1 Jahr haben.
Patienten mit niedrig/intermediär-1 IPSS, mit Hämoglobinwerten < 100 g / l, Serum-Erythropoietin-Werten < 200 - 500 U/L, EK transfusionsunabhängig oder mit < 2 EKs/Monat sind Kandidaten für eine Behandlung mit Erythropoietin Stimulierenden Agenzien (ESA) (29, 33, 34). Zwischen den verschiedenen ESA-Produkten (rekombinantes humanes Erythropoietin (rHuEPO) Alpha und Beta oder Darbepoietin Alpha) wurden keine relevanten Unterschiede festgestellt. Die erforderlichen EPO-Dosierungen sind höher als bei Patienten mit Niereninsuffizienz und man beginnt in der Regel mit 30 000 U / Woche rHuEPO sc (ca. 150 μg Darbepoietin alpha). Bei fehlendem Ansprechen wird die Dosis nach 6-8 Wochen verdoppelt. Der zusätzliche Einsatz von Granulozyten-Colony Stimulaing Factor (G-CSF 3 x 300 - 480 ug / Woche sc) bei Patienten, die nicht genügend auf ESA ansprechen, ist kontrovers und wird in der Schweiz selten eingesetzt (29, 35, 36). Die Einhaltung eines strikten ESA/G-CSF-Substitutionsregimes ist wichtig, um frühzeitig refraktäre Patienten zu identifizieren, die eine schlechtere Prognose haben und allfällige Kandidaten für weiterführende Behandlungen sein könnten.
Thrombopoietin-stimulierende Agenzien (TSA) (Romiplostim, Eltrombopag) wurden in klinischen Studien bei Patienten mit Thrombozytopenie und niedrig/intermediär-1 IPSS getestet. TSA zeigen eine positive Wirkung auf die Thrombozytenzahl und können Blutungen reduzieren haben aber keinen Einfluss auf das Gesamtüberleben (37, 38). Es ist wichtig zu beachten, dass die Behandlungen mit Wachstumsfaktoren bei MDS-Patienten grundsätzlich zugelassen, aber «off-limitatio» sind und daher eine Kostengutsprache von der zuständigen Krankenkasse notwendig ist.
Die immunmodulatorische und anti-angiogenetische Wirkung von Thalidomid wurde schon früher bei MDS Patienten eingesetzt, um den Transfusionsbedarf zu reduzieren (39). Aufgrund der neurologischen Nebenwirkung von Thalidomid, wurde ein 4-Amino-glutarimid Derivat, das Lenalidomid (Revlimid®) ohne diesen ungünstigen Nebeneffekt entwickelt. Mit Lenalidomid lässt sich eine anhaltende Transfusionsunabhängigkeit und zytogenetische Remission bei etwa der Hälfte aller MDS-Patienten mit niedrig / intermediär-1 IPSS und isoliert del(5q) erreichen (40). In einer Phase-3-Studie fand man zudem auch in ca. einem Viertel der Nicht-del(5q)-MDS-Patienten mit niedrig/intermediär-1 IPSS eine Transfusionsunabhängigkeit, während Mutationen in TP53 mit Resistenz und einer Krankheitsprogression assoziiert waren (41, 42) und daher bei fehlendem Ansprechen Abklärungen in Richtung einer Transplantation rechtfertigen. 10% der MDS-Patienten präsentieren sich mit hypoplastischem Knochenmark und sind potentielle Kandidaten für eine immunsuppressive Behandlung mit Antithymozytenglobulin (ATG) in Kombination mit Cyclosporin A (CyA) mit Ansprechraten von etwa einem Drittel (43). Eine Kombination CyA/ATG mit dem TSA Eltrombopag hat einen zusätzlichen Nutzen bei Patienten mit aplastischer Anämie gezeigt, ist aber für hypoplastische MDS Patienten nicht zugelassen (44).
MDS-Patienten mit höherem Risiko
Hypomethylierende Agenzien
MDS-Patienten mit Blastenexzess oder mit höherem Risiko, welche für eine intensive Chemotherapie und allo-HSCT nicht in Frage kommen, sind Kandidaten für eine palliative Behandlung mit hypomethylierende Agenzien (HMAs). Die Pyrimidin-Nukleosid-Analoga, 5-Azacytidin (AZA) und 5 - Aza - 2’ - desoxycytidin / Decitabine (DEC), wurden in Phase-3-Studien an MDS Patienten mit höherem Risiko untersucht (45,46). HMAs sind im Allgemeinen gut verträglich und zeigten signifikant höhere partielle und vollständige Remissionen im Vergleich zu best supportive care, einschliesslich Hydroxyurea und niedrigdosiertem Cytosin Arabinosid (AraC). HMAs bleiben jedoch der intensivierten Induktions-Chemotherapie gefolgt von allo-HSCT unterlegen, für welche jedoch nur eine Minderheit der älteren MDS Patienten in Frage kommt. MDS mit komplexem Karyotyp, sollten aufgrund der niedrigeren Raten vollständiger Remissionen und der höheren Toxizität mit intensiven Chemotherapien, bevorzugt mit HMA behandelt werden (47). Es gibt keine allgemein anerkannten prädiktiven molekularen Marker für das Ansprechen auf HMA und auch die Dauer der Behandlung bleibt unklar. Derzeit gibt es keine etablierte Behandlung nach Versagen von HMAs, diese Patienten sollten daher vorzugsweise auf klinische Studien behandelt werden.
Intensive Induktionschemotherapie
Die AML-basierte Induktions-Chemotherapie, gefolgt von einer allo-HSCT ist MDS Patienten mit höherem Risiko vorbehalten, die für eine intensive Therapie genügend fit sind. Jüngeres Alter, guter Leistungsstatus und günstige Zytogenetik sind unabhängige prognostische Faktoren, die mit einem besseren Überleben assoziiert sind (48). Patienten mit ungünstigen oder komplexen zytogenetischen Veränderungen sowie Mutationen oder Deletionen in TP53 haben ein schlechteres Ansprechen auf eine intensive Chemotherapie und können von einer Behandlung mit HMA mit oder ohne anschliessender allo-HSCT profitieren (47, 49). Die derzeitige Datenlage ist aber im Allgemeinen noch nicht ausreichend, um HMA für die Induktion vor allo-HSCT ausserhalb klinischer Studien zu empfehlen (8).
Die allo HSCT bleibt die einzige kurative Option, ist aber nur für MDS-Patienten geeignet, die genügend fit sind. Die Beurteilung der Komorbiditäten ist wichtig für die Entscheidungsfindung, welche Patienten für eine allo-HSCT in Frage kommen und der HCT-CI Score wird oft für diesen Zweck verwendet (26). Für eine allo HSCT kommen MDS Patienten mit intermediär-2/hoch IPSS in Frage. Das Alter ist der wichtigste prädiktive Faktor für das Gesamtüberleben. Eine retrospektive Analyse der Europäischen Gruppe für Blut- und Knochenmarktransplantation (EBMT) ergab eine behandlungsassoziierte Mortalität von 30% bei Patienten < 20 Jahre, 43% bei Patienten zwischen 20 und 40 Jahren und 50% bei Patienten> 50 Jahre. Reduktion der Intensität der Konditionierung und sorgfältige Auswahl der Patienten haben jedoch gezeigt, dass eine allo-HSCT auch bei Patienten zwischen 60 und 70 Jahren möglich ist. Für MDS-Patienten, die aufgrund von Komorbiditäten nicht für eine myeloablative Konditionierung qualifizieren, kann eine Konditionierung mit reduzierter Intensität in Betracht gezogen werden, vorzugsweise innerhalb klinischer Studien.
