Die stetig wachsende Zahl älterer Menschen, insbesondere der über 80-Jährigen, stellt unsere Gesellschaft vor grosse soziale, wirtschaftliche und natürlich medizinische Herausforderungen. Altern erhöht das Risiko für Multimorbidität und in der Folge auch für Polypharmazie. Dabei treten chronische Krankheitsverläufe in den Vordergrund. Es kommt zu funktionellen Einschränkungen und die Bewältigung des Alltags wird beeinträchtigt, ambulante und möglicherweise auch stationäre Hilfs- und Pflegebedürftigkeit sind vor allem im höheren Alter die Folge (1). Von politischer Seite wiederum wird einerseits aus Kostengründen eine Reduktion stationärer Pflegeplätze angestrebt und eine ambulante Pflege vorgezogen (2). Dabei wird aber auch der Tatsache Rechnung getragen, dass die heutige Seniorengeneration das Leben im Alter revolutioniert und völlig neue selbstbestimmte Lebensformen gestaltet (3). Diese Entwicklung hat bedeutende Auswirkungen auch auf die orale Gesundheit im Alter.
Abstract: The steadily growing number of older people, especially those over 8o years of age, confronts our society with major social, economic and, of course, medical challenges. Ageing increases the risk of multimorbidity and consequently also of polypharmacy. Chronic courses of disease come to the fore. Functional limitations occur and the ability to cope with everyday life is impaired; the need for outpatient and possibly also inpatient assistance and care is the consequence, especially in old age (1). Politicians, on the other hand, are striving to reduce the number of inpatient care places for cost reasons and prefer outpatient care (2). However, this also takes into account the fact that today’s senior generation is revolutionising life in old age and creating completely new self-determined ways of living (3). This development also has significant implications for oral health in old age. Key Words: Oral health, elderly people
Die erfolgreiche orale Prävention in früheren Lebensjahrzehnten hat dazu geführt, dass Menschen mit immer mehr eigenen Zähnen immer älter werden. Hinzu kommen orale Implantate, die es erlauben, verloren gegangene Zähne zu ersetzen. Zudem hat die enorme Entwicklung technischer Möglichkeiten dazu geführt, dass die Menschen mit immer komplexerem, zunehmend festsitzendem und seltener abnehmbarem Zahnersatz altern (4). Auf der anderen Seite bleiben im Alter Zahnverlust, Karies, Gingivitis, Parodontitis und andere orale Infektionen sowie orale Präkanzerosen und Malignome hoch prävalent. Hinzu kommt bei älteren Menschen eine häufige, in erster Linie medikamentös bedingte Hyposalivation hinzu, mit oft sehr rasch sich entwickelnden, fatalen Folgen für die oralen Hart- und Weichgewebe. Orale Erkrankungen sind somit ein oft auftretender Teil der Multimorbidität im Alter und können den allgemeinen Gesundheitszustand beeinträchtigen bzw. mit systemischen Erkrankungen interagieren (5, 6). Ein typisches Beispiel hierzu ist der Diabetes mellitus Typ II, der auf der einen Seite die Progredienz einer Parodontitis beschleunigen kann. Auf der anderen Seite vermag die Parodontitis die Einstellbarkeit des Diabetes zu beeinträchtigen (7).
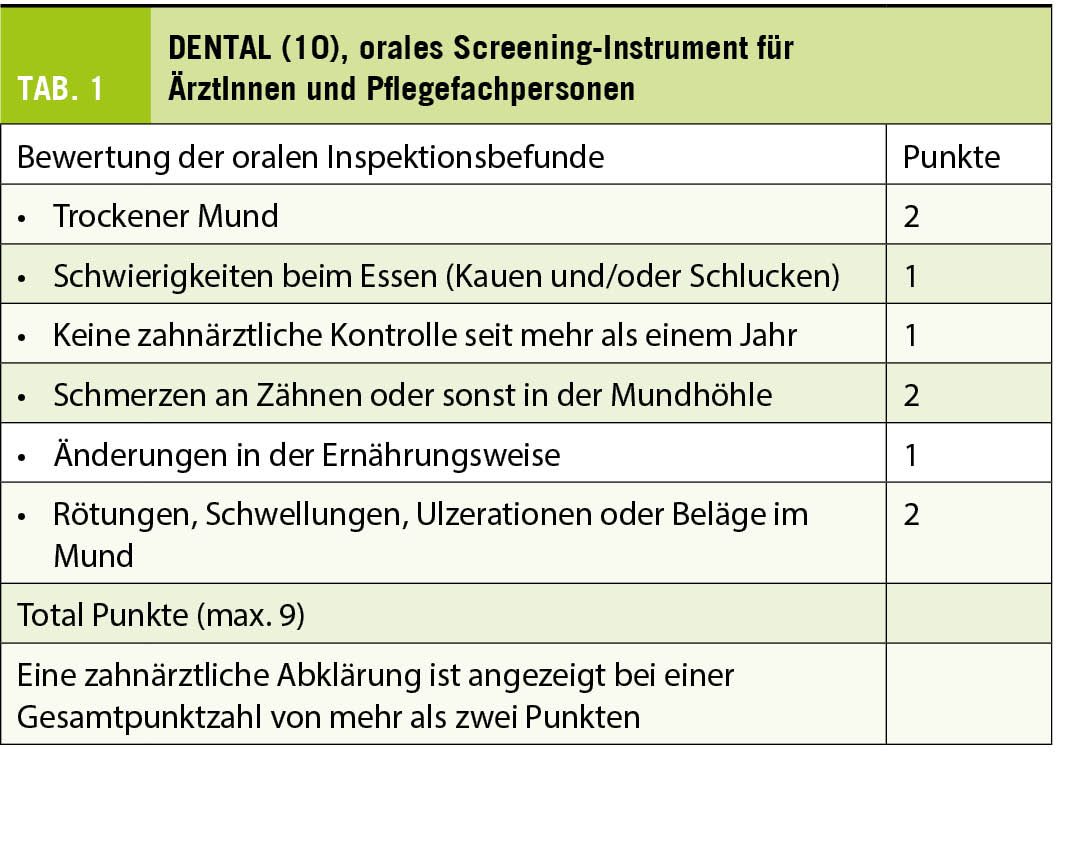
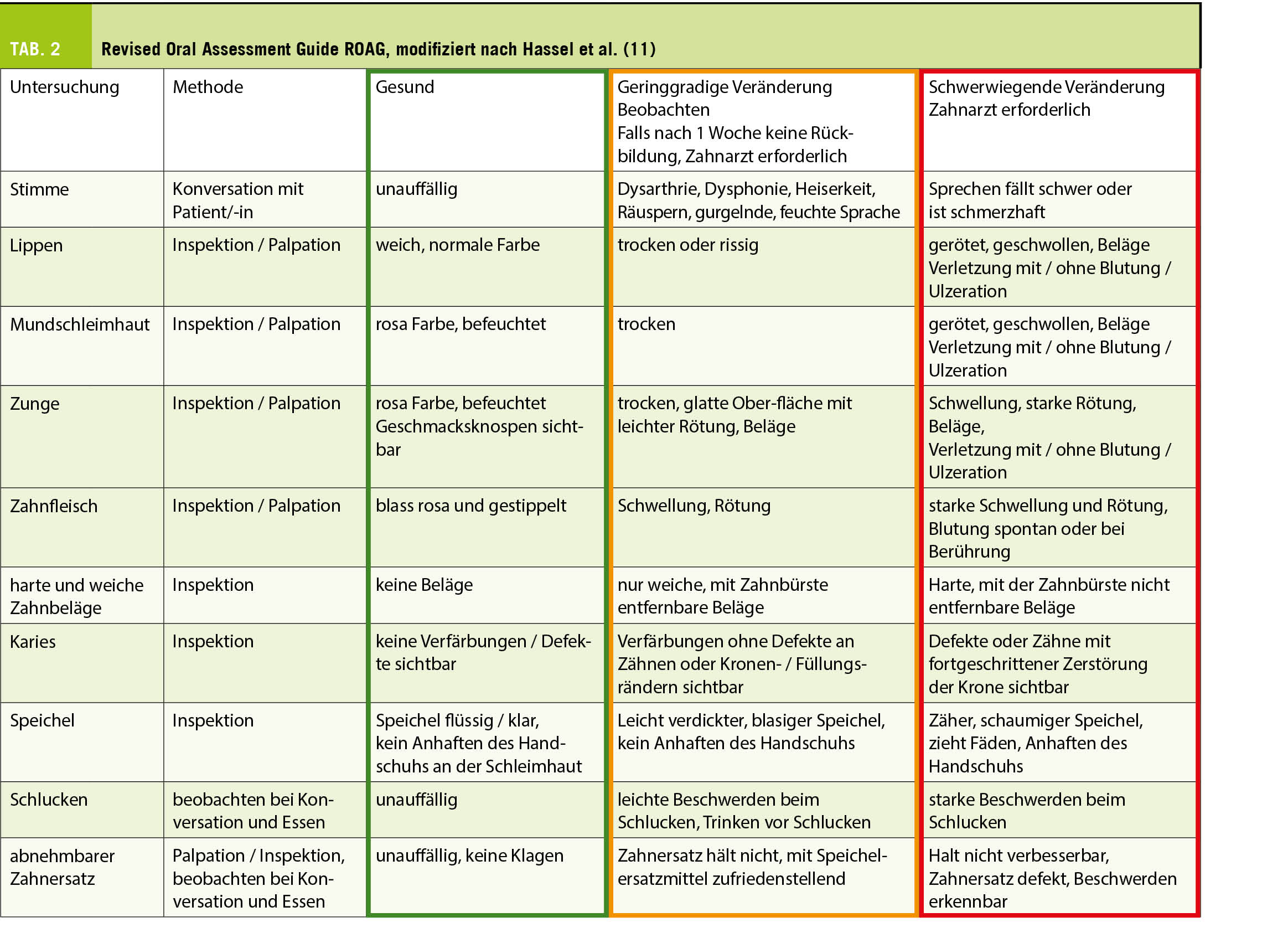
Aus oben Erwähntem geht deutlich hervor, dass alternde Menschen einen wachsenden Bedarf an präventiver, diagnostischer und kurativer zahnmedizinscher Betreuung haben (8). Während die Inanspruchnahme von ärztlichen Leistungen mit zunehmendem Alter in erster Linie durch die Anzahl gleichzeitig bestehender Erkrankungen bestimmt wird und an Häufigkeit zunimmt, geht dagegen die Zahl und Regelmässigkeit zahnärztlicher Konsultationen ab Ende des fünften Lebensjahrzehnts stetig zurück (9). Es darf angenommen werden, dass durch die zunehmenden Herausforderungen, die das Alter mit sich bringen kann, die orale Prävention für die Menschen an Dringlichkeit verliert, obwohl sie um deren Bedeutung wissen und in den vorausgehenden Lebensjahrzehnten von dieser profitiert haben. Eine wiederkehrende Inspektion der Mundhöhle durch den Hausarzt oder Spezialärztinnen, die von älteren Menschen gehäuft konsultiert werden, wäre somit sehr empfehlenswert. Diese ist insbesondere dann angebracht, wenn die letzte zahnmedizinische Kontrolle anamnestisch mehr als ein Jahr zurückliegt. Für die effiziente Beurteilung der Notwendigkeit eines zahnärztlichen Konsiliums stehen ÄrztInnen und Pflegekräften geeignete und gut validierte Screening-Instrumente zur Verfügung, wie beispielsweise das DENTAL (10) (Tab. 1) oder der Revised Oral Assessment Guide ROAG (11) (Tab. 2).
Eine engere interdisziplinäre Zusammenarbeit zwischen Humanmedizinerinnen und Zahnmedizinern wäre auch deshalb von grossem Nutzen, indem beide Seiten neue Diagnosen unmittelbarer kommunizieren würden. Aus zahnmedizinischer Sicht ist z.B. die sofortige Kenntnis der Diagnose einer Demenz von zentraler Bedeutung,
weil sie grundlegende Implikationen für die orale Prävention, Therapie und langfristige Betreuung hat (siehe Merkblatt Mund-/Zahngesundheit für Angehörige und Pflegende von Menschen mit Demenz; am Online-Beitrag). Bleibt diese strategische Anpassung an die absehbar sich verschlechternden Betreuungsbedingungen aus, notabene in einer Krankheitsphase, in der zahnmedizinische Interventionen meist gut durchführbar wären und die Betroffenen sich noch an orale Veränderungen anpassen könnten, so kommt es in der Regel zu einem Ausscheren aus der zahnärztlichen Betreuung. Orale Erkrankungen sind die Folge und schreiten rasch fort. In diesem Zusammenhang ist die Erkenntnis von zentraler Bedeutung, dass orale Erkrankungen im Alter nicht erst nach Übertritt in eine Institution auftreten, sondern bereits als Teil der Multimorbidität mitgebracht werden und die Pflege sehr häufig vor kaum mehr lösbare Herausforderungen bei der Zahn- und Mundhygiene stellen (12).
Die hohe Prävalenz oraler Erkrankungen bei pflegebedürftigen Betagten konfrontiert auch die zahnmedizinischen Behandlungsteams mit sehr schwierigen Situationen. Der institutionelle Kontext erschwert zum einen die diagnostischen und therapeutischen Prozesse wesentlich. Zum anderen fehlen häufig eine geeignete Behandlungsinfrastruktur und eine adäquate mobile Ausrüstung. Zudem überfordern die zum Lebensende hin oft komplexen medizinischen Problemstellungen. Schliesslich ist die teure Praxisinfrastruktur während der externen Patientenbetreuung nicht ausgelastet und werden die zahnmedizinischen Leistungen in Institutionen nur ungenügend honoriert. Dies führt dazu, dass die zahnmedizinische Versorgung von Menschen, die in Institutionen leben, nach wie vor der grossen Nachfrage nicht zu entsprechen vermag (13). Vor dem Hintergrund der Favorisierung ambulanter Pflege dürfte sich dieser Versorgungsnotstand noch weiter akzentuieren.
Es ist für die Erhaltung der oralen Gesundheit von zentraler Bedeutung, dass durch eine engere interdisziplinäre Vernetzung zwischen Human- und Zahnmedizin neue Diagnosen unmittelbar kommuniziert und konsiliarische Abklärungen gefördert werden. Dadurch können bereits bei den zu Hause lebenden SeniorInnen Prävention, Therapie sowie Langzeitbetreuung frühzeitig den sich verändernden Lebensbedingungen laufend angepasst werden und mögliche Wechselwirkungen zwischen systemischen und oralen Erkrankungen erkannt werden. Aus zahnmedizinscher Sicht bedeutet dies, dass die oralen Risiken umso mehr reduziert werden müssen, je stärker die soziobiologischen Risiken ansteigen, damit auch unter sich erschwerenden Bedingungen orale Prävention, Therapie und Langzeitbetreuung nicht nur durch das zahnmedizinische Team, sondern auch durch Pflegende nachhaltig möglich bleiben (13).