Zukunftsperspektiven
Die Entdeckung von RSDM bei Patienten mit myeloischen Neoplasien eröffnet eine ungeahnte Palette neuer diagnostischer und therapeutischer Möglichkeiten. Eine klonale Evolution kann damit bereits in frühen Stadien einer klonalen Hämatopoese indentifiziert werden und erlaubt möglicherweise auch die Identifikation von Patienten, welche von einer früheren Intervention profitieren können. Es konnte zum Beispiel gezeigt werden, dass Mutationen in TP53, EZH2, ETV6, RUNX1 und ASXL1 mit einer schlechten Prognose assoziiert sind und ein molekulares Scoring-System wird derzeit entwickelt (IPSS-R Mole) (50). Bei MDS-Patienten mit niedrigem Risiko und RS- oder SF3B1-Mutationen (MDS-RS-SLD / MLS) führt der TGF-beta ligand-trap Luspatercept in zwei Drittel der Patienten zu einem erythroiden Ansprechen und Transfusionsfreiheit. Luspatercept scheint die Erythropoese durch Mechanismen zu verbessern, die unabhängig von EPO sind. Basierend auf der Annahme, dass eine niedrig dosierte und längere Exposition mit HMAs zu einer Verbesserung der Differenzierung führen kann, werden zur Zeit orale AZA-Formulierungen in klinischen Studien an MDS-Patienten mit niedrigem Risiko getestet (51). Weitere Medikamente, die an MDS Patienten untersucht werden, umfassen Toll-like-Rezeptor-2-Antikörper, CD95-Ligand(FAS-Ligand)-Hemmer, Multikinase-Inhibitoren (z. B. Rigosertib), Checkpoint-Inhibitoren (z. B. Nivolumab, Durvalumab), Telomerase-Inhibitoren (z. B. Imetelstat) und Inhibitoren von mutiertem IDH1 / 2 (zB AG - 120 / AG-221).
Darüber hinaus ist die Verbesserung der Behandlungsallokation basierend auf Wirksamkeit, Verträglichkeit, Nutzen und Richtlinien-Konformität ein aktives Forschungsgebiet der Versorgungsforschung. Daher ist der Einschluss von MDS-Patienten in longitudinale Kohortenstudien sehr zu begrüssen, so wie es die SAKK 33/18 («I-CARE for MDS») Studie verfolgen wird.
Weiter werden longitudinale Kohorten und Biobanking von biologischem Material von Patienten mit MDS oder frühen Formen einer klonalen Hämatopoese (CCUS) sehr nützlich sein, um detaillierte, gesundheitsbezogene Daten mit Biomarkern zu verknüpfen. Diese Plattform steht seit 2016 mit dem Swiss MDS Regsitry/Biobank zur Verfügung und soll in Zukunft helfen, unser Verständnis der MDS Biologie zu verbessern, um damit auch im Sinne einer «Präzisionsmedizin» die Prognose und das Therapieansprechen der Patienten besser abschätzen zu können.
Zusammenfassung
Die Myelodysplastischen Syndrome (MDS) bilden eine heterogene Gruppe von klonalen Erkrankungen des Blutes mit einer zunehmenden Inzidenz in der älteren Bevölkerung. Aufgrund der demographischen Alterung unserer Gesellschaft werden MDS eine wachsende Bedeutung in unserem Gesundheitswesen haben. MDS werden durch Genmutationen in den hämatopoetischen Stammzellen verursacht und sind gekennzeichnet durch eine ineffektive Hämatopoiese mit Zytopenien und Dysplasien sowie einer Neigung zur Progression in eine sekundäre akute myeloische Leukämie (sAML). Eine exakte Diagnose und Risikostratifizierung sind für eine korrekte Behandlung entscheidend. Patienten mit niedrigem Risiko haben ein medianes Überleben von 3 bis 8 Jahren und sterben vorwiegend an zytopenieassoziierten Komplikationen (kardiovaskuläre Ereignisse, Infektionen und Blutungen). Die Ziele einer Behandlung von Patienten mit niedrigem Risiko liegen daher vor allem in der Verbesserung der Lebensqualität, Verringerung von zytopenieassoziierten Komplikationen und einem Hinauszögern eines Progresses in ein höhergradiges MDS. Bei MDS Patienten mit hohem Risiko liegt hingegen die mediane Überlebenszeit nur bei 1 bis 3 Jahren und die AML-assoziierte Mortalität steht im Vordergrund. Bei diesen Patienten sollte die Behandlung darauf ausgerichtet sein, die Progression in eine AML hinauszuzögern und das Gesamtüberleben zu verbessern. Die allogene hämatopoetische Stammzelltransplantation bleibt die einzige kurative Option für Patienten mit hohem Risiko. Jedoch ist nur eine Minderheit der meistens älteren und polymorbiden MDS Patienten geeignet für eine solche intensive Behandlung. Daher werden die meisten Patienten mit supportiven Massnahmen und palliativen Behandlungen, wie zum Beispiel Wachstumsfaktoren, Immunmodulatoren und hypomethylierenden Agenzien behandelt. Da ältere Patienten mit chronischen Zytopenien häufig in der allgemein internistischen Praxis gesehen werden, ist die Kenntnis über mögliche Präsentationsformen und angemessene Behandlungsoptionen wichtig für alle Ärzte aus der Grundversorgung.
PD Dr. med. Nicolas Bonadies
Leitender Arzt und Koordinator Swiss MDS Study Group
Universitätsklinik für Hämatologie und Hämatologisches Zentrallabor
Inselspital Bern
Freiburgstrasse 18
3010 Bern
Nicolas.Bonadies@insel.ch
Der Autor hat deklariert, keine Interessenskonflikte in Zusammenhang mit diesem Artikel zu haben.
Die Myelodysplastischen Syndrome (MDS) bilden eine heterogene Gruppe von klonalen Erkrankungen des Blutes mit einer zunehmenden Inzidenz in der älteren Bevölkerung
MDS werden durch Genmutationen in den hämatopoetischen Stammzellen verursacht und sind gekennzeichnet durch eine ineffektive Hämatopoiese mit Zytopenien und Dysplasien sowie einer Neigung zur Progression in eine akute myeloische Leukämie (AML).
Eine exakte Diagnose und Risikostratifizierung sind für eine korrekte Behandlung entscheidend
Patienten mit niedrigem Risiko haben ein medianes Überleben von 3 bis 8 Jahren und sterben vorwiegend an zytopenieassoziierten Komplikationen (kardiovaskuläre Ereignisse, Infektionen und Blutungen).
Die Ziele einer Behandlung von Patienten mit niedrigem Risiko liegen daher vor allem in der Verbesserung der Lebensqualität, Verringerung von Zytopenie-assoziierten Komplikationen und einem Hinauszögern eines Progresses in ein höhergradiges MDS.
Bei MDS Patienten mit hohem Risiko liegt hingegen die mediane Überlebenszeit nur bei 1 bis 3 Jahren und die AML-assoziierte Mortalität steht im Vordergrund. Bei diesen Patienten sollte die Behandlung darauf ausgerichtet sein, die Progression in eine AML hinauszuzögern und das Gesamtüberleben zu verbessern.
Die allogene hämatopoetische Stammzelltransplantation bleibt die einzige kurative Option für Patienten mit hohem Risiko. Jedoch ist nur eine Minderheit der meistens älteren und polymorbiden MDS Patienten geeignet für eine solche intensive Behandlung
Take-Home Message
Les syndromes myélodysplasiques (MDS) sont un groupe hétérogène de maladies clonales du sang avec une incidence croissante chez les personnes âgées.
Les MDS sont causés par des mutations génétiques dans les cellules souches hématopoïétiques et se caractérisent par une hématopoïèse inefficace avec une cytopénie et une dysplasie et une tendance à la progression vers la leucémie myéloïde aiguë (LMA).
Le diagnostic exact et la stratification des risques sont cruciaux pour un traitement correct.
Les patients à faible risque ont une survie médiane de 3 à 8 ans et meurent principalement de complications associées à la cytopénie (événements cardiovasculaires, infections et saignements).
Les principaux objectifs du traitement des patients à faible risque sont donc d’améliorer la qualité de vie, de réduire les complications associées à la cytopénie et de retarder l’évolution vers un MDS de grade supérieur.
Chez les patients atteints de MDS à risque élevé, cependant, le temps de survie médian n’est que de 1 à 3 ans et la mortalité associée à la LMA est au premier plan. Chez ces patients, le traitement doit être conçu de manière à retarder la progression vers la LMA et à améliorer la survie globale.
La greffe de cellules souches hématopoïétiques allogéniques demeure la seule option curative pour les patients à haut risque. Cependant, seule une minorité des patients atteints de MDS généralement âgés et polymorbides convient à ce type de traitement intensif.
1. Bonadies N, Feller A, Rovo A, et al. Trends of classification, incidence, mortality, and survival of MDS patients in Switzerland between 2001 and 2012. Cancer Epidemiol 2017;46:85-92.
2. Dinmohamed AG, Visser O, van Norden Y, et al. Trends in incidence, initial treatment and survival of myelodysplastic syndromes: a population-based study of 5144 patients diagnosed in the Netherlands from 2001 to 2010. Eur J Cancer 2014;50:1004-12.
3. Roman E, Smith A, Appleton S, et al. Myeloid malignancies in the real-world: Occurrence, progression and survival in the UK’s population-based Haematological Malignancy Research Network 2004-15. Cancer Epidemiol 2016;42:186-98.
4. Neukirchen J, Schoonen WM, Strupp C, et al. Incidence and prevalence of myelodysplastic syndromes: data from the Dusseldorf MDS-registry. Leuk Res 2011;35:1591-6.
5. Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JC, Schimmer AD. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer 2007;7:118-29.
6. Bejar R, Levine R, Ebert BL. Unraveling the molecular pathophysiology of myelodysplastic syndromes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29:504-15.
7. Malcovati L, Hellstrom-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood 2013;122:2943-64.
8. Abdel-Wahab O, Figueroa ME. Interpreting new molecular genetics in myelodysplastic syndromes. Hematology American Society of Hematology Education Program 2012;2012:56-64.
9. Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011;364:2496-506.
10. Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011;478:64-9.
11. Kon A, Shih LY, Minamino M, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 2013;45:1232-7.
12. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391-405.
13. Valent P, Horny HP. Minimal diagnostic criteria for myelodysplastic syndromes and separation from ICUS and IDUS: update and open questions. Eur J Clin Invest 2009;39:548-53.
14. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477-87.
15. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488-98.
16. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9-16.
17. Rollison DE, Howlader N, Smith MT, et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001-2004, using data from the NAACCR and SEER programs. Blood 2008;112:45-52.
18. Santini V. Treatment of low-risk myelodysplastic syndromes. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program 2016;2016:462-9.
19. Fenaux P, Ades L. How we treat lower-risk myelodysplastic syndromes. Blood 2013;121:4280-6.
20. Komrokji RS. Current State of the Art: Management of Higher Risk Myelodysplastic Syndromes. Clinical lymphoma, myeloma & leukemia 2016;16 Suppl:S39-43.
21. Garcia-Manero G. Myelodysplastic syndromes: 2015 Update on diagnosis, risk-stratification and management. American journal of hematology 2015;90:831-41.
22. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997;89:2079-88.
23. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012;120:2454-65.
24. Malcovati L, Germing U, Kuendgen A, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2007;25:3503-10.
25. Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis 1987;40:373-83.
26. Sorror ML, Maris MB, Storb R, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood 2005;106:2912-9.
27. Sorror ML, Storb RF, Sandmaier BM, et al. Comorbidity-age index: a clinical measure of biologic age before allogeneic hematopoietic cell transplantation. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2014;32:3249-56.
28. Szczepiorkowski ZM, Dunbar NM. Transfusion guidelines: when to transfuse. Hematology American Society of Hematology Education Program 2013;2013:638-44.
29. Malcovati L, Hellstrom-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood 2013;122:2943-64.
30. Gattermann N, Finelli C, Della Porta M, et al. Hematologic responses to deferasirox therapy in transfusion-dependent patients with myelodysplastic syndromes. Haematologica 2012;97:1364-71.
31. List AF, Baer MR, Steensma DP, et al. Deferasirox reduces serum ferritin and labile plasma iron in RBC transfusion-dependent patients with myelodysplastic syndrome. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2012;30:2134-9.
32. Mitchell M, Gore SD, Zeidan AM. Iron chelation therapy in myelodysplastic syndromes: where do we stand? Expert Rev Hematol 2013;6:397-410.
33. Park S, Grabar S, Kelaidi C, et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood 2008;111:574-82.
34. Hellstrom-Lindberg E, Negrin R, Stein R, et al. Erythroid response to treatment with G-CSF plus erythropoietin for the anaemia of patients with myelodysplastic syndromes: proposal for a predictive model. British journal of haematology 1997;99:344-51.
35. Balleari E, Rossi E, Clavio M, et al. Erythropoietin plus granulocyte colony-stimulating factor is better than erythropoietin alone to treat anemia in low-risk myelodysplastic syndromes: results from a randomized single-centre study. Ann Hematol 2006;85:174-80.
36. Casadevall N, Durieux P, Dubois S, et al. Health, economic, and quality-of-life effects of erythropoietin and granulocyte colony-stimulating factor for the treatment of myelodysplastic syndromes: a randomized, controlled trial. Blood 2004;104:321-7.
37. Kantarjian H, Fenaux P, Sekeres MA, et al. Safety and efficacy of romiplostim in patients with lower-risk myelodysplastic syndrome and thrombocytopenia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2010;28:437-44.
38. Mavroudi I, Pyrovolaki K, Pavlaki K, et al. Effect of the nonpeptide thrombopoietin receptor agonist eltrombopag on megakaryopoiesis of patients with lower risk myelodysplastic syndrome. Leuk Res 2011;35:323-8.