Copyright bei Aerzteverlag medinfo AG
Prof. Dr. med. dent. Christian E. Besimo
Riedstrasse 9
6430 Schwyz
christian.besimo@bluewin.ch
Der Autor hat keine Interessenskonflikte im Zusammenhang mit diesem Artikel.
1 Höpflinger F, Bayer-Oglesby L, Zumbrunn A. Pflegebedürftigkeit und Langzeitpflege im Alter. Aktualisierte Szenarien für die Schweiz. Huber, Bern 2011.
2 Gesundheitsdepartement des Kantons Basel-Stadt. Gesundheitsversorgungsbericht über die Spitäler, Pflegeheime, Tagespflegeheime und Spitex-Einrichtungen im Kanton Basel-Stadt. Werner Druck & Medien, Basel November 2019.
3 Höpflinger F, Hugentobler V, Spini D. Wohnen in den späten Lebensjahren. Grundlagen und regionale Unterschiede. Age Report V. Seismo, Zürich 2019.
4 Schneider C, Zemp E, Zitzmann NU. Oral health improvements in Switzerland over 20 years. Eur J Oral Sci 2017;125:55–62.
5 Petersen PE, Kandelman D, Arpin S, Ogawa H. Global oral health of older people – Call for publiic health action. Community Dental Health 2010;27(Suppl. 2):257-268.
6 Marchini L, Ettinger R. Personalized dental caries management for frail older adults and persons with special needs. Dent Cllin N Am 2019;63:631-651.
7 Preshaw PM, Alba AL, Herrera D, Jepsen S, Konstantinidis A, Makrilakis K, Taylor R. Periodontitis and diabetes: A two-way relationship. Diabetologia. 2012;55:21–31.
8 Han P, Suarez-Durall P, Mulligan R. Dry mouth: A critical topic for older adult patients. J Prosthodont Res 2015;59:6-19.
9 Biffar R, Klinke-Wilberg T. Gesundheit der Älterwerdenden und Inanspruchnahme ärztlicher Dienste – Zahnmedizinische Konsequenzen und Aufgaben. Senioren-Zahnmedizin 2013;1:35-42.
10 Busch LA. D-E-N-T-A-L: A rapid self-administered screening instrument to promote referrals for further evaluation in older adults. J Am Geriatr Soc 1996;44:979-981.
11 Hassel AJ, Leisen J, Rolko C, Rexroth W, Ohlmann B, Rammelsberg P. Clinical assessment of oral health between physician and dentist – A pilot study on inter-examiner reliability. Z Gerontol Geriatr 2008;41:132-138.
12 Besimo C, Besimo-Meyer RH. Zahn- und Mundgesundheit – Ein Stiefkind in der Betreuung von Menschen mit Demenz. Prophylaxe Impuls 2016;20:124-128.
13 Wocke C, Eckardt R. Multimorbidität im Alter. Senioren-Zahnmedizin 2013;1:9-13.
14 Besimo, C. Paradigmenwechsel zugunsten einer besseren oralen Gesundheit im Alter. Swiss Dental Journal 2015;125:599-604.
Kardiovaskuläres Risiko und renales Risiko?
Der Hausarzt benötigt zur Beantwortung dieser Fragen Kenntnisse der neusten wichtigsten Studie (Selbststudium, Weiterbildungen). Was sind die Richtlinien zu Hypertonie, Lipiden und Diabetes? Was sind die Werte der Patientin, welche wichtigen Fragen sind zu stellen? Welche Medikamente oder Kombinationen werden von den Krankenkassen rückvergütet?
HbA1c als Kontrollwert ein wichtiges Therapieziel
Ein gutes HbA1c ist immer noch wichtig, um mikro- und makrovaskuläre Komplikationen zu vermeiden. < 7.0 % bei Patienten ohne Sulfonylharnstoffe und/oder Insulin, am besten <6.5 % (möglichst normal) ohne Gefahr für Hypoglykämie. <8.0 % bei älteren Patienten (> 80 Jahre) und/oder bei bereits manifesten Komorbiditäten (Niereninsuffizienz, Herzinsuffizienz, kardiovaskulären Erkrankungen) und Insulintherapie.
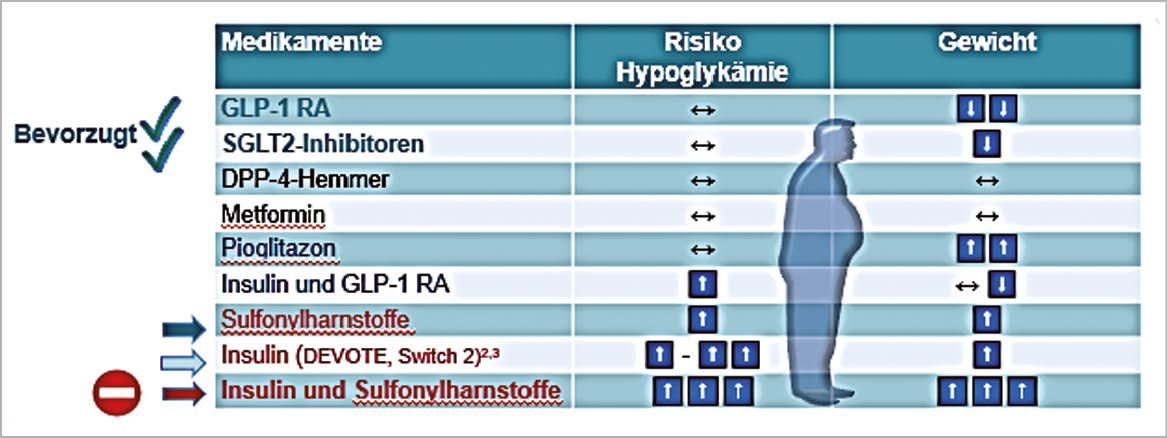
Therapie des T2DM: Bedenken der Patienten in Bezug auf Hypoglykämien und Gewichtszunahme
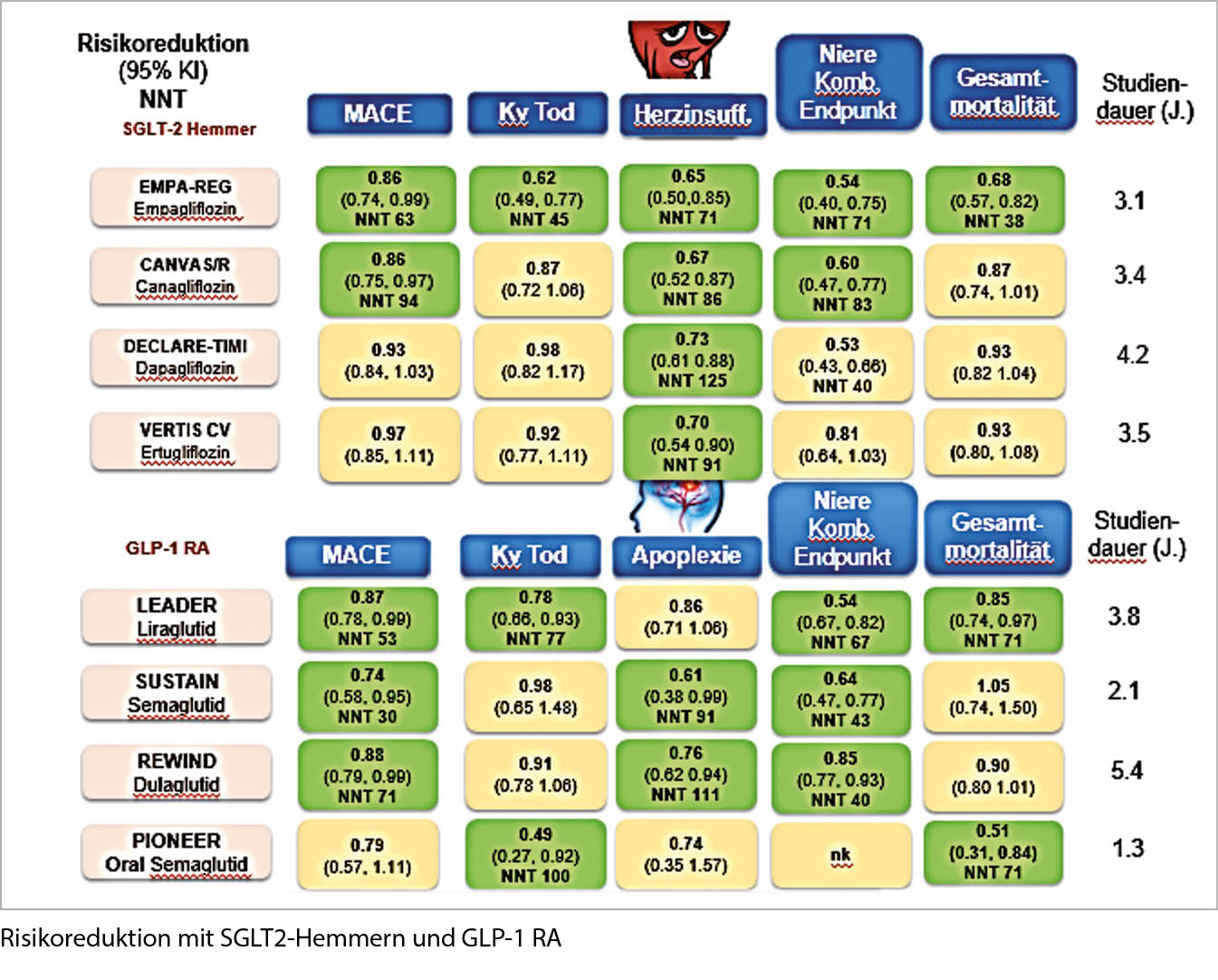
Prävention einer Nierenschädigung und Auftreten von Herzinfarkt und Schlaganfall bei T2DM durch GLP-1-RA und SGLT2-Hemmer. Die GLP-1-RA erhöhen die Insulinausschüttung und vermindern die Glukagonausschüttung, bewirken eine Appetit-Hemmung und führen zum Gewichtsverlust. Nebenwirkungen sind Übelkeit und Erbrechen. Die SGLT2-Hemmer reduzieren die Glukoserückresorption in der Niere und erhöhen dadurch die Glukoseausschüttung (-70g/Tag). Nebenwirkungen der SGLT2-Hemmer sind Infektionen des Genitaltrakts, 300-500ml grösseres Herzvolumen und Ketoazidose (Insulinmangel).
Situation in der Schweiz 2021
1. Insulinmangel? Besteht bei ca. 25 % aller Patienten. Wichtigste Frage, sollte immer gestellt werden!
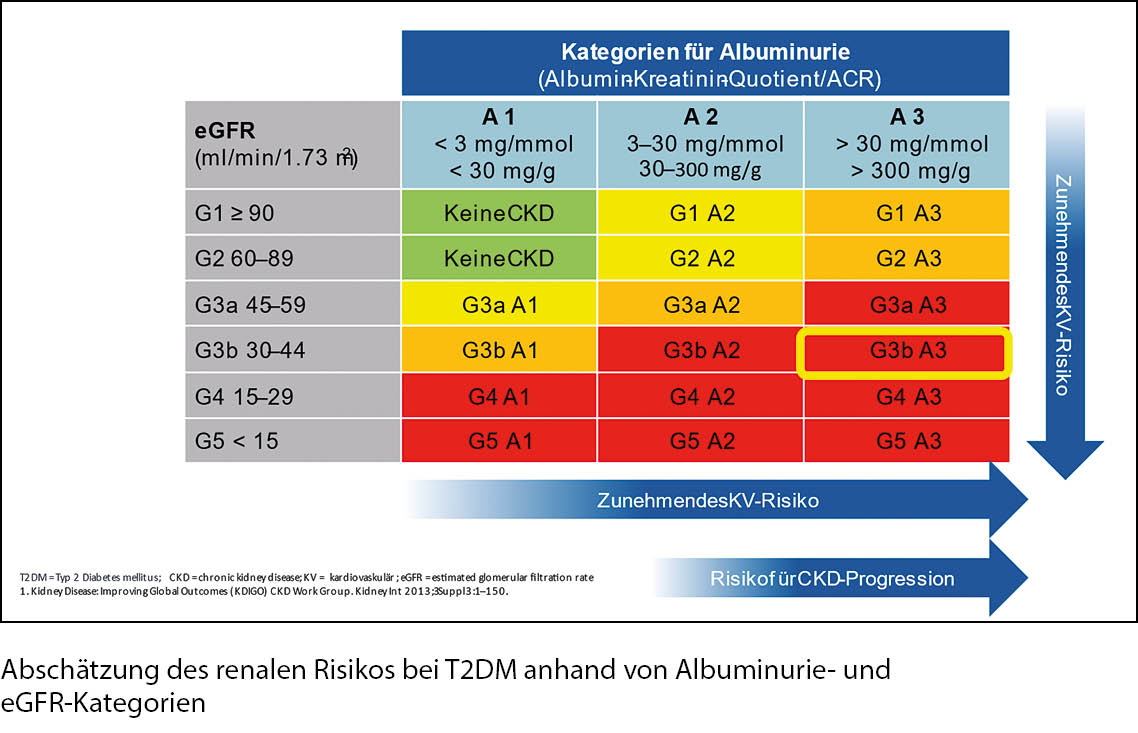
2. eGFR < 60ml/min: ca. 25 % aller Patienten. Nephroprotektion durch SGLT2-Hemmer, GLP-1-RA
3. Symptomatische kardiovaskuläre Erkrankung 25-32 %, asymptomatisch ca. 25 %.
4. Herzinsuffizienz ca. 10 % aller Patienten, bei ca. 25 % asymptomatisch, Diagnose in der Praxis schwierig, SGLT2-Hemmer. (HFrEF ¼, HFpEF ¾).
Welche initiale Therapie bei Diabetes? Metformin + SGLT2-Hemmer oder Metformin + GLP-1-RA?
Bezüglich MACE haben beide Kombinationen die gleichen Vorteile, die Nephroprotektion ist bei den SGLT2-Hemmern etwas ausgeprägter, im Hinblick auf Schlaganfall gibt es nur bei den GLP-1-RA einen Vorteil, bei Herzinsuffizienz nur bei den SGLT2-Hemmern. Der Gewichtsverlust ist bei den GLP-1-RA ausgeprägter als bei den SGLT2-Hemmern, eine orale Therapie existiert nur für die GLP-1-RA. Alle Vorteile sprechen für eine Kombination GLP-1-RA + SGLT2-Hemmer.
Stellenwert der DPP-4-Hemmer: Die Zusammenfassung der primären Endpunkte der Studien mit DPP-4-Hemmern ergibt keinen Effekt auf MACE.
Warum werden DPP-4-Hemmer trotzdem so häufig eingesetzt?
DPP-4 Hemmer senken HbA1c zuverlässig. Sie senken damit auch mikro- und makrovaskuläre Komplikationen (über einen längeren Zeitraum). DPP-4-Hemmer haben keine Nebenwirkungen und können einfach verschrieben werden. Sie verursachen keine Hypoglykämien und keine Gewichtszunahme. Sie sind eine Alternative (2. Wahl) für GLP-1-RA (BMI <28). Sie sollten langsam durch GLP-1-RA ersetzt werden (auch oral).