39. Raza A, Meyer P, Dutt D, et al. Thalidomide produces transfusion independence in long-standing refractory anemias of patients with myelodysplastic syndromes. Blood 2001;98:958-65.
40. Fenaux P, Giagounidis A, Selleslag D, et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood 2011;118:3765-76.
41. Santini V, Almeida A, Giagounidis A, et al. Randomized Phase III Study of Lenalidomide Versus Placebo in RBC Transfusion-Dependent Patients With Lower-Risk Non-del(5q) Myelodysplastic Syndromes and Ineligible for or Refractory to Erythropoiesis-Stimulating Agents. J Clin Oncol 2016;34:2988-96.
42. Jadersten M, Saft L, Smith A, et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29:1971-9.
43. Passweg JR, Giagounidis AA, Simcock M, et al. Immunosuppressive therapy for patients with myelodysplastic syndrome: a prospective randomized multicenter phase III trial comparing antithymocyte globulin plus cyclosporine with best supportive care–SAKK 33/99. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29:303-9.
44. Olnes MJ, Scheinberg P, Calvo KR, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med 2012;367:11-9.
45. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009;10:223-32.
46. Lubbert M, Suciu S, Baila L, et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29:1987-96.
47. Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015;126:291-9.
48. Ossenkoppele GJ, Graveland WJ, Sonneveld P, et al. The value of fludarabine in addition to ARA-C and G-CSF in the treatment of patients with high-risk myelodysplastic syndromes and AML in elderly patients. Blood 2004;103:2908-13.
49. Knipp S, Hildebrand B, Kundgen A, et al. Intensive chemotherapy is not recommended for patients aged >60 years who have myelodysplastic syndromes or acute myeloid leukemia with high-risk karyotypes. Cancer 2007;110:345-52.
50. Nazha A, Narkhede M, Radivoyevitch T, et al. Incorporation of molecular data into the Revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia 2016.
51. Saunthararajah Y. Key clinical observations after 5-azacytidine and decitabine treatment of myelodysplastic syndromes suggest practical solutions for better outcomes. Hematology American Society of Hematology Education Program 2013;2013:511-21.
Der Begriff Immuntherapie wird heutzutage sehr weit gefasst und beinhaltet viele verschiedene Ansätze. Fokussiert man sich auf Antikörper-basierte Konzepte, so besitzen monoklonale Antikörper bereits einen festen Platz in der Myelombehandlung und Antikörpervarianten wie Immuntoxine, bispezifische Antikörper oder auch Antikörper-beladene T-Zellen befinden sich in der klinischen Erprobung bzw. Zulassung. Allen Ansätzen gemeinsam ist eine möglichst optimale Aktivierung des patienteneigenen Immunsystems gegen Myelomzellen. Damit könnte das Multiple (MM) Myelom in Zukunft eine heilbare Erkrankung werden.
Le terme immunothérapie est aujourd’hui compris dans un sens très large et comprend de nombreuses approches différentes. Si l’accent est mis sur les concepts basés sur les anticorps, les anticorps monoclonaux ont déjà une place ferme dans le traitement du myélome et les variantes d’anticorps telles que les immunotoxines, les anticorps bispécifiques ou les cellules T chargées d’anticorps sont en cours d’essai clinique ou d’approbation. Toutes les approches ont en commun la meilleure activation possible du système immunitaire du patient contre les cellules myélomateuses. Cela pourrait faire du myélome multiple (MM) une maladie guérissable à l’avenir
Monoklonale Antikörper
Die passive Immuntherapie mittels monoklonaler Antikörper (Mab) hat sich in der Lymphomtherapie schon seit fast 20 Jahren und in der Myelomtherapie nun in den letzten Jahren erfolgreich etabliert. Dabei werden in der Myelom-Behandlung vornehmlich Antikörper mit Spezifität für das CS1 Antigen (z.B. Elotuzumab) bzw. das CD38 Antigen (z.B. Daratumumab) verwendet. Diese monoklonalen Antikörper erkennen spezifisch ihr Zielantigen und lösen über eine Komplementaktivierung (CDC) oder Rekrutierung zytotoxischer Zellen (ADCC) eine Immunantwort aus. MAb können als Monotherapie oder in Kombination mit bereits etablierten Therapeutika eingesetzt werden. Den grössten Stellenwert besitzen zur Zeit CD38 spezifische MAb wie Daratumumab, da sie sowohl als Monotherapie als auch in Kombinationstherapien eine hohe Aktivität gegenüber Plasmazellen aufweisen.
Therapie mit dem CD38 spezifischen Antikörper Daratumumab
Daratumumab ist ein humaner CD38-IgG1κ Antikörper, der an zwei β-stranghaltige Aminosäuren 233-246 und 267-280 des CD38 Antigens bindet. Seine Wirkung entfaltet er u.a. über den Fc Anteil des Antikörpers mit Komplementaktivierung im Sinne einer effizienten CDC als auch einer ADCC. Auf Grundlage verschiedener Studien wurde die optimale Dosierung von 16 mg/kg KG mit einer zunächst wöchentlichen (8 Gaben), dann zweiwöchentlichen (8 Gaben) und anschliessend monatlichen Applikation als Standard definiert. Die Gesamtansprechrate (RR) für Patienten mit weit fortgeschrittenem bzw. refraktärem MM beträgt ca. 31% mit einem medianen OS von 20 Monaten (1). Die Daratumumab Monotherapie ist in der Schweiz zugelassen und kassenpflichtig.
Kombinationstherapien Daratumumab plus Velcade/Dexamethason bzw. Revlimid/Dexamethason
Es gibt aktuell 2 grosse, randomisierte Phase III Studien, die Daratumumab (D) + Revlimid/Dexamethason (Rd) bzw. Velcade/Dexamethason (Vd) bei MM Patienten ab der 2. Therapielinie getestet haben. In der CASTOR-Studie (DVd vs. Vd) mit 498 Patienten, die im Median zwei Therapielinien durchlaufen hatten, wurde in beiden Armen nach 8 Zyklen die Therapie mit Vd gestoppt und Daratumumab im experimentellen Arm bis zur Progression weitergegeben. Nach einem medianen Follow-Up von 7,4 Monaten war das mediane PFS im Daratumumab-Arm deutlich länger als im Kontrollarm (nicht erreicht vs. 7,16 Monate, HR 0,39, p<0,0001) (2). Die zusätzliche Gabe von Daratumumab führte zu einer Verdopplung an qualitativ sehr guten Remissionen (VGPR oder CR). In der kürzlich präsentierten Aktualisierung (medianes Follow-up 19 Monate) betrug das PFS für alle Patienten 17 vs. 7 Monate. Auch die Kombination DRd führte im Vergleich zu Rd (POLLUX-Studie) zu einer signifikanten Verlängerung des medianen PFS (nicht erreicht vs. 18 Monate, HR 0,37, p<0,0001) (3). In beiden Studien traten Infusionsreaktionen fast ausschliesslich (>90%) im ersten Behandlungszyklus auf und nur sehr wenige waren Grad 3 (5-10%) Reaktionen, Grad 4 Reaktionen wurden nicht beobachtet. Auch profitierten in beiden Studien alle vordefinierten Subgruppen von der Daratumumab Therapie. Besonders hervorzuheben ist die hohe Rate an MRD Negativität (für DRd im Standardrisikoarm bis 30% und im Hochrisikoarm bis 21%). Diese tiefe Krankheitskontrolle könnte eventuell einen ersten Schritt Richtung Krankheitselimination andeuten.