Essentielle Empfehlungen für Allgemeininternisten (SGED/SSED 2020)
Die Motivation für Veränderungen der Lebensgewohnheiten ist sehr wichtig. Die Behandlung soll multifaktoriell erfolgen. Als Erstlinientherapie wird Metformin + GLP-1-RA oder Metformin + SGLT2-Hemmer empfohlen, als zweite Linie + SGLT2-Hemmer (bei Erstlinie Metformin + GLP-1-
RA) bzw. + GLP-1-RA. Als dritte Linie in beiden Fällen + Basalinsulin oder Sulfonylharnstoffe (Gliclazid). Darauf Basal-Bolus Insulin oder Mischinsulin. Weiterfahren mit Metformin, SGLT2-Inhibitoren, GLP-1-RA. Stopp Sulfonylharnstoffe und DPP-4-Hemmer (bei Patienten mit tiefem bis mässigem kardiovaskulärem Risiko oder ohne Risikofaktoren können DPP-4-Hemmer oder Sulfonylharnstoffe (Gliclazid bevorzugt) angewendet werden).
Es sollen aber auch die weiteren Risikofaktoren, Hypertonie und Lipide behandelt werden. Im Vergleich mit Hypertonie und Diabetes findet LDL-Cholesterin viel weniger Beachtung (Zielerreichung 72 %, 81 % und 20 %). Eine LDL-Cholesterinsenkung über den Zeitraum von 50 Jahren (Mendel’sche Randomisierungsstudien) reduziert das relative Risiko für ein kardiovaskuläres Ereignis um ca. 50 – 55 % pro mmol/l LDL-Cholesterin.
Die Behandlung einer Dyslipidämie umfasst eine Ernährungsumstellung (weniger Kohlenhydrate und Alkohol), mehr Bewegung und falls notwendig den Einsatz eines Statins. Die Ernährungsumstellung und die Alkoholeinschränkung reduziert vor allem die Triglyzeride. Die Richtlinien der ESC empfehlen bei moderatem Risiko einen LDL-C Zielwert <2.6 mmol/l (I/A)), bei hohem Risiko < 1.8 mmol/l und mindestens 50 % LDL-Cholesterinsenkung (I/A). Bei sehr hohem Risiko <1.4 mmol/l und 50 % LDL-Cholesterinsenkung (I/B). Statine sind bevorzugte 1. Linientherapie (I/A), falls Ziel nicht erreicht, Zugabe von Ezetimibe (I/B) und bei sehr hohem Risiko bei max. Statindosis und Ezetimibe oder bei Statinunverträglichkeit die Zugabe eines PCSK9-Hemmers (I/A).
Die optimale Therapie bei unserer Patientin mit Typ-2-Diabetes mellitus umfasst einen ACE-Hemmer + Ca-Blocker (Coveram® 10/10 1-0-0), Statin + Ezetimibe (Rosuvastatin / Ezetimibe 20/10 1-0-0, Stopp DPP-4-Hemmer, Metformin + SGLT2-Inhibitor (Xigduo® XR 10/1000 1-0-0, Jardiance® Met 5/500 1-0-1, GLP-1-
RA (Ozempic® 1 mg/Woche).
Resultate nach 4 Monaten:
Blutdruck 137/76 mmHg 🙂
LDL-C 1.5 mmol/l 🙂
HbA1c 6.9 % 🙂
Gewichtsabnahme 6 kg 🙂
Der Autor deklariert Teilnahme an Advisory Boards und Referentenhonorare von Novo Nordisk, Sanofi, MSD, Boehringer Ingelheim, Servier und Astra Zeneca.
Dank Erfolgen in der translationalen und klinischen Forschung mit Fokus auf der Pathophysiologie (spezifische Tyrosinkinase-Hemmer, TKI) gerichtet auf das Onkoprotein BCR-ABL1) der chronischen myeloischen Leukämie (CML) können heute Patienten zielgerichtet und personalisiert therapiert werden. Kenntnisse zu therapieinduzierten molekularen Verläufen, deren Interpretation und Konsequenzen, der Umgang mit potenziellen Nebenwirkungen und Resistenzen auf die modernen Therapieoptionen sowie das mögliche längerfristige Ziel der therapiefreien Remission (TFR) stehen heute bei der CML im Vordergrund.
Grâce aux succès de la recherche translationnelle et clinique axée sur la pathophysiologie de la leucémie myéloïde chronique (LMC) (inhibiteurs spécifiques de tyrosine kinase (ITK) ciblant l’oncoprotéine BCR-ABL1), les patients peuvent aujourd’hui bénéficier d’un traitement ciblé et personnalisé. Les connaissances sur les évolutions moléculaires induites par le traitement, leur interprétation et leurs conséquences, la gestion des effets secondaires potentiels et des résistances aux options thérapeutiques modernes ainsi que l’objectif possible à long terme de la rémission sans traitement sont aujourd’hui au premier plan dans la LMC.
CML ist eine seltene hämatologische Erkrankung mit einer jährlichen Inzidenz von 1.0-1.5 auf 100 000 Einwohner (1). Patienten mit CML, die gut auf eine der modernen Therapieoptionen mit TKI ansprechen, haben heute eine zur Normalbevölkerung vergleichbare Lebenserwartung (2). Dadurch wird sich die Prävalenz von Patienten mit CML weltweit und auch in der Schweiz bis 2050 vervierfachen (3). Der einzige bekannte Risikofaktor, eine CML zu entwickeln, sind ionisierende Strahlen, wie sie einige Jahre nach Reaktorunfällen beobachtet wurden (4).
Die CML ist eine klonale, hämatopoietische Stammzell-Erkrankung, deren Zellen durch eine Translokation zwischen den langen Armen der Chromosomen 9 und 22 [t(9;22)(q34;q11.2)] charakterisiert ist. Zytogenetisch wird das verkürzte Chromosom 22 als Philadelphia-Chromosom (Ph) (nach dem Entdeckungsort) genannt. Die Translokation t(9;22) führt zur Fusion des ABL1 Gens mit dem BCR Gen, was zur Aktivierung der Tyrosinkinase des onkogenen Fusionsproteins BCR-ABL1 führt mit der Folge von erhöhter Proliferation, reduzierter Apoptose, abnormaler Adhäsion und Migration der klonalen Zellen sowie deren genetischer Instabilität (5). Auch wenn bei der CML noch andere Signalwege eine Rolle spielen und aktiviert sind, ist BCR-ABL1 allein ausreichend für die Entwicklung einer CML.
Diagnose und initiales Work-up
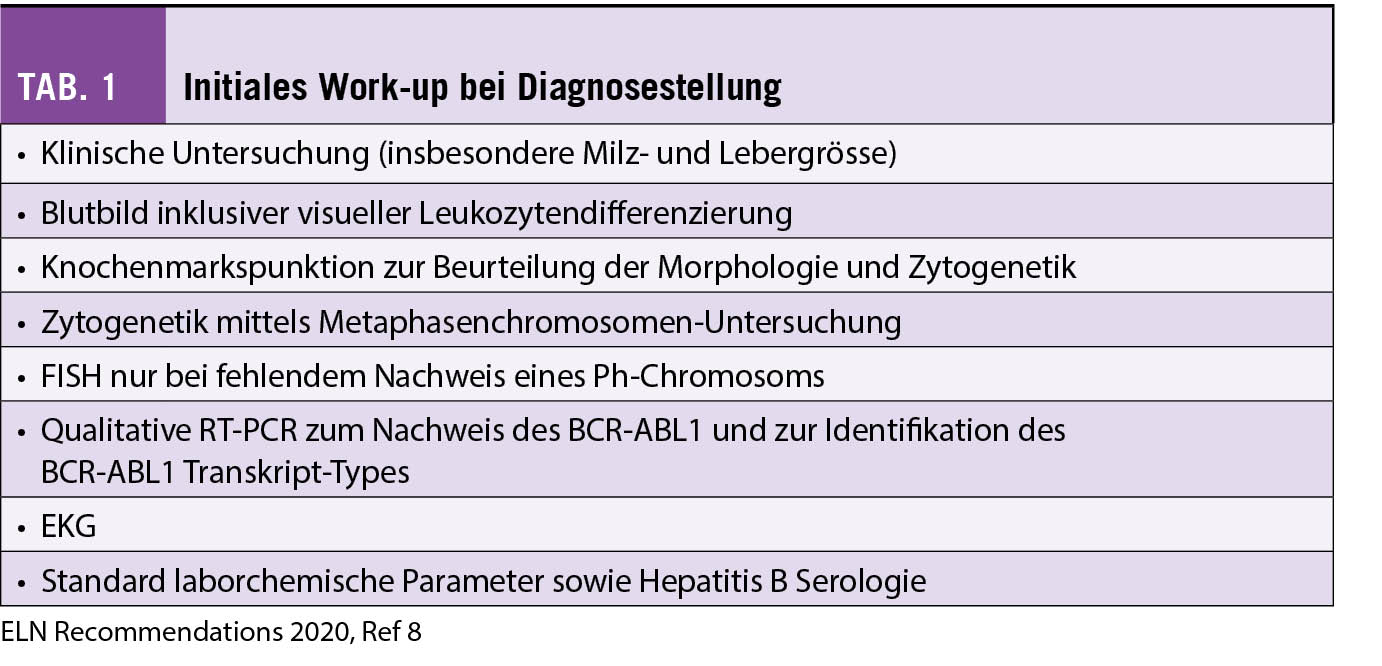
Über 50% der Patienten mit der Diagnose CML werden heute zufällig entdeckt. Symptome bei CML sind eher unspezifisch wie vermehrte Müdigkeit, Gewichtsverlust, nächtliches Schwitzen sowie bei Milzvergrösserung Völlegefühl und abdominelle Beschwerden. Ein Verdacht auf eine CML muss aufkommen, wenn sich im Blutbild eine Leukozytose mit zahlreichen myeloiden Vorstufen, eine milde Anämie und meist eine Thrombozytose zeigen. Sehr typisch ist eine Basophilie und z.T. eine Eosinophilie. Die Funktion der hämatopoietischen Zellen ist in der Regel erhalten, weshalb Patienten mit CML in einer chronischen Phase (CP) nicht ein höheres Infektions- oder Blutungsrisiko aufweisen. Zur Prognosestellung (siehe nächster Abschnitt), ist es relevant, die Blutwerte und die Grösse der Milz (in cm unter dem untersten Rippenbogen) vor Beginn der Therapie zu erfassen. Die Diagnosestellung einer CML kann mittels qualitativer RT-PCR (reverse transcription polymerase chain reaction) für BCR-ABL1 Transkripte (primär e13a2, auch b2a2 genannt, oder e14a2, auch b3a2 genannt) aus dem peripheren Blut erfolgen. Zur Bestimmung der Krankheitsphase (CP oder akzelerierte Phase (AP) oder Blastenkrise (BC)) braucht es initial eine Knochenmarkspunktion. Zur Einschätzung des Krankheitsrisikos braucht es zudem eine Zytogenetik mittels Metaphasenkaryotypisierung (mindestens 20 Metaphasen sind aus Qualitätsgründen gefordert) aus dem Knochenmark oder, bei fehlendem Aspirat, aus dem peripheren Blut (siehe Tabelle 1). In der Knochenmarksuntersuchung geht es um die Anzahl der Blasten und Promyelozyten, die Basophilie und Fibrose zur Abgrenzung einer CML in CP (in über 95% der Fälle) gegenüber der AP und BC oder anderer myeloproliferativer Erkrankungen (6). Bei der Zytogenetik geht es nebst dem Ph-Chromosom auch um zusätzliche klonale zytogenetische Aberrationen, welche prognostisch eine Bedeutung (z.B. Trisomie 8, 7q-, zusätzliches Ph-Chromosom, Isochromosom 17q mit schlechter Prognose) haben können (7). Handelt es sich um eine CML in AP oder BC, wird auch eine BCR-ABL1 Mutationsanalyse und allenfalls andere myeloide Marker empfohlen, da diese gehäuft vorkommen und relevant sind für die Prognose (8).
Risikostratifizierung
Verschiedene Risiko-Scores (Sokal, Euro, Eutos, ELTS) basierend auf initial erhobenen Befunden (Alter, Milzgrösse, Basophilie, Blastenzahl, Thrombozytose, Eosinophilie) wurden in den letzten Jahren etabliert (8). Auf der Website: https://www.kompetenznetz-leukaemie.de/content/aerzte/cml/scores/eutos_score/ und https://www.kompetenznetz-leukaemie.de/content/aerzte/cml/scores/elts/ des Kompetenznetzwerk Leukämien können diese durch Eingabe der Parameter einfach ermittelt werden. Der ELTS Score wurde basierend auf Daten von CML Patienten unter Therapie mit TKI etabliert und berücksichtigt, dass die Patienten heute meist nicht an CML-bedingten Ursachen sterben. Als Prognose-Score wird der ELTS Score deshalb primär empfohlen.
Therapieoptionen
BCR-ABL1 spezifische TKIs gelten heute als Standardtherapie für die Behandlung von neu diagnostizierten Patienten mit CML. Aktuell stehen 5 verschiedene TKIs (Imatinib, Nilotinib, Dasatinib, Bosutinib, Ponatinib) in der Schweiz zur Verfügung. Bei Imatinib handelt es sich um die Erstgeneration, Nilotinib, Dasatinib und Bosutinib gehören zur Zweitgeneration und Ponatinib zur Drittgeneration von TKIs. Die verschiedenen TKIs unterscheiden sich in ihrer Wirkungsstärke, Wirkung auf andere Kinasen, Aktivität gegen ABL1 Mutanten, Pharmakokinetik sowie dem Nebenwirkungsprofil.
Erstgeneration TKI Imatinib
Durch die IRIS Studie (International Randomized Study of Interferon and STI571) um die Jahrtausendwende, welche Imatinib 400 mg/d peroral (p.o.) mit der damaligen Standardtherapie, einer Kombination von IFN-α mit niedrig dosiertem Cytarabine, verglich, revolutionierte sich die Therapie der CML, da sich sowohl das Überleben als auch die Lebensqualität der Patienten enorm verbesserten (9). Aufgrund verschiedener Studien (CML IV, SPIRIT) zeigte sich, dass eigentlich Imatinib 600mg/d p.o. die optimale Dosis wäre, was für die Behandlung von Patienten mit CML in AP und BP auch empfohlen wird, nicht aber für Patienten in CP (400mg/d Imatinib p.o.) (10, 11). Unter Imatinib können bei einem Teil der Patienten niedrig-gradige Nebenwirkungen (z.B. Hautödeme, Muskelkrämpfe, Knochenschmerzen, Leberwerterhöhungen) auftreten, welche auf die Dauer die Lebensqualität beeinträchtigen. Diese sollten aktiv erfragt und angegangen werden; zusätzliche Langzeitnebenwirkungen unter Imatinib haben sich nicht ergeben (12). Imatinib ist zudem heute in generischer Form erhältlich und dadurch billiger als das Original.
Zweitgeneration TKI Nilotinib
Die Behandlung mit Nilotinib, ein selektiverer und potenterer Inhibitor von BCR-ABL1, führte bei Patienten mit CML in CP und AP, die auf Imatinib nicht angesprochen hatten, zu eindrücklichem Erfolg. 2005 wurde Nilotinib (2 x 400mg/d p.o.) deshalb als Zweitlinientherapie bei vorangegangener Resistenz oder Intoleranz auf TKI zugelassen (13). Aufgrund der guten Resultate von Nilotinib 2 x 300mg/d und 2 x 400mg/d im Vergleich zu Imatinib 400mg/d in der ENESTnd Studie (Evaluating Nilotinib Efficacy and Safety in Clinical Trails – Newly Diagnosed) erhielt die Therapie mit Nilotinib 2 x 300mg/d die Zulassung auch als Erstlinientherapie (14). Bei dieser Studie verbesserten sich das molekulare Ansprechen und das progressionsfreie Überleben unter beiden Dosierungen, das Gesamtüberleben aber nur mit der 2 x 400mg/d Dosierung.
Unter Nilotinib muss ein Anstieg der Leber- und Bauchspeicheldrüsenwerte, der Blutzuckerspiegel und der Lipide beachtet werden. Zudem kann es längerfristig zu arteriellen Gefässverschlüssen kommen. Deshalb ist Nilotinib weniger geeignet für Patienten mit Diabetes oder Hyperlipidämien sowie bei Prädisposition zu oder vorliegender arteriellen Gefässerkrankung (12).
Zweitgeneration TKI Dasatinib
Dasatinib, ein potenter BCR-ABL1 und SRC Hemmer, ausgetestet in verschiedenen Dosen (70 mg/d, 100 mg/d, 140 mg/d) bei Patienten mit CML, die auf Imatinib ungenügend angesprochen haben oder intolerant waren, zeigte ebenfalls erstaunlich gute Ansprechraten. Dabei kristallisierte sich eine Dosis von 100mg/d p.o. für CML in CP und 2 x 70 mg/d p.o. für CML in AP/BC und für die Zweitlinientherapie bei CML in CP heraus. Durch die Phase 3 DASISION Studie (Dasatinib versus Imatinib Study in treatment-Naive CML Patients), bei welcher 100mg/d Dasatinib mit 400 mg/d Imatinib bei neu diagnostizierten CML Patienten in CP verglichen wurde, ergab sich die Zulassung für Dasatinib 100mg/d respektive 2 x 70 mg/d in der Erstlinie für CML in CP respektive in AP/BC. Die Studie zeigte eine deutliche Verbesserung des zytogenetischen und molekularen Ansprechens, jedoch nicht des Gesamtüberlebens (15).
Dasatinib ist assoziiert mit einem gewissen Risiko für pleurale Effusionen, und gelegentlich kann auch eine pulmonal arterielle Hypertonie auftreten, weshalb Patienten mit CML und pulmonalen Vorerkrankungen eher nicht mit Dasatinib therapiert werden sollten (12).
Zweitgeneration TKI Bosutinib
Bosutinib, ein potenter BCR-ABL1 und SRC Hemmer, wurde zuerst als Drittlinientherapie in einer Dosierung von 500 mg/d p.o. zugelassen. In der Studie für die Erstlinientherapie, welche Bosutinib 500 mg/d im Vergleich zu Imatinib 400 mg/d prüfte, wurde der primäre Endpunkt, ein komplettes zytogenetisches Ansprechen, trotz des besseren molekularen Ansprechens nicht erreicht, weshalb Bosutinib dafür vorerst nicht zugelassen wurde. Möglicherweise haben ungenügende Vorkehrungen zur Milderung von vor allem gastrointestinalen Nebenwirkungen zu vermehrtem Absetzen von Bosutinib geführt und damit zu einer Unterschätzung der Unterschiede zwischen den beiden Therapiearmen. Beim Vergleich (BEFORE Studie: Bosutinib Trial in First Line Chronic Myelogenous Leukemia Treatment) von 400 mg/d Bosutinib zu 400 mg/d Imatinib ergab sich eine deutliche Verbesserung des molekularen Ansprechens nach 12 Monaten, aber keine Verbesserung im Gesamt- und progressionsfreien Überleben (16).
Bosutinib kann als Nebenwirkung zu Diarrhoe, Lebertoxizität und Niereninsuffizienz führen, weshalb Patienten mit CML und Leber- oder Nierenvorerkrankungen nicht primär mit Bosutinib behandelt werden sollten (12).
Drittgeneration TKI Ponatinib
Ponatinib wurde spezifisch zur Therapie von Patienten mit einer T315I Mutation, welche unter Therapie mit Erst- und Zweitgenerationen TKI entstehen kann und zu einer hohen Therapieresistenz führt, entwickelt. Ponatinib ist der potenteste BCR-ABL1 Hemmer mit langer Halbwertszeit, jedoch weniger selektiv, weshalb auch andere Kinasen gehemmt werden. Initial wurde Ponatinib 45 mg/d p.o. bei Patienten mit rezidivierter oder refraktärer CML in allen Phasen sowie bei rezidivierter und refraktärer Ph+ akuter lymphatischer Leukämie (ALL) ausgetestet. Das Ansprechen war stabil für die CML in CP, jedoch labiler und transienter in höheren Phasen. Aufgrund von kardiovaskulären Nebenwirkungen unter fortgesetzter Therapie mit Ponatinib 45mg/d als Erstlinientherapie in der Phase 3 PACE Studie (Ponatinib Ph+ALL and CML Evaluation), wurde diese vorzeitig sistiert (17). Die Toxizität von Ponatinib ist dosis-abhängig und deshalb wird heute eine Dosisreduktion empfohlen, sobald ein molekulares Ansprechen des BCR-ABL1 auf 1% gemäss IS (internationaler Standardisierung) erreicht wird. Diese Empfehlung basiert auf Daten aus der OPTIC (Optimizing Ponatinib Treatment In CML) Studie, die gut aufzeigen konnte, dass ein Start mit 45 mg/d Ponatinib mit einer Reduktion auf 15mg/d (ausser bei Vorliegen einer T315I Mutation) nach Erreichen eines molekularen Ansprechens des BCR-ABL1 auf 1% IS, die kardiovaskulären Nebenwirkungen deutlich reduziert bei erhaltener Wirkung (18).
Wahl des Erstlinien TKI
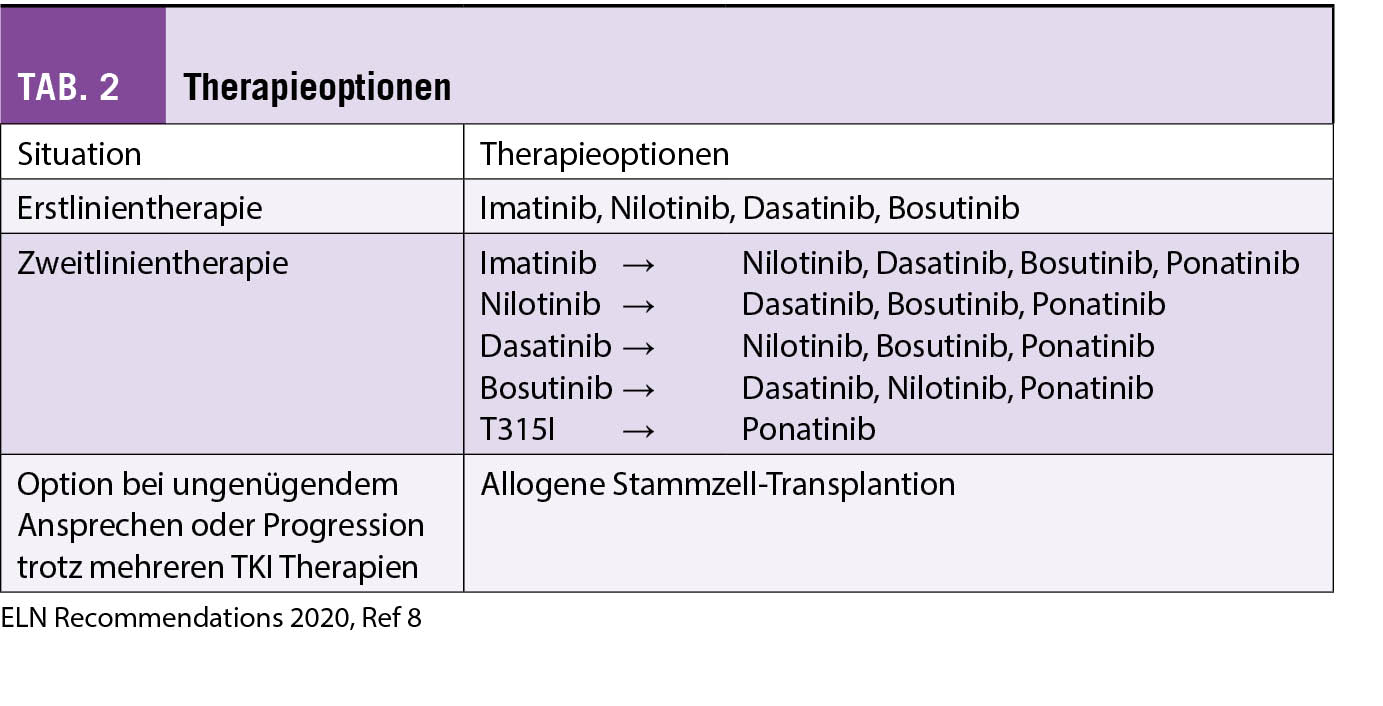
Die Wahl des Erstlinien-TKI bei Patienten mit neu diagnostizierter CML richtet sich primär nach dem Alter, dem Ziel der Therapie, den Komorbiditäten und dem potenziellen Nebenwirkungsprofil. Gemäss ELN 2020 Empfehlungen sind alle 4 TKI für die Erstlinientherapie geeignet (siehe Tabelle 2), wobei für Patienten mit einem hohen Risiko (siehe Risikostratifizierung und Zytogenetik) der Beginn mit einem Zweitgenerationen-TKI empfohlen wird (8).
Ziele, Monitoring, Meilensteine
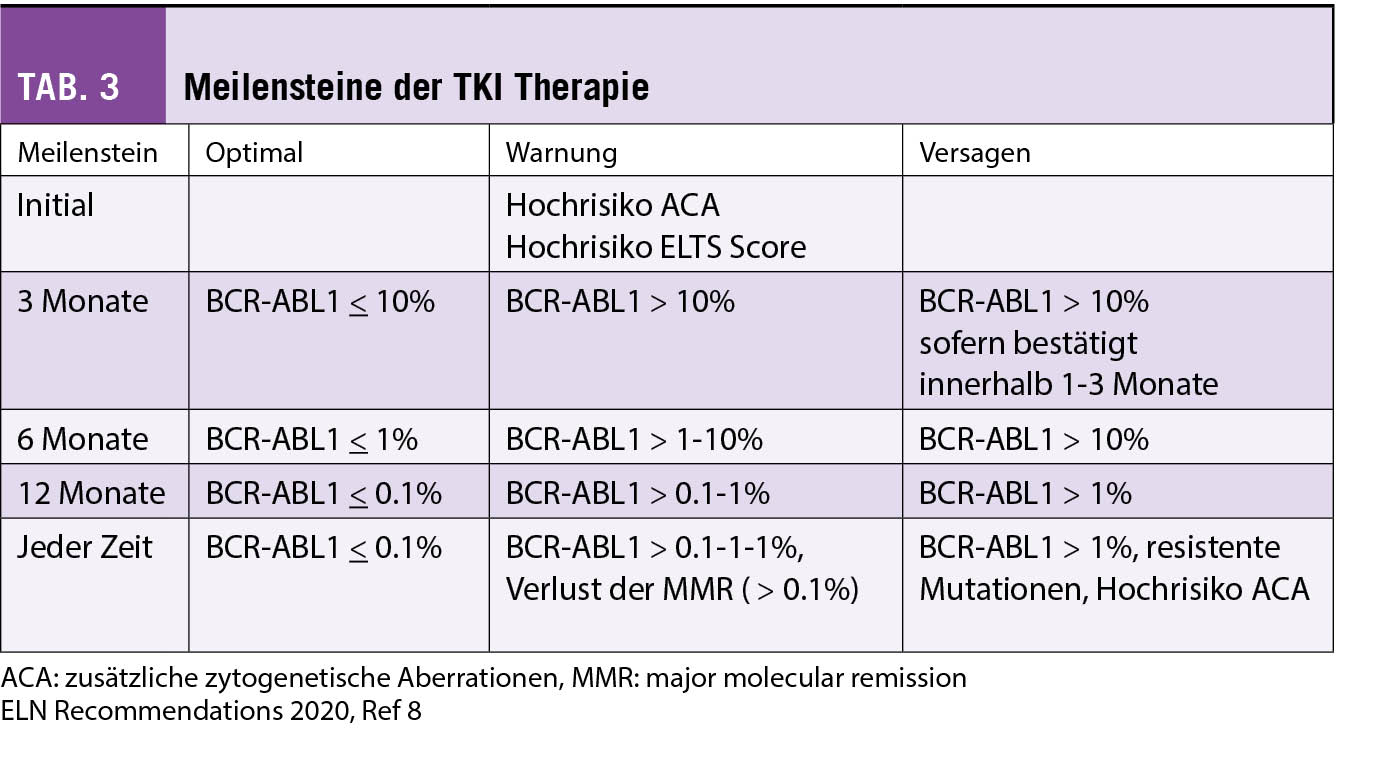
Nach Beginn der Erstlinientherapie mit einem TKI müssen das hämatologische und molekulare Ansprechen alle 3 Monate erfasst werden, damit beurteilt werden kann, ob der Patient die geforderten Ziele pro Meilenstein optimal erreicht oder ob die Therapie angepasst oder geändert werden muss. Vor einem Wechsel des TKI sollte jeweils eine ungenügende Medikamenteneinnahme ausgeschlossen werden. Nebenwirkungen, Interaktionen mit anderen Medikamenten oder Phytotherapeutika sowie vergessene Einnahmen können zu ungenügendem Therapieansprechen führen. In Tabelle 3 sind die Meilensteine und geforderten hämatologischen und molekularen Ziele dargestellt (8).
TKI-Resistenz
Spricht der Patient von Beginn weg nicht auf die TKI Therapie an, handelt es sich um eine primäre Resistenz; verliert der Patient hingegen das Ansprechen nach initialer Wirkung dann spricht man von sekundärer oder akquirierter Resistenz. Molekular kann der Verlust des Ansprechens auf Ebene der BCR-ABL1 Tyrosinkinaseaktivität (Reaktivierung) liegen oder es können BCR-ABL1-unabhängige Mechanismen dafür verantwortlich sein. Eine TKI-Resistenz ist mit einer schlechteren Prognose assoziiert.