Monokolonale Antikörper sind in der Regel passive Immuntherapeutika, d.h. ihre Aktivität ist nicht langfristig im Organismus nach Applikation vorhanden und sobald der Wirkspiegel im Blut unterhalb der jeweils erforderlichen Mindestkonzentration fällt (in der Regel nach 2-3 Halbwertszeiten), ist auch kein Effekt mehr vorhanden. Zur Steigerung der Mab Aktivität kann dieser z.B. an ein Zellgift gekoppelt werden (sog. Immuntoxin). Damit wird der Antikörper als Trägermolekül verwendet und führt ein in der Regel hochpotentes Zellgift direkt an die gewünschte Zielzelle.
Immuntoxine
Immuntoxine (IT) machen sich zwei Eigenschaften von Antikörpern zunutze: die Spezifität für das Zielmolekül und die Fähigkeit, nach Bindung an der Zelloberfläche in die Zelle zu internalisieren und dort das gekoppelte Toxin frei zu setzen (sog. Antibody-Drug-Conjugate(ADC) (Abb.1). Prototyp ist Brentuximab Vedotin (BV) als Konjugat eines CD30-spezifischen Antikörpers mit Monomethylauristatin E, das die mitotische Spindel zerstört und damit Apoptose in Lymphomzelle auslöst. Ähnliche Ansätze werden auch in der Myelombehandlung verfolgt und am erfolgversprechendsten sind Konstrukte mit Spezifität für das B cell maturation antigen (BCMA). BCMA wird in späten Stadien der B Zelldifferenzierung membranständig exprimiert und ist für das Überleben langlebiger Plasmazellen erforderlich. Monomethyl-Auristatin-F gekoppelt an einen BCMA spezifischen Antikörper wird nach Bindung an der Myelomzelle rasch internalisiert und setzt sein aktives Toxin frei. In einer offenen Phase-I Studie bei Patienten mit rezidiviertem / refraktärem MM (rrMM) wurde das IT alle 3 Wochen intravenös bei guter Verträglichkeit infundiert. Bei keinem der 24 MM-Patienten gab es unerwünschte Ereignisse, die zum Abbruch der Behandlung führten. Bei vier Patienten war eine Dosisreduktion aufgrund von Nebenwirkungen des Toxins erforderlich: okuläre Toxizität (n = 1), Hornhauterkrankung / okuläre Toxizität (n = 1), trockene Augen (n = 1) und Keratitis (n = 1). Es wurden jedoch keine DLTs berichtet und damit kann auf eine insgesamt akzeptable Verträglichkeit geschlossen werden (4). Sehr rudimentäre Daten existieren bzgl. der Wirksamkeit: Bei 0,24 mg / kg 1 MR und 1 VGPR, bei Dosen ≥ 0,96 mg / kg 3 PR und 1 MR. Damit ergibt sich eine sog. clinical benefit rate (CBR) von 25% und es müssen sicherlich noch grössere Studien durchgeführt werden, um den klinischen Benefit eindeutig zu belegen.
Immunmodulierende Antikörper
Dieser recht neue Immuntherapieansatz verwendet zumeist auch Mab (Abb. 1). Diese erkennen aber nicht zwangsläufig ein Tumor-antigen, sondern schalten das Immunsystem im Sinne einer aktiven Immunisierung an und können zudem durch die Etablierung sog. Gedächtniszellen eine langfristige Immunität – ähnlich einer Impfung – erzielen. Paradebeispiel sind Checkpoint Inhibitoren (CPI) in der Behandlung solider Tumoren, vornehmlich CTLA4 oder PD1/PDL-1 blockierende MAbs. Deren Einsatz hat sich beim Multiplen Myelom (MM) insbesondere aufgrund einer recht hohen (pulmonalen) Toxizität bisher nicht durchgesetzt. Erfolgsversprechend sind derzeit jedoch zwei neue Ansätze basierend auf bispezifischen Antikörpern und Tumor-spezifischen T-Zellen.
Bispezifische Antikörper
Bispezifische Antikörper (BiMab) entstehen durch die Kombination zweier Antikörperdomänen (Abb.1): Eine erkennt ein Zielantigen auf der Myelomzelle (z.B. BCMA), und die zweite erkennt ein Aktivierungsantigen (z.B. CD3) auf T Zellen. Damit werden T Zellen an Myelomzellen gebunden und lokal aktiviert. Paradebeispiel ist auch hier wieder ein Konstrukt (CD3-CD19, Blinatumumab) zur Behandlung von B Zell Leukämien und Lymphomen. In der Myelombehandlung befindet sich diese Technik noch in der präklinischen Entwicklung, zeigt aber z.B. für einen BCMA-CD3 spezifischen BiMab hoffnungsvolle Resultate. So konnte für die Ko-Kultivierung autologer Myelom- und T Zellen in Präsenz eines BCMA-CD3 Konstrukts unabhängig vom Krankheitsstatus ein hoher Grad der T Zellaktivierung mit entsprechender Tumorzelllyse beobachtet werden. In ersten Primatenversuchen wurde neben der guten Verträglichkeit eine Elimination BCMA-positiver Plasmazellen im Knochenmark verzeichnet (5). Daher befinden sich derzeit mehrere BiMAb Studien in der Planung und werden in Kürze aktiviert.
Tumorspezifische T-Zellen
Die Umprogrammierung autologer oder allogener T Zellen durch viralen Gentransfer tumorspezifischer Antikörpersequenzen (sog. CAR T Zell-Technologie) stellt momentan eines der interessantesten klinischen Forschungsgebiete dar. Bisher werden dafür Abwehrzellen des jeweiligen Patienten aus dem Blut entnommen, im Labor genetisch manipuliert und durch Einschleusung einer Antikörperdomäne (z.B. mit BCMA Spezifität) gegen Myelomzellen gerichtet (Abb.1). Nach Rückgabe in den Patienten können diese modifizierten Abwehrzellen die Myelomzellen aufspüren und zerstören. Hauptnebenwirkung ist das sog. cytokine release syndrome (CRS), das typischerweise zwischen Tag 4 und 8 nach Zellrückgabe entstehen kann. Inzwischen sind aber Massnahmen zur Therapie eines einsetzenden CRS etabliert und damit die Verträglichkeit gegeben. Von den vier bisher präsentierten klinischen BCMA CAR T Zellstudien lässt sich folgern, dass hohe Ansprechraten (bis zu 100%) in den zum Teil doch intensiv vorbehandelten Patienten (median bis zu 7 Vortherapien) mit CR Raten von bis zu 40% erreicht werden können (6-9).
Eine vorgängige Lymphodepletion mittels Chemotherapie (z.B. Cyclophosphamid oder Fludarabin) erwies sich als vorteilhaft, da dadurch eine bessere Verträglichkeit bei höheren Zelldosen erzielt werden konnte Langfristige Remissionen sind trotz der bereits genannten multiplen Vortherapien beobachtet worden, wobei man wahrscheinlich noch einige Jahre warten muss, um zu beurteilen, ob auch die erhofft hohen Heilungsraten ein realistisches Ziel sind. Die Kosten einer Zelltherapie stellen derzeit sicherlich noch eine schwer zu nehmende Hürde dar und es ist ungewiss, wann diese Technologie auch in der Schweiz verfügbar sein wird.
Prof. Dr. med. Christoph Renner
Onkozentrum Hirslanden Zürich und Onkozentrum Zürich
Witellikerstrasse 40
8032 Zürich
Der Autor hat keine Interessenskonflikte im Zusammenhang mit diesem Beitrag deklariert.
Monoklonale Antikörper sind 40 Jahre nach ihrer Erstbeschreibung Eckpfeiler einer Immuntherapie bei vielen Tumorerkrankungen und konnten auch beim Multiplen Myelom einen festen Platz im Behand-lungskonzept einnehmen.
Antikörpervarianten werden noch effektiver und gezielter Myelom-
zellen abtöten können und damit hoffentlich die Effektivität als auch Verträglichkeit der Therapie verbessern können.
Die neuartigen Zelltherapien versprechen zudem eine langfristige zelluläre Kontrolle der Erkrankung und lassen sogar Heilungen als mögliches Therapieziel erscheinen
Message à retenir
40 ans après leur description initiale, les anticorps monoclonaux sont la pierre angulaire de l’immunothérapie pour de nombreuses maladies tumorales et font désormais partie intégrante du concept de traitement du myélome multiple.
Les variantes d’anticorps seront capables de tuer les cellules myélomateuses encore plus efficacement et spécifiquement, ce qui, nous l’espérons, améliorera l’efficacité et la tolérabilité de la thérapie.
Les nouvelles thérapies cellulaires promettent également un contrôle cellulaire à long terme de la maladie et font même apparaître la guérison comme un objectif thérapeutique possible.
1 Usmani SZ, Weiss BM, Plesner T et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood. 2016 Jul 7;128(1):37-44.
2. Palumbo A, Chanan-Khan A, Weisel K et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N Engl J Med. 2016 Aug 25; 375(8):754-66.
3. Dimopoulos MA, Oriol A, Nahi H et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N Engl J Med. 2016 Oct 6; 375(14):1319-1331.
4. Trudel S, MD, Lendvai N, Popat R, et al. Deep and durable responses in patients (Pts) with relapsed/refractory multiple myeloma (MM) treated with monotherapy GSK2857916, an antibody drug conjugate against B-cell maturation antigen (BCMA): preliminary results from part 2 of study BMA117159. Presented at: ASH Annual Meeting and Exposition; Dec. 9-12, 2017; Atlanta, Georgia. Abstract 741.
5. Hipp S, Tai YT, Blanset D et al. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia. 2017 Aug; 31(8): 1743-1751.
6. Berdeja JG, Lin Y, Raje NS, et al. First-in-human multicenter study of bb2121 anti-BCMA CAR T-cell therapy for relapsed/refractory multiple myeloma: updated results [abstract]. J Clin Oncol. 2017;35(suppl 15). Abstract 3010.
7. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma [abstract]. J Clin Oncol. 2017;35(suppl 18). Abstract LBA3001.
8. Cohen AD, Garfall AL, Stadtmauer EA, et al. B-cell maturation antigen (BCMA)-specific chimeric antigen receptor T cells (CART-BCMA) for multiple myeloma (MM): initial safety and efficacy from a phase I study [abstract]. Blood. 2016;128(22). Abstract 1147.
9. Ali SA, Shi V, Maric I, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128(13):1688-1700.
Als Grundversorger wird man häufig mit dem Problem «Harninkontinenz» konfrontiert. Mit einer guten Anamnese und einer einfachen klinischen Untersuchung können die Arbeitsdiagnose suffizient geklärt und konservative Therapien begonnen werden. Nach der Darstellung der Abklärungsschritte im ersten Teil in „der informierte arzt“ im Mai dieses Jahres folgt hier der zweite Teil, welcher den Aspekten der Behandlung gewidmet ist.
Nach Ausschluss einer Retention führen die Anamnese und klinische Untersuchung zwangsläufig zu einer vernünftigen Arbeitsdiagnose. Der Grundversorger kann problemlos konservative Therapien beginnen. Weil er die Lebenssituation und die internistische Krankengeschichte der Patientin gut kennt, wird er die Behandlung der Inkontinenz in einen Gesamtkontext bringen können.
Einzig der Leidensdruck der Patientin bestimmt das Tempo und Akzeleration innerhalb der Therapiemodalitäten.
Behandlung Belastungsinkontinenz (SUI)
Die Belastungsinkontinenz ist durch eine gestörte Anatomie bedingt und erfordert demzufolge eine Korrektur derselben. Physiotherapie: Im Rahmen der konservativen Erstlinienbehandlung der SUI ist die Physiotherapie am wichtigsten. Es ist darauf zu achten, die Patientin in eine beckenbodenorientierte Physiotherapie zu überweisen, die neben Beckenbodentraining auch Biofeedback und Elektrostimulation anbietet. Auf der Webseite der Pelvisuisse, dem Verein der in Beckenboden-Rehabilitation spezialisierten Physiotherapeutinnen, sind Fachfrauen gelistet. Mit einer Verbesserung der Kontinenz kann man in ca. 50% rechnen (1). Vor allem bei offener Familienplanung ist ein physiotherapeutischer Ansatz sinnvoll. Die Physiotherapie verbessert kompensatorisch die Beckenbodenmuskulatur, doch kann sie die muskulären und bindegewebigen Abrisse und Überdehnungen nicht heilen. Pessare: Ist die Familienplanung nicht abgeschlossen und leidet die Patientin unter einer Belastungsinkontinenz, sind Pessare geeignet, die Zeit bis zu einer Inkontinenzoperation zu überbrücken. Pessare stützen den abgeflachten urethrovesikalen Übergang. Ein gut sitzendes Pessar wird der Patientin wieder erlauben, Tennis zu spielen oder zu Joggen. Es gibt wiederverwendbare Pessare aus weichem Silikon und Einmalpessare aus Schaumstoff. Für die SUI werden typischerweise Ring- oder Schalenpessare mit einer suburethralen Verstärkung verwendet (Abbildung 1). Von den Einmalpessaren existieren praktische Probesets mit verschiedenen Pessargrössen (zum Beispiel RecaFEM® Probepackung). Sie werden wie normale Tampons angewendet, müssen aber vor dem Einführen nass gemacht werden. Bestätigt man der Patientin eine Inkontinenz, hat sie Anrecht auf eine definierte Vergütung pro Jahr für Inkontinenzmaterialien. Indikation für die Überweisung: Ist die Belastungsinkontinenz sehr störend und führen konservative Massnahmen nicht zu einer Besserung oder wünscht die Patientin eine unmittelbare Lösung, haben wir mit den suburethralen Bandoperationen (z.B. TVT™) einfache und hoch effektive Eingriffe zur Verfügung, die auch langfristig Erfolge zeigen (Abb. 2) (2). Eine abgeschlossene Familienplanung ist günstig.