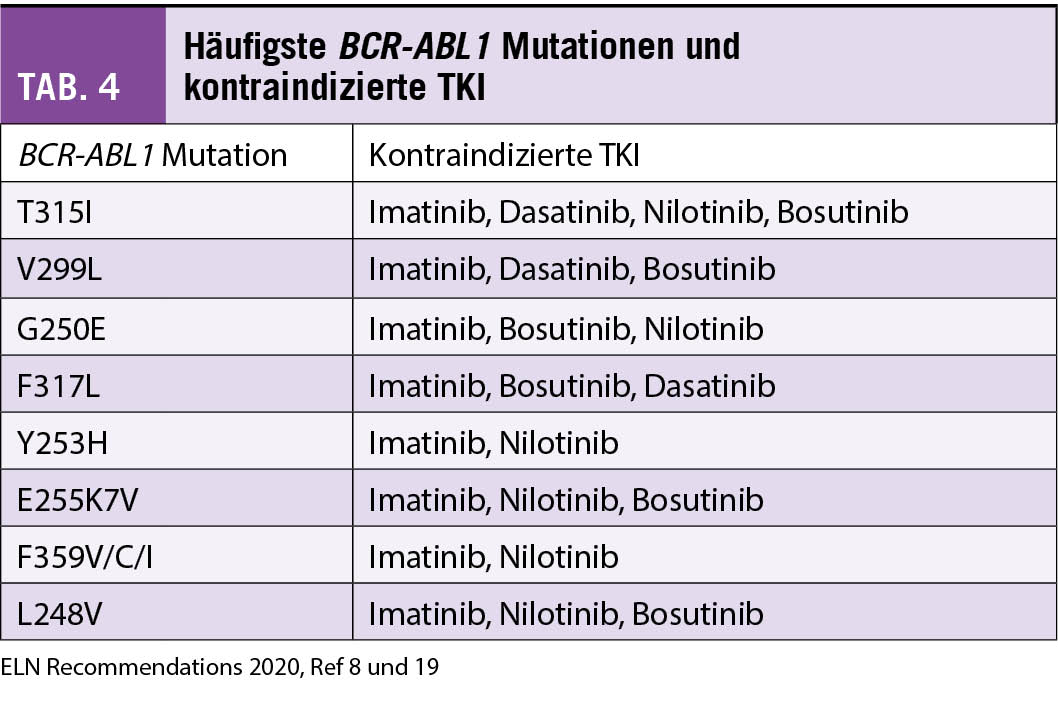
Die häufigste Ursache einer TKI-Resistenz sind Mutationen in der Kinase-Domäne des BCR-ABL1, welche die Medikamentenbindung behindern. Die häufigsten sind in Tabelle 4 aufgelistet, ebenso die jeweilig kontraindizierten TKI (8, 19). Mutationen werden häufiger bei sekundärer TKI-Resistenz oder bei CML in AP und BC gefunden. Solche müssen bei ungenügendem Ansprechen, bei Rezidiv oder Progression von einer chronischen in eine höhergradige Phase der CML gesucht werden. Diese Mutationssuche sollte mittels der sensitiveren «Next-Generation Sequencing (NGS)» Technologie erfolgen. Unter Imatinib ergibt sich das breiteste Spektrum von Mutationen; bei den Zweitgenerationen-TKI ist das Mutationsspektrum bereits eingeschränkter, allerdings sind die wenigen Mutationen oft mit höherer TKI-Resistenz assoziiert. Insbesondere ist die hoch-resistente T315I Mutation zu erwähnen, welche nur durch eine Therapie mit dem dafür entwickelten Ponatinib angegangen werden kann (siehe Tabelle 2).
Therapieoptionen bei Versagen
Bei inadäquater Wirkung oder Verlust des Ansprechens auf einen TKI sollten durch eine eingehende Anamnese sowie durch eine TKI-Plasmaspiegelbestimmung eine allfällig ungenügende Einnahme des TKI, Medikamenten-Interaktionen oder tiefere TKI-Plasma-Spiegel ausgeschlossen werden. Zudem gehört eine Standortbestimmung mit klinischer Untersuchung, Differentialblutbild, Knochenmarksuntersuchung, Zytogenetik mit Metaphasen-Karyotyp aus dem Knochenmark sowie eine BCR-ABL1 Mutationssuche dazu. Bei einer TKI-Resistenz auf Imatinib kann auf einen Zweitgenerationen-TKI gewechselt werden solange der Patient in CP ist und keine T315I Mutation aufweist. Die Wahl des Zweitgenerationen-TKI hängt bei allfälligen Mutationen von deren Profil ab. Ponatinib ist die einzige medikamentöse Option bei einer T315I Mutation; zudem sollte eine Evaluation für eine allogene hämatopoietische Stammzell-Transplantation diskutiert und evaluiert werden.
Nach Wechsel der TKI-Therapie ist das Ansprechen wiederum regelmässig gemäss den vorgesehenen Meilensteinen zu kontrollieren. Wenn Patienten eine TKI-Resistenz auf einen Zweitgenerationen-TKI entwickeln, kann auf einen anderen Zweitgenerationen-TKI gewechselt werden. Allerdings sinkt die potenzielle Ansprechrate, und ein Wechsel auf den Drittgenerationen-TKI mit möglichem besserem Ansprechen sollte in Betracht gezogen werden. Der Nachteil von Ponatinib sind die gefürchteten potenziellen vaskulären Nebenwirkungen, die allerdings deutlich reduziert werden können durch eine dosis-adaptierte Strategie (initial 45mg/d, bei Erreichen eines BCR-ABL1 Wertes von 1% IS Reduktion auf 15mg/d, ausser bei Vorliegen einer T315I Mutation) (18). Die Auswahl an medikamentösen Therapien könnte sich bei TKI-Resistenz auf einen Zweitgenerationen-TKI bald um eine Option erweitern: Asciminib (ABL001) – siehe zukünftige Therapien.
Therapie-freie Remission
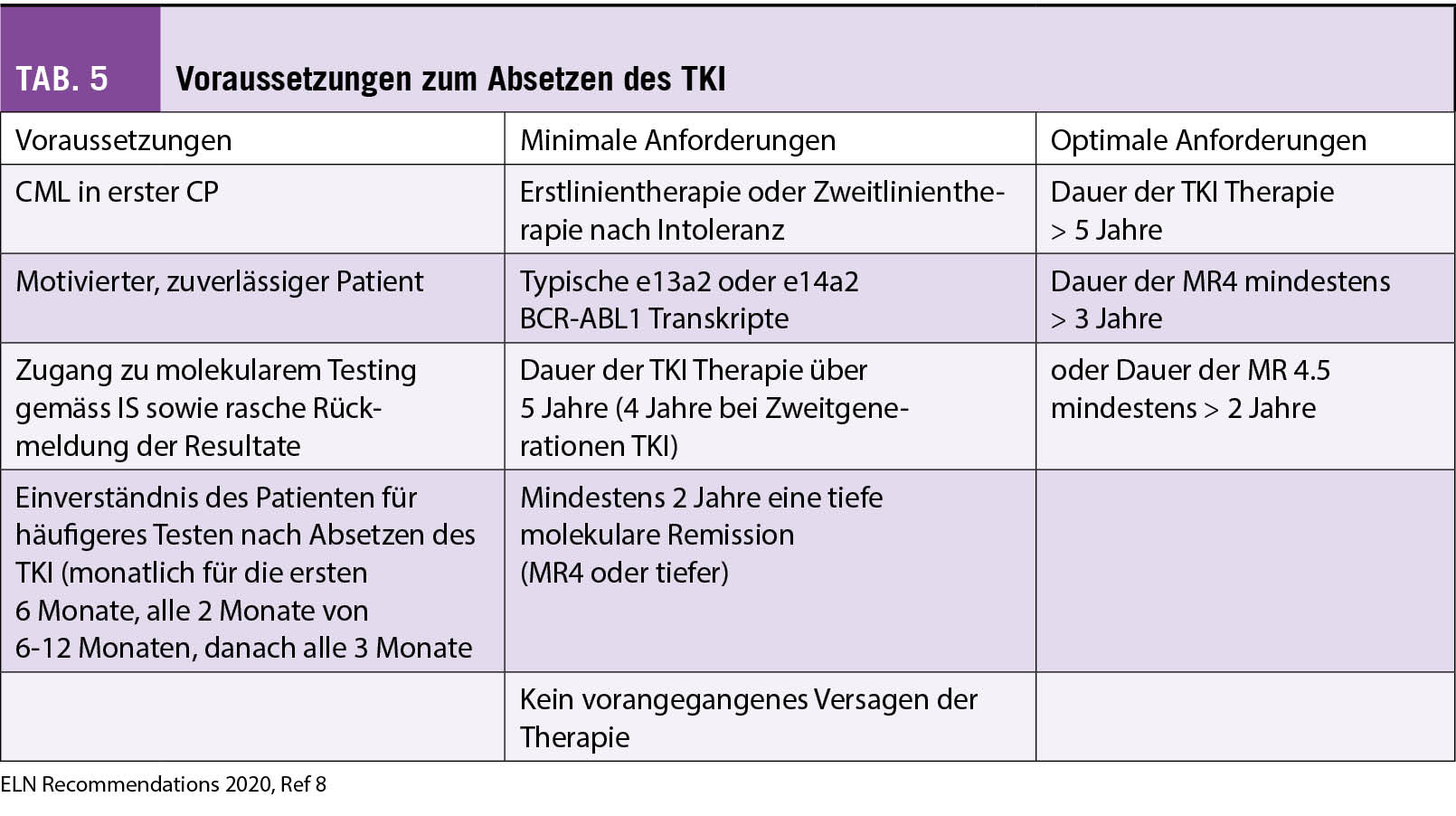
Vor allem für jüngere Patienten und um Langzeit-Nebenwirkungen unter TKI Therapie zu vermeiden, wird heute das Ziel einer TFR angestrebt (8). Durch die STIM Studien (Stopp Imatinib Studien) und weiteren Stopp TKI Studien in der Folge haben wir gelernt, dass ca. 40-60% der Patienten, welche ein BCR-ABL1 von mindestens einer MR4-4.5 IS über 2 Jahre erreicht haben, rezidiv frei bleiben (20). Längere TKI-Exposition sowie längere Dauer eines sehr tiefen molekularen Ansprechens erhöht die Wahrscheinlichkeit für eine TFR, wohingegen Patienten mit einem initial hohen Risiko-Score (Sokal) ein höheres Rezidivrisiko aufweisen. In Tabelle 5 sind die Voraussetzungen und Kriterien gemäss ELN Empfehlungen für ein TKI-Absetzen aufgelistet (8). Zweitgenerationen TKI als Erstlinientherapie führen schneller zu einer höheren Rate an Patienten mit tieferem molekularem Ansprechen, was beim Ziel TFR für den Einsatz der Zweitgenerationen-TKI spricht, jedoch können diese mit gewissen Risiken und Nebenwirkungen verbunden sein, so dass für den einzelnen Patienten eine Risiko-Nutzen Abwägung durchgeführt werden sollte. Die meisten Rückfälle nach Absetzen des TKI erfolgen in den ersten 6-12 Monaten, und bei Rezidiv führt der Wiederbeginn des entsprechenden TKI in der Regel wieder zu demselben molekularen Ansprechen. Zu beachten ist, dass ca. 20-30% der Patienten, insbesondere solche mit einer vorbestehenden Arthritis, ein TKI-Absetzsyndrom mit muskulo-skelettalen Schmerzen entwickeln können. Dieses kann mit antiinflammatorischen Schmerzmitteln und kurzzeitigem Einsatz von Steroiden behandelt werden (21).
Zukünftige Therapien
Phase 1 und 2 Studien in der Erstlinientherapie sowie Phase 3 Studien in höheren Therapielinien zeigen für Asciminib (ABL001), ein STAMP-Inhibitor (Specifically Targeting the BCR-ABL1 Myristoyl Pocket), der die Myristate-bindende Tasche des BCR-ABL1 blockiert und damit die inaktive Konformation der Tyrosinkinase stabilisiert, vielversprechende Wirksamkeit mit sehr geringer Toxizität (22). Eine Zulassung dieser Substanz ist für 2022 vorgesehen.
Bei diesem Artikel handelt es sich um einen Zweitabdruck des in «der informierte arzt» 11-2021 erschienenen Originalartikels.
Copyright bei Aerzteverlag medinfo AG
Prof. Dr. med. G. M. Baerlocher, EMBA
Assoziierte Professorin der Universität Bern
FMH Innere Medizin und Hämatologie
FAMH Hämatologie, inkl. DNA/RNA
Murtenstrasse 40
3008 Bern
gabriela.baerlocher@hematology.ch
Incyte Corporation: Beratertätigkeit, Unterstützung eines Projektes zur Fortbildung von Medizinalpersonal.
◆ Der Nachweis von BCR-ABL1 aus dem peripheren Blut bestätigt die Diagnose einer CML, die Knochenmarksuntersuchung und -zytogenetik sind für die Definition der CML Phase und des Risikoprofils nötig
◆ Die Wahl des BCR-ABL1 TKI als Erstlinientherapie hängt vom Alter und den Komorbiditäten des Patienten sowie der CML Phase und des Risikoprofils ab
◆ Relevant sind ein regelmässiges hämatologisches und molekulares Monitoring sowie eine Anpassung der Therapie bei ungenügendem Ansprechen oder Rezidiv
◆ Bei TKI Resistenz können BCR-ABL1 Mutationen vorliegen; Ponatinib ist der einzige zugelassene TKI für die Behandlung bei Vorliegen einer T315I Mutation; dabei sollte kardiovaskulären Risikofaktoren eine besondere Beachtung geschenkt werden
◆ Allfällige Kandidaten für einen TKI Absetzversuch sind optimalerweise Patienten mit einer TKI Therapie von mindestens 5 Jahren, welche eine ≤ MR4 für mehr als 3 Jahre aufweisen.
Messages à retenir
◆ La détection de BCR-ABL1 dans le sang périphérique confirme le diagnostic de LMC, l’examen de la moelle osseuse et la cytogénétique sont nécessaires pour définir la phase de la LMC et le profil de risque.
◆ Le choix de l’ITK BCR-ABL1 comme traitement de première ligne dépend de l’âge et des comorbidités du patient ainsi que de la phase de la LMC et du profil de risque.
◆ Un suivi hématologique et moléculaire régulier ainsi qu’une adaptation du traitement en cas de réponse insuffisante ou de récidive sont pertinents.
◆ En cas de résistance aux ITK, des mutations BCR-ABL1 peuvent être présentes ; le ponatinib est le seul ITK autorisé pour le traitement en présence d’une mutation T315I ; dans ce cas, les facteurs de risque cardiovasculaires doivent faire l’objet d’une attention particulière.
◆ Les éventuels candidats à un essai d’arrêt des ITK sont, de manière optimale, des patients ayant suivi un traitement par ITK pendant au moins 5 ans et présentant une rémission moléculaire profonde ≤ MR4 pendant plus de 2 ans.
1. Siegel, R.L., et al, Cancer Statistics, 2017. CA Cancer J Clin, 2017. 67(1): p. 7-30.
2. Bower, H., et al., Life Expectancy of Patients With Chronic Myeloid Leukemia Approaches the Life Expectancy of the General Population. J Clin Oncol, 2016. 34(24): p. 2851-7.
3. Delord, M., et al., The rising prevalence of chronic myeloid leukemia in France. Leuk Res, 2018. 69: p. 94-99.
4. Little M.P. , et al, Risks of leukemia in Japanese atomic bomb survivors, in women treated for cervical cancer, and in patients treated for ankylosing spondylitis. Radiat Res 1999 Sep;152(3):280-92.
5. Chereda B, et al, Natural course and biology of CML. Ann Hematol 2015 Apr;94 Suppl 2:S107-21. doi: 10.1007/s00277-015-2325-z.
6. Thiele J, et al, et al. Bone marrow features and clinical findings in chronic myeloid leukemia–a comparative, multicenter, immunohistological and morphometric study on 614 patients. Leuk Lymphoma 2000 Jan;36(3-4):295-308.
7. Fabarius A., et al, Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML Study IV. Blood 2011 Dec 22;118(26):6760-8. doi: 10.1182/blood-2011-08-373902. Epub 2011 Oct 28.
8. Hochhaus A., et al, European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020 Apr;34(4):966-984. doi: 10.1038/s41375-020-0776-2. Epub 2020 Mar 3.
9. Druker B.J. et al, Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001 Apr 5;344(14):1031-7. doi: 10.1056/ NEJM200104053441401.
10. Hoffmann V.S., et al, Systematic review and meta-analysis of standard-dose imatinib vs. high-dose imatinib and second generation tyrosine kinase inhibitors for chronic myeloid leukemia. J Cancer Res Clin Oncol 2017 Jul;143(7):1311-1318. doi: 10.1007/s00432-017-2385-7.
11. Preudhomme C, et al, Imatinib plus peginterferon alfa-2a in chronic myeloid leukemia. N Engl J Med 2010 Dec 23;363(26):2511-21.
12. Steegmann J.L., et al, European LeukemiaNet recommendations for the management and avoidance of adverse events of treatment in chronich myeloid leukaemia. Leukemia 2016;30(8):1648-71
13. Kantarjian H., et al, Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med 2006 Jun 15;354(24):2542-51. doi: 10.1056/NEJMoa055104.
14. Saglio G., et al, Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010 Jun 17;362(24):2251-9. doi: 10.1056/NEJMoa0912614.
15. Kantarjian H.M., et al, Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION). Blood 2012 Feb 2;119(5):1123-9. doi: 10.1182/blood-2011-08-376087. Epub 2011 Dec 9.
16. Cortes J.E., et al, Bosutinib Versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia: Results From the Randomized BFORE Trial. J Clin Oncol 2018 Jan 20;36(3):231-237.
17. Lipton J.H., et al, Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: an international, randomised, open-label, phase 3 trial, Lancet Oncol 2016 May;17(5):612-21. doi: 10.1016/S1470-2045(16)00080-2. Epub 2016 Apr 12.
18. Kantarjian H.M., et al, Efficacy and safety of ponatinib (PON) in patient with chronic-phase chronic Myeloid Leukemia (CP-CML) who failed one ore more second-generation (2G) tyrosine kinase inhibitors (TKIs): Analyses based on PACE and OPTIC. Blood 2020;136(Supplement 1):43-4
19. Shah N.P., et al, Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia, Cancer Cell 2002 Aug;2(2):117-25. doi: 10.1016/s1535-6108(02)00096-x.
20. Mahon F.X., et al, Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial, Lancet Oncol 2010 Nov;11(11):1029-35. doi: 10.1016/S1470-2045(10)70233-3.
21. Richter J., et al, Muskuloskeletal pain in patients with chronic myeloid leukemia after discontinuation of imatinib: a tyrosine kinase inhibitor withdrawal syndrome?, J Clin Oncol 2014;32(25):2821-3
22. Hughes T.P., et al, Asciminib in chronic Myeloid Leukmeia after ABL kinase inhibitor failure. N Engl J Med 2019;381(24):2315-26
Die intestinale Mikrobiota, das «Mikrobiom», ist in den
Fokus des medizinischen, wissenschaftlichen und öffentlichen Interesses gerückt. Die Darm-Mikrobiota spielt eine grosse Rolle bei verschiedenen Erkrankungen, nicht nur des Intestinums, sondern auch in der psychiatrischen, kardiologischen, rheumatologischen und onkowlogischen Praxis. Im Folgenden werden aktuelle Studienergebnisse zu den Einflüssen der Ernährung auf die Mikrobiota-Zusammensetzung des menschlichen Darms beleuchtet.
Le microbiote intestinal, le «microbiome», est devenu un centre d’intérêt médical, scientifique et public. Les microbiotes intestinaux jouent un grand rôle dans diverses maladies, non seulement intestinales, mais aussi psychiatriques, cardiologiques, rhumatologiques et oncologiques. Les résultats d’études récentes sur l’influence de l’alimentation sur la composition du microbiote intestinal humain sont examinés ci-dessous.
Die Zusammensetzung der Darm-Mikrobiota ist bei einer Vielzahl von Erkrankungen verändert. Dies ist nicht nur bei Darmerkrankungen, wie z.B. dem Reizdarmsyndrom oder chronisch entzündlichen Darmerkrankungen der Fall, sondern auch bei psychiatrischen Erkrankungen, wie z.B. Depression oder Autismus, bei kardialen Erkrankungen wie bei der koronaren Herzkrankheit oder der Herzinsuffizienz, bei Erkrankungen des rheumatologischen Formenkreises und bei Tumorerkrankungen. Eine grosse Zahl an Laboratorien hat die Zeichen der Zeit erkannt und bietet «Stuhlanalysen» oder «Mikrobiota-Analysen» zu teilweise erheblichen Kosten an. Es werden dann häufig gleichzeitig mit der Analyse der Mikrobiota-Veränderungen Ernährungsempfehlungen an die Patientinnen weitergegeben, die eine diagnostizierte «Dysbiose» wieder ins Gleichgewicht bringen soll. Wie ist aber die Evidenz dafür, dass die Mikrobiota-Zusammensetzung durch Ernährung beeinflusst werden kann? Besteht die Möglichkeit, durch Ernährungsumstellungen gezielt in das intestinale Mikrobiota-Ökosystem einzugreifen?
Mikrobiota und Mikrobiom, Mycobiom, Virom – einige Fakten
Die normale Darm-Mikrobiota des Menschen besteht neben den Bakterien aus eukaryontischen Pilzen, Viren und einigen Archaeen, die vorwiegend den unteren Darmtrakt besiedeln (1). Während die Zusammensetzung der Bakterien inzwischen recht gut untersucht ist, wissen wir noch wenig über das Virom, über die Bakteriophagen und das Mycobiom. Bis zu 100 Billionen (1014) Mikroorganismen pro Mensch besiedeln den Darm und machen etwa 2 kg des Körpergewichts aus (2). Bei den heutigen Bakterienkultur-unabhängigen Analyse-Methoden werden hauptsächlich Variationen von Genen (meist der 16sRNA) sequenziert, die einerseits bei allen Bakterien gemeinsam sind und in der Evolution stark konserviert wurden, andererseits jedoch mit speziesspezifischen Unterschieden behaftet sind (3-6). Wenn wir durch Sequenzierung die genetische Zusammensetzung der Mikrobiota (Mikrobiom = Gesamtheit aller Bakteriengene im Darm) bestimmen, wissen wir allerdings noch gar nichts über die Funktion der Bakterien in einem spezifischen, individuellen, menschlichen Darm-Ökosystem. Dieselbe Bakterienspezies kann in unterschiedlichen Menschen unterschiedlich Funktionen übernehmen. Das metabolische Profil verschiedener Bakterienspezies hängt von deren Umgebung und der Funktion anderer Spezies in der Umgebung ab. Daher sagen die allermeisten der kommerziell durchgeführten Mikrobiota-Untersuchungen überhaupt nichts darüber aus, wie sich jemand ernähren sollte. Die meisten Stuhlbakterien-Analysen werden nach unkontrollierten Entnahmebedingungen und unkontrollierten Transportbedingungen durchgeführt. Die allermeisten Spezies der menschlichen Darmmikrobiota sind obligate Anaerobier, die rasch sterben, während fakultative Anaerobier und Aerobier auch bei Raum(Transport-)Temperatur weiterwachsen können. Entsprechende Empfehlungen sind nicht Evidenz-basiert.
Neben genetischen Einflüssen sind Umwelteinflüsse wie die Ernährung von Bedeutung für die Zusammensetzung der menschlichen Mikrobiota (7-11). Die am häufigsten vorkommenden Phyla (Stämme) des menschlichen Gastrointestinaltraktes sind Actinobacteria, Bacteriodetes, Firmicutes und Proteobacteria (12). Während diese grossen Phyla allen Menschen gemeinsam sind, gibt es ein hohes Mass an interindividueller Variabilität der Darm-Mikrobiota-Zusammensetzung auf niedrigeren taxonomischen Ebenen.
Einflüsse der Ernährung auf die Mikrobiota-Zusammensetzung in der frühen Kindheit
Es wird derzeit diskutiert, ob nicht bereits in utero diätetische Einflüsse auf die Zusammensetzung der Darmmikrobiota vorhanden sind (12, 13). Die Ernährung von Müttern in der Schwangerschaft hat einen signifikanten Einfluss auf das Risiko einer Dysbiose oder von Stoffwechselerkrankungen der Nachkommen (13). Die Darmmikrobiota wird dabei als potenzieller vermittelnder Faktor angesehen, zumal die intrauterine Umgebung nicht steril ist, was darauf hindeutet, dass eine mütterlich-fetale Übertragung von Mikrobiota-Komponenten während der Schwangerschaft auftreten kann (12, 13). So wurden die Bakterien der Phyla Firmicutes, Tenericutes, Proteobacteria, Bacteriodetes und Fusobacteria in der menschlichen Plazenta gefunden (14).
Nach der Geburt unterscheidet sich die Darm-Mikrobiota von gestillten Säuglingen deutlich von derjenigen von nicht gestillten Babys. Gesunde Säuglinge, die in den ersten 6 Lebensmonaten gestillt wurden, zeigten eine signifikante Zunahme von Actinobacteria (wie z.B. Bifidobacterium) und Proteobacteria (Enterobacteriaceae) (15). Dies wurde in mehreren Untersuchungen bestätigt: Bei gestillten Säuglingen findet sich in den ersten Lebenswochen eine starke Zunahme von Bifidobacterien, während mit Säuglingsnahrung ernährte Kinder hohe Anteile von Klebsiella und Serratia aufwiesen (16). Bei Frühgeborenen auf einer Neugeborenen-Intensivstation zeigte sich ein erhöhter Staphylokokken-Anteil an der Dammikrobiota im Vergleich zu gesunden Säuglingen, und die Besiedlung mit Bifidobacteriaceae war verzögert (17). Eine längere Stilldauer begünstigt die Proliferation von Bifidobacterium und Veillonella und verringert die Menge Lachnospiraceae, Ruminococcaceae, und anderen selteneren Bakterien (18). Neben der Ernährung hat allerdings auch die Art der Geburt einen grossen Einfluss auf die Mikrobiota-Zusammensetzung der Säuglinge. Bei vaginaler Geburt wird schnell das intestinale Mikrobiom der Mutter aufgenommen und im Dickdarm etabliert. Eine Sectio stört die Übertragung der Mikrobiota von der Mutter auf den Säugling, eine fäkale Mikrobiota Übertragung (FMT) kann die «natürliche» Darmmikrobiota Sectio-geborener Säuglinge postnatal wiederherstellen (19).
Mit der Einführung fester Nahrung verändert sich die Darmmikrobiota bei Säuglingen wieder. Die mikrobielle Diversität ist höher bei 9 Monate alten Kleinkindern mit einer protein- und ballaststoffreichen und relativ fettarmen Diät (18). Eine positive Korrelation wurde zudem zwischen Proteinaufnahme und Vorhandensein von Lachnospiraceae gefunden, während eine negative Korrelation mit Bifidobacteriaceae zu existieren scheint. Nach Beendigung des Stillens geht der relative Anteil Bifidobacteriaceae zurück (18). Die Aufnahme von Ballaststoffen war positiv mit der Häufigkeit von Pasteurellaceae korreliert (18). Diese Daten legen nahe, dass die Zunahme der Darm-Mikrobiota-Diversität vom Säuglingsalter zur Kindheit stark durch Änderungen der Ernährung beeinflusst wird (12).
Einflüsse von spezifischen Diätmustern auf die Mikrobiota-Zusammensetzung beim Erwachsenen
Auch beim Erwachsenen wurden signifikante Einflüsse der Zusammensetzung der Ernährung auf die Zusammensetzung der Mikrobiota berichtet. Die Darmmikrobiota reagiert schnell auf Ernährungsumstellungen. Es wurde postuliert, dass die Ernährung in der Lage ist, fast 60% der Mikrobiota zu ändern (20). Beim Menschen werden auch die dominanten Phyla Bakterioides, Firmicutes und Actinobacteria beeinflusst (21). Es werden drei «Enterotypen» im Hinblick auf die Zusammensetzung der Mikrobiota unterschieden (Tab. 1): Der Enterotyp 1 ist ein Bacteroides-dominanter Typ. Eine Diät, die vor allem tierische Proteine und gesättigte Fette enthält (was auch häufig mit dem Terminus «Westernized Diet» in der Literatur umschrieben wird), führt zu diesem Enterotyp, der wie erwähnt von Bacteroides dominiert wird (22). Eine Ernährung, die vor allem auf Kohlenhydrate aufbaut, führt zu dem Prevotella-dominanten Enterotyp 2 (22). Als Enterotyp 3 wird eine Ruminococcus-dominante Darmmikrobiota bezeichnet. Diese Mikroorganismen spalten effizient Zucker und Muzine. Normalerweise machen die Phyla Firmicutes und Bacteroidetes 90% der Darmmikrobiota aus. Das Phylum Firmicutes besteht hauptsächlich aus den Gattungen Clostridium, Enterococcus, Lactobacillus und Ruminococcus. Die Hauptgattungen des Phylums Bacteroides sind Prevotella und Bacteroides. Während eine «Westernized Diet» vorwiegend tierisches Protein und gesättigte Fettsäuren enthält, ist eine mediterrane Diät durch eine hohe Menge an Ballaststoffen, ungesättigten Fettsäuren und auch von Polyphenolen gekennzeichnet (für die eine präbiotische Wirkung auf bestimmte Bakterien-Stämme postuliert wird) (23-29). Das NU-AGE-Projekt untersuchte daher, ob eine einjährige mediterrane Diät-Intervention die Darmmikrobiota verändern kann und gesundheitsfördernd wirkt. Bei 612 Probanden in fünf europäischen Ländern (UK, Frankreich, Niederlande, Italien und Polen) wurde die Mikrobiota-Zusammensetzung vor und nach einer 12-monatigen mediterranen Diät analysiert. Eine hohe Diät-Adhärenz war mit spezifischen Mikrobiom-Änderungen assoziiert (29). Bakterielle Taxa, die durch die Einhaltung der Diät angereichert wurden waren positiv mit mehreren Gesundheits-Markern und verbesserten kognitiven Funktionen verbunden, und waren negativ mit Entzündungsmarkern, wie C-reaktivem Protein und Interleukin-17 assoziiert (29). Die Analyse der mikrobiellen Metaboliten zeigte, dass die ernährungsmodulierte Mikrobiomveränderung mit einer Zunahme der kurz-/verzweigtkettigen Fettsäureproduktion und einer geringeren Produktion von sekundären Gallensäuren, p-Cresolen, Ethanol und Kohlendioxid verbunden war (29). Ähnliche Studien kamen zu vergleichbaren Resultaten (28, 30-33). Bei Übergewichtigen führte eine mediterrane Diät zu einer Zunahme des Ballaststoff-abbauenden Bakteriums Faecalibacterium prausnitzii und zur vermehrten Bildung von kurzkettigen Fettsäuren (32). Eine mediterrane Diät scheint also mit einer günstigen Änderung der Mikrobiota verbunden zu sein, was ihre gesundheitsfördernden Eigenschaften teilweise erklären mag. Während sich die Literatur zu den Effekten einer mediterranen Diät weitgehend einig ist, ist das bei einer vegetarischen Diät weniger der Fall: Die Mikrobiota-Zusammensetzung von Vegetariern scheint sich aber gegenüber der von Omnivoren zu unterscheiden (zumindest in den meisten Studien, siehe 34-37). Menschen, die längere Zeit (> 3 Monate) eine vegetarische Diät einhalten, weisen z.B. einen grösseren relativen Anteil von Bacteriodes-Prevotella, Bacteriodes thetaiotamicron und Clostridium clostridioforme, aber weniger Clostridium coccoides im Stuhl im Vergleich zu Omnivoren auf (37). Allerdings konnte eine systemtische Übersichtsarbeit keine konsistente Korrelation zwischen veganer Ernährung oder vegetarischer Ernährung und der Mikrobiota-Zusammensetzung im Vergleich zu Omnivoren finden (38). In einer eigenen Studie bei Patienten mit chronisch entzündlichen Darmerkrankungen sahen wir signifikante Unterschiede zwischen Patienten mit vegetarischer Ernährung, Gluten-freier Diät und omnivorer Ernährung (39). Diese waren aber nicht mit einem besseren Krankheitsverlauf oder einer Steigerung des Wohlbefindens bzw. einer Reduktion von depressiven Verstimmungen assoziiert (38). Wie erwähnt sagt das Vorhandensein bestimmter Bakterienarten nichts über deren jeweilige spezifische Funktion aus.