Behandlung überaktive Blase (OAB)
Die krankheitstypische Anamnese mit Urge und der Ausschluss von Resturin genügen, um die Arbeitsdiagnose OAB zu stellen und eine Therapie einzuleiten. Die überaktive Blase wird nur letztinstanzlich invasiv behandelt. Die Diagnose OAB ist eine Ausschlussdiagnose, die Häufigkeit der OAB nimmt mit dem Alter kontinuierlich zu und die Grenzen zwischen symptomatischen Formen und der idiopathischen OAB sind fliessend. Darum stehen Therapieansätze, die Verhalten, Trophik der Schleimhäute oder Harnwegsinfektionen berücksichtigen, an erster Stelle: Trinkverhalten: Eine angepasste Flüssigkeitsaufnahme mit Trinken vorzugsweise am Tag und weniger am Abend ist vor allem bei älteren Menschen eine geeignete Strategie, um die Anzahl nächtlicher Miktionen zu reduzieren. Änderungen im Lifestyle: Gewichtsreduktion bei Übergewicht, Verzicht auf blasentonisierende Getränke wie Kaffee und Cola, Verzicht auf CO2-versetzte Getränke, moderate sportliche Aktivität und Verzicht auf Nikotingenuss können die Kontinenz positiv beeinflussen und sind wichtige Erstlinienempfehlungen (3).
Physiotherapie hat auch bei der Behandlung der überaktiven Blase einen hohen Stellenwert. Einerseits wird die Verschlussfunktion durch Beckenbodentraining gestärkt, andererseits durch Elektrostimulation über Reflexbögen die Überaktivität des Detrusors gedämpft, auch wenn der Mechanismus hier nicht wirklich verstanden ist.
Der Östrogenentzug in der Menopause führt zu einer atrophen, verletzlichen und sensitiven Schleimhaut und in der Folge zu Symptomen des unteren Harntraktes wie Dysurie und häufigem Harndrang (4). Lokale Östrogenapplikation als Estradiol(E2)- oder Estriol(E3)- haltige Crèmen, Vaginaltabletten oder Ovula in einer Langzeittherapie ist eine einfache und meist sehr effektive Massnahme. Eine gute Datenlage existiert zur Prophylaxe rezidivierender Harnwegsinfekte: In der Menopause reduziert lokale Östrogenisierung die Infekthäufigkeit um einen Faktor 10 (5) (Tabelle 1).
Es gibt Phytotherapeutika, die rezeptfrei erhältlich sind. Beispielsweise verhindert der Einfachzucker D-Mannose (Femannose® N, Hänseler D-Mannose) rezidivierende Harnwegsinfekte ähnlich effektiv wie eine Antibiotikaprophylaxe (6). Bryophyllum Kautabletten 50% in einer Dosis von 3 x 2 Tbl. täglich kann bei der Behandlung der OAB eingesetzt werden (7).
Die überaktive Blase ist die Krankheit des alternden Menschen. Gerade bei fragilen polymorbiden Patientinnen ist die Polypharmazie häufig und die Medikamenteninteraktionen schwieriger kontrollierbar, so dass es sinnvoll sein kann, die Medikamentenliste auch unter dem Aspekt der OAB einzudämmen anstatt auszuweiten. So können beispielsweise Diuretika, β-Blocker,
Kalziumantagonisten, NSAR, Antiparkinsonmittel, Psychopharmaka, Barbiturate oder Opiate Symptome des unteren Harntraktes negativ beeinflussen.
Eine medikamentös schlecht eingestellte Herzinsuffizienz oder ein schlecht eingestellter Diabetes mellitus können Symptome der überaktiven Blase verstärken. Antimuskarinika sind medikamentöse Grundpfeiler der OAB Therapie (Tabelle 2). Sie können mit anderen konservativen Therapiemassnahmen kombiniert werden. Einige Produkte erlauben eine Dosisakzeleration. Es ist allerdings nicht bei jeder Patientin günstig, die Medikamentenliste mit einem blasenspezifischen Medikament zu ergänzen (Polypharmazie, siehe oben). Antimuskarinika reduzieren im Vergleich zu Plazebo in bescheidenem Mass die Anzahl Miktionen und Inkontinenzepisoden pro Tag. Die verschiedenen Antimuskarinika sind ähnlich effektiv, sie unterscheiden sich vor allem im Nebenwirkungsprofil. Prinzipiell erfolgt die Wirkung nicht selektiv an der Blase, häufige Nebenwirkungen werden durch Blockade der parasympathischen Innervation an anderen Organen verursacht. Typisch sind Mundtrockenheit, Obstipation, Tachykardie, reduzierte Akkommodationsfähigkeit, Erhöhung des Augeninnendruckes bis hin zu kognitiven Störungen. Die Nebenwirkungen können zu einer schlechten Compliance führen. Das quaternäre Amin Trospiumchlorid (Spasmo-Urgenin® Neo oder Spasmex®) sollte die Bluthirnschranke nicht passieren und ist darum bei betagten Menschen günstig.
β - 3 - Agonisten relaxieren den Detrusor durch Stimulation der sympathischen β-Adrenorezeptoren und wirken damit über einen anderen Pfad als die Antimuskarinika (Abbildung 3). Entsprechend sind sympathische Nebenwirkungen ein Problem: Eine unkontrollierte arterielle Hypertonie ist eine Kontraindikation, unter Therapie mit Mirabegron soll der Blutdruck kontrolliert werden. Dafür fehlen die typischen anticholinergen Nebenwirkungen (8). Aktuell gibt es nur ein Präparat auf dem schweizerischen Markt: Mirabegron (Betmiga®). Mirabegron wurde bezüglich Wirksamkeit (Häufigkeit von Miktion, Häufigkeit von Inkontinenzepisoden) gegenüber Placebo (9) untersucht, dem Medikament wurde eine den Antimuskarinika vergleichbare Wirkung attestiert.
Indikation für die Überweisung:
Ist die Inkontinenz therapierefraktär oder persistieren auffällige Befunde wie Mikrohämaturie, ist eine Überweisung an die UrogynäkologIn sinnvoll, welche die Diagnostik fortsetzt: Die Zystoskopie kann relevante Befunde wie einen Blasentumor, Fremdmaterial oder Hunner’sche Ulcera (interstitielle Zystitis) aufdecken und die Urodynamik die Diagnose «OAB» objektivieren, indem eine hypersensitive hypokapazitive oder auch instabile Blase gemessen werden kann.
Invasive Therapieformen der OAB: Die intravesikale Injektion von Botulinumtoxin (Botox®) ist hierbei die Therapie der Wahl, weil sie sehr effektiv und minimal invasiv ist und sie bei Bedarf hinaufdosiert und repetiert werden kann (10). Botulinumtoxin führt zu einer reversiblen chemischen Denervation der Blase. Konzeptionell wird die parasympathische Innervation isoliert am gewünschten Ort «Blase» ausgeschaltet, was gegenüber einer medikamentösen anticholinergen Therapie mit den systemischen Nebenwirkungen von grossem Vorteil ist. Gerade für polymorbide Patientinnen mit langer Medikamentenliste oder Patientinnen mit dementieller Entwicklung ist die intravesikale Botoxinjektion eine naheliegende Lösung. Botoxinjektionen sind nicht nur bei neurogenen Blasenstörungen, sondern auch bei der idiopathischen OAB eine Kassenleistung, vorausgesetzt, die Patientin ist bezüglich konservativer Massnahmen therapierefraktär.