Unterschiedliche Diäteinflüsse je nach Grunderkrankung?
Zu bedenken ist zudem, dass Ernährungsinterventionen oder spezifische Diäten je nach Grunderkrankung unterschiedliche Effekte auf die Mikrobiota-Zusammensetzung haben können. Bei Zöliakie-Patienten findet sich eine Reduktion probiotischer Spezies, wie z.B. Lactobacillus und Bifidobakterien, und eine relative Zunahme entzündungsfördernder Bakterien der Gattung Veillonaceae (40). Zöliakie-Patienten müssen eine lebenslange Gluten-freie Diät einhalten. Eine Gluten-freie Diät ist aber aufgrund von populärwissenschaftlichen Hypothesen nun auch bei Menschen mit NCGS (nicht-Zöliakie-Glutensensitivität) und bei Gesunden weit verbreitet. Wenn man diese drei Gruppen untersucht, zeigt sich, dass die Gluten-freie Diät bei allen den Bakterienreichtum reduziert und gleichzeitig die Zusammensetzung der Darmmikrobiota je nach Grunderkrankung (asymptomatische Probanden) und Krankheitszustand (Zöliakie und NCGS) unterschiedlich beeinflusst (40): Bei gesunden Probanden führt eine Gluten-freie Diät zur Depletion nützlicher Spezies, z.B. Bifidobakterien, zugunsten opportunistischer Pathogene, z.B. Enterobacteriaceae und Escherichia coli (40). Im Gegensatz dazu führt eine Gluten-freie Diät bei Zöliakie und NCGS zur Wiederherstellung der Diversität der Mikrobiota-Population und zur Verringerung entzündungsfördernder Spezies (40).
Zusammenfassung
Wir stehen erst am Anfang des Verständnisses der komplexen Interaktionen zwischen unserer Mikrobiota und unseren Körperfunktionen. Schon jetzt ist aber klar, dass die Mikrobiota-Zusammensetzung unseres Darmes und deren Produktion von Metaboliten Einfluss auf Gesundheitsrisiken, entzündliche Erkrankungen, kardiovaskuläre Erkrankungen und auch psychiatrische Erkrankungen hat. Die Zusammensetzung der Darm-Mikrobiota wird durch Umweltfaktoren bestimmt. Daher liegt es nahe, den Einfluss der Ernährung auf das Mikrobiom zu untersuchen. Viele solcher Einflüsse wurden festgestellt. Stillen nach einer vaginalen Entbindung führt zu einem diverseren Mikrobiom. Eine mediterrane Diät führt zu einer diverseren und günstigeren Mikrobiota. Dagegen hat eine vegetarische oder vegane Diät keinen konsistenten Einfluss. Wichtig ist darüber hinaus, dass eine Ernährungsintervention je nach Grunderkrankung unterschiedliche Auswirkungen haben kann. Eine Gluten-freie Diät führt bei Zöliakie-Patienten zu einer «Verbesserung» der Mikrobiota, bei Gesunden jedoch zu negativen Auswirkungen. Diätempfehlungen, wie sie von einschlägigen Labors auf der Basis einer individuellen Stuhlprobensequenzierung gemacht werden, sind nicht evidenzbasiert und können im Einzelfall durchaus schädlich sein. Patientinnen sollte abgeraten werden, für diese Untersuchungen sinnlos Geld auszugeben.
Copyright bei Aerzteverlag medinfo AG
Prof. Dr. med. Dr. phil. Gerhard Rogler
Klinik für Gastroenterologie und Hepatologie
UniversitätsSpital Zürich
Rämistrasse 100
8091 Zürich
gerhard.rogler@usz.ch
Consulting und Advisory Boards für Abbvie, Arena, Astra Zeneca, Augurix, BMS, Boehringer, Calypso, Celgene, FALK, Ferring, Fisher, Genentech, Gilead, Janssen, MSD, Novartis, Pfizer, Phadia, Roche, UCB, Takeda, Tillots, Vifor, Vital Solutions und Zeller; Vortragshonorare von Astra Zeneca, Abbvie, BMS, FALK, Janssen, MSD, Pfizer, Phadia, Takeda, Tillots, UCB, Vifor und Zeller; Grant Support von Abbvie, Ardeypharm, Augurix, Calypso, FALK, Flamentera, MSD, Novartis, Pfizer, Roche, Takeda, Tillots, UCB und Zeller. Co-Founder von PharmaBiome, einem Start-up zur Mikrobiomtherapie.
◆ Die Zusammensetzung der Darm-Mikrobiota wird durch Umweltfaktoren, wie z.B. unsere Diät mitbestimmt.
◆ Eine mediterrane Diät ist jedoch die einzige Ernährungsform, bei der sich die Studien einig sind, dass sie zu einer günstigen Mikrobiota-Zusammensetzung führt (Enterotyp 2).
◆ Gesunde sollten keine restriktiven Diäten (wie z.B. Gluten-freie Ernährung) durchführen. Dies kann – im Gegensatz zu Patienten – zu einer Dysbiose des Mikrobioms führen.
◆ Mikrobiomuntersuchungen durch kommerzielle Anbieter sind häufig aufgrund der Entnahmebedingungen wenig aussagekräftig. Darauf basierende Ernährungsempfehlungen sind nicht sinnvoll und entbehren einer soliden Evidenz.
Messages à retenir
◆ La composition du microbiote intestinal est en partie déterminée par des facteurs environnementaux, tels que notre régime alimentaire.
◆ Cependant, un régime méditerranéen est le seul type d’alimentation pour lequel les études s’accordent à dire qu’il conduit à une composition favorable du microbiote (entérotype 2).
◆ Les personnes en bonne santé ne devraient pas suivre de régime restrictif (comme par exemple un régime sans gluten). Cela peut – contrairement aux patients – entraîner une dysbiose du microbiote.
◆ Les analyses du microbiome effectuées par des prestataires commerciaux sont souvent peu pertinentes en raison des conditions de prélèvement. Les recommandations nutritionnelles basées sur ces résultats n’ont pas de sens et manquent de preuves solides.
1. Hollister EB, Gao C, Versalovic J. Compositional and functional features of the gastrointestinal microbiome and their effects on human health. Gastroenterology 2014;146:1449-58.
2. Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science 2005;308:1635-8.
3. Scanlan PD, Marchesi JR. Micro-eukaryotic diversity of the human distal gut
microbiota: qualitative assessment using culture-dependent and -independent analysis of faeces. ISME J 2008;2:1183-93.
4. Scanlan PD, Shanahan F, O’Mahony C, et al. Culture-independent analyses of temporal variation of the dominant fecal microbiota and targeted bacterial subgroups in Crohn’s disease. J Clin Microbiol 2006;44:3980-8.
5. Dekio I, Hayashi H, Sakamoto M, et al. Detection of potentially novel bacterial components of the human skin microbiota using culture-independent molecular profiling. J Med Microbiol 2005;54:1231-1238.
6. Sakata S, Tonooka T, Ishizeki S, et al. Culture-independent analysis of fecal
microbiota in infants, with special reference to Bifidobacterium species. FEMS
Microbiol Lett 2005;243:417-23.
7. Rehman A, Sina C, Gavrilova O, et al. Nod2 is essential for temporal development of intestinal microbial communities. Gut 2011;60:1354-62.
8. Backhed F, Ley RE, Sonnenburg JL, et al. Host-bacterial mutualism in the human intestine. Science 2005;307:1915-20.
9. Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science 2001;292:1115-8.
10. Olivares M, Laparra JM, Sanz Y. Host genotype, intestinal microbiota and inflammatory disorders. Br J Nutr 2013;109 Suppl 2:S76-80.
11. Ventura M, Turroni F, Motherway MO, et al. Host-microbe interactions that facilitate gut colonization by commensal bifidobacteria. Trends Microbiol 2012;20:467-76.
12. Shock T, Badang L, Ferguson B, et al. The interplay between diet, gut microbes, and host epigenetics in health and disease. J Nutr Biochem 2021;95:108631.
13. Chu DM, Meyer KM, Prince AL, et al. Impact of maternal nutrition in pregnancy and lactation on offspring gut microbial composition and function. Gut Microbes 2016;7:459-470.
14. Aagaard K, Ma J, Antony KM, et al. The placenta harbors a unique microbiome. Sci Transl Med 2014;6:237ra65.
15. Eggesbo M, Moen B, Peddada S, et al. Development of gut microbiota in infants not exposed to medical interventions. APMIS 2011;119:17-35.
16. Ku HJ, Kim YT, Lee JH. Microbiome Study of Initial Gut Microbiota from Newborn Infants to Children Reveals that Diet Determines Its Compositional Development.
J Microbiol Biotechnol 2020;30:1067-1071.
17. Tauchi H, Yahagi K, Yamauchi T, et al. Gut microbiota development of preterm infants hospitalised in intensive care units. Benef Microbes 2019;10:641-651.
18. Laursen MF, Andersen LB, Michaelsen KF, et al. Infant Gut Microbiota Development Is Driven by Transition to Family Foods Independent of Maternal Obesity. mSphere 2016;1.
19. Korpela K, Helve O, Kolho KL, et al. Maternal Fecal Microbiota Transplantation in Cesarean-Born Infants Rapidly Restores Normal Gut Microbial Development: A Proof-of-Concept Study. Cell 2020;183:324-334 e5.
20. Zhang C, Zhang M, Wang S, et al. Interactions between gut microbiota, host
genetics and diet relevant to development of metabolic syndromes in mice.
ISME J 2010;4:232-41.
21. Merra G, Noce A, Marrone G, et al. Influence of Mediterranean Diet on Human Gut Microbiota. Nutrients 2020;13.
22. Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome. Nature 2011;473:174-80.
23. Andre P, Pais de Barros JP, Mj Merle B, et al. Mediterranean diet and prudent diet are both associated with low circulating esterified 3-hydroxy fatty acids, a proxy of LPS burden, among older adults. Am J Clin Nutr 2021.
24. Bailey MA, Holscher HD. Microbiome-Mediated Effects of the Mediterranean
Diet on Inflammation. Adv Nutr 2018;9:193-206.
25. Bifulco M. Mediterranean diet: the missing link between gut microbiota and inflammatory diseases. Eur J Clin Nutr 2015;69:1078.
26. Cani PD, Van Hul M. Mediterranean diet, gut microbiota and health: when age and calories do not add up! Gut 2020;69:1167-1168.
27. Childs CE. From the Mediterranean Diet to the Microbiome. J Nutr 2018;148:819-820.
28. De Filippis F, Pellegrini N, Vannini L, et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2016;65:1812-1821.
29. Ghosh TS, Rampelli S, Jeffery IB, et al. Mediterranean diet intervention alters the gut microbiome in older people reducing frailty and improving health
status: the NU-AGE 1-year dietary intervention across five European countries. Gut 2020;69:1218-1228.
30. Gutierrez-Diaz I, Fernandez-Navarro T, Salazar N, et al. Adherence to a Mediterranean Diet Influences the Fecal Metabolic Profile of Microbial-Derived Phenolics in a Spanish Cohort of Middle-Age and Older People. J Agric Food Chem 2017;65:586-595.
31. Gutierrez-Diaz I, Fernandez-Navarro T, Sanchez B, et al. Mediterranean diet and faecal microbiota: a transversal study. Food Funct 2016;7:2347-56.
32. Meslier V, Laiola M, Roager HM, et al. Mediterranean diet intervention in overweight and obese subjects lowers plasma cholesterol and causes changes in the gut microbiome and metabolome independently of energy intake. Gut 2020;69:1258-1268.
33. Piazzi G, Prossomariti A, Baldassarre M, et al. A Mediterranean Diet Mix Has
Chemopreventive Effects in a Murine Model of Colorectal Cancer Modulating
Apoptosis and the Gut Microbiota. Front Oncol 2019;9:140.
34. Barrett HL, Gomez-Arango LF, Wilkinson SA, et al. A Vegetarian Diet Is a Major Determinant of Gut Microbiota Composition in Early Pregnancy. Nutrients 2018;10.
35. Djekic D, Shi L, Brolin H, et al. Effects of a Vegetarian Diet on Cardiometabolic Risk Factors, Gut Microbiota, and Plasma Metabolome in Subjects With Ischemic Heart Disease: A Randomized, Crossover Study. J Am Heart Assoc 2020;9:e016518.
36. Kim MS, Hwang SS, Park EJ, et al. Strict vegetarian diet improves the risk factors associated with metabolic diseases by modulating gut microbiota and reducing
intestinal inflammation. Environ Microbiol Rep 2013;5:765-75.
37. Matijasic BB, Obermajer T, Lipoglavsek L, et al. Association of dietary type with fecal microbiota in vegetarians and omnivores in Slovenia. Eur J Nutr 2014;53:1051-64.
38. Trefflich I, Jabakhanji A, Menzel J, et al. Is a vegan or a vegetarian diet associated with the microbiota composition in the gut? Results of a new cross-sectional study and systematic review. Crit Rev Food Sci Nutr 2020;60:2990-3004.