Die sakrale Neuromodulation (Blasenschrittmacher) ist ein etabliertes und effektives Verfahren in der Behandlung der OAB und gegen medikamentöse Therapien und gegen Botulinumtoxin geprüft (11, 12), wird aber, weil invasiver und teurer, in der Praxis deutlich seltener eingesetzt.
Behandlung «Inkontinenz bei chronischer Retention»
Selten ist eine Inkontinenz bei der Frau durch eine Überlaufsituation bedingt. Ist die Retention als Inkontinenzursache erkannt (hohe Restharnmenge), ist die primäre fachärztliche Abklärung angezeigt.
In der Urogynäkologie können ein massiver Genitaldescensus oder eine Urethralstenose obstruktiv wirken. Die Detrusorkontraktilität kann durch chirurgische (onkologische Chirurgie im kleinen Becken) oder «internistische» Denervierung (z.B. autonome Polyneuropathie bei Diabetes mellitus) verursacht sein. Meist bleiben die intravesikalen Drücke tief und die Nieren werden nicht durch Reflux kompromittiert. (Komplexe neuro-urologische Krankheitsbilder wie echte Detrusor-Sphinkter-Dyssynergie sind nicht Gegenstand dieses Artikels.)
Mittels «Miktion nach Uhr» kann man eine Kontinenz erreichen: Die Patientin wird prophylaktisch und konsequent nach Zeitplan z.B. alle 1½ Stunden auf die Toilette begleitet/geschickt. Medikamentös kann der schlaffe Detrusormuskel mit Cholinergica tonisiert werden, zum Beispiel mit Ubretid®. Die häufigste Nebenwirkung sind Darmkoliken. Den glatten Sphinkter der Urethra öffnet man komplementär mit einem α - 1 - Rezeptor-Blocker, zum Beispiel Pradif 400®. Die häufigste Nebenwirkung ist Schwindel bei Therapiebeginn.
Für das Erlernen des Selbskatheterismus muss man die Patientin an fachspezifische Pflegefachkräfte überweisen, eine gewisse Agilität und Fingerfertigkeit seitens der Patientin ist Voraussetzung.
Auch wenn nicht erwünscht, kann in einer Pflegeeinrichtung eine Versorgung mit einem Dauerkatheter oder einer suprapubischen Harnableitung die ideale Lösung für eine auf Retention basierenden Inkontinenz sein. Wird der Katheter mit einem Ventil bestückt, erspart man der Patientin den Urinbeutel.
Dr. med. Gabriella Stocker
Frauenklinik Stadtspital Weid und Triemli
Birmensdorferstrasse 501
8063 Zürich
Frauenklinik Stadtspital Weid und Triemli
Birmensdorferstrasse 501
8063 Zürich
daniel.passweg@triemli.zuerich.ch
Die Autoren haben in Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
Ist die Patientin therapierefraktär oder persistieren auffällige Befunde wie Mikrohämaturie, ist die Überweisung an die FachärztIn angezeigt.
Erstlinientherapie der Belastungsinkontinenz ist Physiotherapie,
Zweitlinientherapie die suburethrale Bandeinlage.
Für die hauptsächlich konservative Behandlung der OAB ist das
hausärztliche Setting ideal.
Inkontinenz bei chronischer Retention gehört primär durch die FachärztIn abgeklärt.
1. Bø K, Talseth T, Holme I. Single blind, randomised controlled trial of pelvic floor exercises, electrical stimulation, vaginal cones, and no treatment in management of genuine stress incontinence in women. BMJ. 1999;318:487-93.
2. Nilsson CG1, Palva K, Rezapour M, Falconer C. Eleven years prospective follow-up of the tension-free vaginaltape procedure for treatment of stress urinary incontinence.
Int Urogynecol J Pelvic Floor Dysfunct. 2008;19:1043-7.
3. Dallosso HM, McGrother CW, Matthews RJ, Donaldson MM; Leicestershire MRC Incontinence Study Group. The association of diet and other lifestyle factors with overactive bladder and stress incontinence: a longitudinal study in women. BJU Int. 2003;92:69-77.
4. Bachmann GA, Nevadunsky NS. Diagnosis and treatment of atrophic vaginitis. Am Fam Physician. 2000;61:3090-3096.
5. Raz R, Stamm WE. A controlled trial of intravaginal estriol in postmenopausal women with recurrent urinary tract infections. N Engl J Med. 1993;329:753-6.
6. Kranjcec B, Papeš D, Altarac S. D-mannose powder for prophylaxis of recurrent urinary tract infections in women: a randomized clinical trial. World J Urol. 2014;32:79-84.
7. Betschart C, von Mandach U, Seifert B, Scheiner D, Perucchini D, Fink D, Geissbühler V. Randomized, double-blind placebo-controlled trial with Bryophyllum pinnatum versus placebo for the treatment of overactive bladder in postmenopausal women. Phytomedicine. 2013;20:351-8.
8. Chapple C, Khullar V, Nitti VW, Frankel J, Herschorn S, Kaper M, Blauwet MB, Siddiqui E. Efficacy of the β3-adrenoceptor agonist mirabegron for the treatment of overactive bladder by severity of incontinence at baseline: a post hoc analysis of pooled data from three randomised phase 3 trials. Eur Urol. 2015;67:11-4.
9. Wagg A, Cardozo L, Nitti VW, Castro-Diaz D, Auerbach S, Blauwet MB, Siddiqui E. The efficacy and tolerability of theβ3-adrenoceptor agonist mirabegron for the treatment of symptoms of overactive bladder in older patients. Age Ageing. 2014;43:666-75.
10. Schmid DM, Sauermann P, Werner M, Schuessler B, Blick N, Muentener M, Strebel RT, Perucchini D, Scheiner D, Schaer G, John H, Reitz A, Hauri D, Schurch B. Experience with 100 cases treated with botulinum-A toxin injections in the detrusormuscle for idiopathic overactive bladder syndrome refractory to anticholinergics. J Urol. 2006;176:177-85.
11. Siegel S, Noblett K, Mangel J, Griebling TL, Sutherland SE, Bird ET, Comiter C, Culkin D, Bennett J, Zylstra S, Berg KC, Kan F, Irwin CP. Results of a prospective, randomized, multicenter study evaluating sacral neuromodulation with InterStim therapy compared to standard medical therapy at 6-months in subjects with mild symptoms of overactive bladder. Neurourol Urodyn. 2015;34:224-30.
12. Amundsen CL, Richter HE, Menefee SA, Komesu YM, Arya LA, Gregory WT, Myers DL, Zyczynski HM, Vasavada S, Nolen TL, Wallace D, Meikle SF. OnabotulinumtoxinA vs Sacral Neuromodulation on Refractory Urgency Urinary Incontinence in Women: A Randomized Clinical Trial. JAMA. 2016;316:1366-1374.