39. Schreiner P, Yilmaz B, Rossel JB, et al. Vegetarian or gluten-free diets in patients with inflammatory bowel disease are associated with lower psychological well-being and a different gut microbiota, but no beneficial effects on the course of the disease. United European Gastroenterol J 2019;7:767-781.
Die konstitutionelle klinische Blutungsneigung ist Realität, diese beruht sowohl auf erworbenen wie auch auf hereditären Veränderungen der Gerinnung. Junge Menschen mit weniger Expositionsrisiken im Vergleich zu den Älteren fallen klinisch nicht sofort auf, deshalb ist eine gründliche, frühe Labordiagnostik sehr hilfreich und sinnvoll.
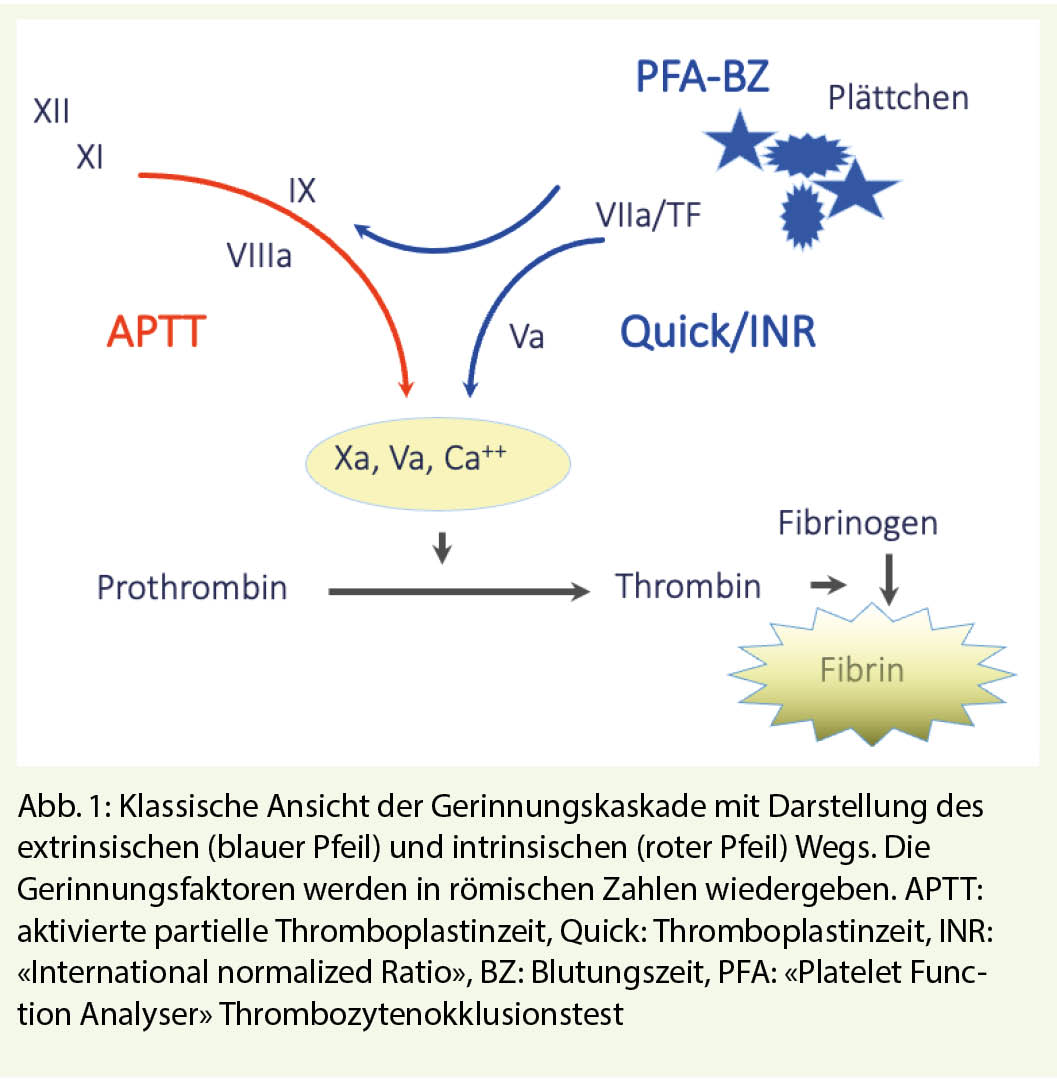
Die hämostatischen Mechanismen sind ein komplexes biologisches System, wo verschiedene lösliche Gerinnungsenzyme in Interaktion mit Zellstrukturen eine kaskadenartige, gegenseitige Aktivierung abspielen. Das System bleibt «eingefangen» im Gefässnetz und befindet sich primär in Ruhezustand, bis eine mechanische oder toxische Verletzung der Gefässwand vorkommt. Die Verletzung bedeutet Blutverlust und ist gleichzeitig der Trigger für den Start der Reaktionskaskade, welche unausweichlich in die Bildung des hämostatischen Thrombus führt. Die Reaktionspartner sind heute bekannt, sowohl in ihrer Struktur wie auch in ihrer Funktion (Abb. 1).
Eine Abweichung aus diesem physiologischen Weg, im Sinne eines Mangels eines Gerinnungsfaktors oder einer Hemmung dessen Funktion, bedeutet Verlangsamung der Gerinnung, klinisch als Blutungsneigung erkannt.
Zeichen der Blutungsneigung sind die Petechien (stecknadelkopfgrosse Flecken), die Ekchymosen (münzengrosse Flecken), die Suffusionen (grossflächige diffuse Blutungen) oder die Hämatome (abgrenzbare Blutungen).
Aus der Art der Blutung kann man nur bedingt die Ursache extrapolieren. Vaskuläre Defekte äussern sich meistens als Hämatome, Hautblutungen oder gastrointestinale Blutungen. Thrombozytäre Defekte lösen Petechien, Zahnfleischblutungen, Nasenblutungen, Gastrointestinal- oder ZNS-Blutungen aus. Störungen der plasmatischen Gerinnung manifestieren sich oft als Hämatome in den Muskellogen, als Hautblutungen, intraartikuläre Blutungen oder Blutungen nach Trauma.
Eine sorgfältige Anamnese, die klinische Untersuchung und die gezielte Laborabklärung führen fast immer zur genauen Identifizierung der Gerinnungsstörung.
Erste Abklärung
Anamnese
Eine gezielte detaillierte Blutungsanamnese ist sehr hilfreich. Da die Patienten die vereinzelten früheren Blutungsepisoden oft vergessen oder verdrängen, sollten sie vor blutigen diagnostischen und therapeutischen Eingriffen gezielt befragt werden (Tabelle 1). Strukturierte Fragebögen im Sinne eines Scores, wie der Blutungserfassungsscore BAT-ISTH“ der Internationalen Gesellschaft für Thrombose und Hämostase ISTH, können auch hilfreich sein. Starke Menstrualblutungen seit der Menarche erfordern Abklärung auf von Willebrand-Krankheit. Je niedriger der von Willebrand Faktor, desto stärker ist die Blutungsneigung.
Objektive Befunde
Bei den milden Gerinnungsstörungen kommt es zu einem Befund erst bei Provokationen (Trauma, Operation, Antikoagulation), bei den mittelschweren oder schweren Defekten können Blutungen auch spontan auftreten. Bei älteren schweren Hämophilen liegen eindrückliche Störungen des Bewegungsapparates (hämophile Arthropathie, Muskel-atrophie, Kontrakturen, Deformitäten etc.) vor, bei jüngeren hingegen fehlen in der Regel wegen der prophylaktischen Substitution solche Residuen. Neben eindeutigen akuten Blutungsmanifestationen müssen auch unklare Beschwerden bei hereditärer Blutungsneigung bis zum Beweis des Gegenteils primär als blutungsbedingt angesehen werden.
Labordiagnostik
Bei Verdacht auf hereditäre oder erworbene Blutungsneigung soll eine Basisdiagnostik verordnet werden. Diese beinhaltet ein Blutbild, die Globaltests der Gerinnung und der Plättchenfunktion (Thrombozytenokklusionstest PFA). Ergänzend werden auch der von Willebrand Faktor und Faktor XIII verlangt, weil diese durch die Globaltests nicht erkannt werden (Tabelle 2). Damit erfasst man die häufigen und seltenen Störungen der Hämostase. Je nach Resultat der Basisdiagnostik wird die Abklärung mit Spezialanalysen erweitert, wie z.B. gezielte Bestimmung der Gerinnungsfaktoren oder Analyse der Thrombozytenfunktion mittels Plättchenaggregation und/oder Immunphänotypisierung (Tabelle 3).
Spezialanalytik
Gezielte Untersuchung der Plättchenaggregation
Die Plättchenaggregation untersucht und klassifiziert Störungen der Plättchenfunktion, und liefert Hinweise auf Defekte der Plättchenmembranrezeptoren. Dabei wird in vitro die Stimulierbarkeit der Plättchen durch verschiedene natürliche Agonisten (ADP, Kollagen, Adrenalin, Ristocetin, Arachidonsäure, Thrombin (TRAP) und Thromboxanrezeptor-Agonist) turbidimetrisch bestimmt.
Gezielte Untersuchung der Plättchenmembranrezeptoren (Immunphänotypisierung)
Diese werden bei V.a. hereditäre Thrombopathien nachbestellt und helfen zur Diagnose und Typisierung von Defekten der Plättchenmembranrezeptoren. Dabei wird mit monoklonalen Antikörpern und Durchflusszytometrie die Rezeptorendichte auf der Plättchenoberfläche vor und nach in-vitro Plättchenaktivierung semi-quantitativ bestimmt.
Von Willebrand Faktor-Diagnostik und Multimere (VWF-MM)
Das von Willebrand Syndrom ist die häufigste hereditäre Blutungsneigung (Inzidenz 1/200-300) und wird durch die Bestimmung des von Willebrand Faktors (Funktion und Antigen) sowie des Faktors VIII erfasst. Die Abklärung wird durch die Analyse der VWF-MM ergänzt. Diese wird bei V.a. von Willebrand-Syndrom jeden Typs nachbestellt und hilft zur Diagnose und Typisierung des vWF-Defektes. Die Methode besteht in der qualitativen und quantitativen Charakterisierung der zirkulierenden vWF-Multimere mittels Gel-Elektrophorese.
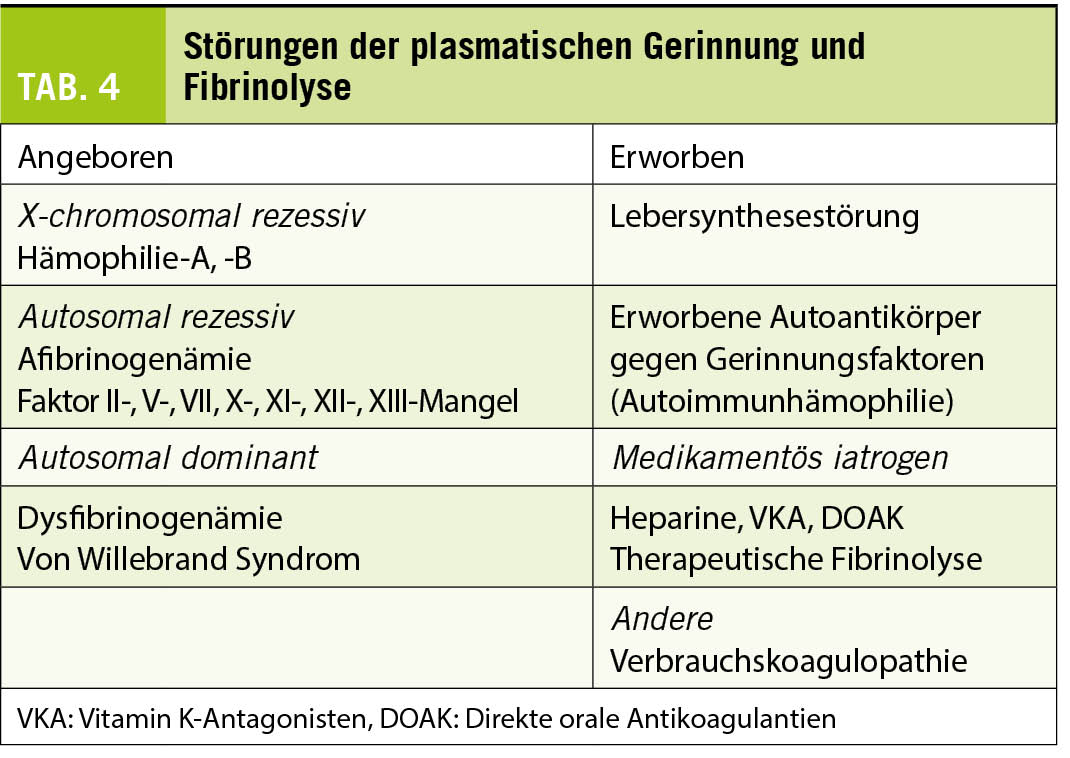
Klinik und Diagnostik der Blutungsneigung (Tabelle 4)
Hämophilie A und B
Die Hämophilie ist eine X-chromosomal vererbte Verminderung der Gerinnungsfaktoren VIII oder IX, Männer sind betroffen, Frauen sind Konduktorinnen. Klinisch sind Gelenksblutungen und Muskelblutungen typisch. Bei Trauma oder Operation ist aber mit ebenso massiven Blutungen zu rechnen.
Bei schwerer und mittelschwerer Hämophilie A oder B ist die APTT eindeutig verlängert, bei milder Hämophilie ist sie nur grenzwertig erhöht oder noch im Normbereich. Eine normale APTT erlaubt daher den Ausschluss einer milden Hämophilie nicht, wenn ein Verdacht aufgrund der Anamnese vorliegt. Die gezielte Bestimmung des Faktors VIII oder IX identifiziert genau den Defekt.
Von Willebrand-Syndrom
Autosomal vererbte Störung des von Willebrand-Faktors (vWF) als Folge einer Vielzahl von Abnormitäten des von vWF-Gens. Der von Willebrand-Faktor ist wichtig für die Adhäsion der Plättchen an die verletzte Gefässintima und ihre Aktivierung. Zudem stabilisiert er den Faktor VIII, mit dem er im Plasma komplex-gebunden zirkuliert. Zahlenmässig überwiegen die milden Formen, die nur bei Provokationen bluten und meistens eine normale Blutungszeit aufweisen. Alle Typen zusammengenommen sollen nach gewissen Schätzungen fast 1% der Bevölkerung betreffen. Die schwerste Form (Typ 3) hat eine Inzidenz von 1 pro Mio.
Die Diagnostik beruht auf der quantitativen Bestimmung des von Willebrand-Faktors (Funktion und Antigen) und der Faktor VIII-Aktivität. Die Typeneinteilung ist dauernd im Fluss, wobei eine Klassierung nach einigen Phänotypen den klinischen Bedürfnissen bisher genügt.
Typ 1: Konkordante Verminderung von funktionellem und immunologisch gemessenem vWF auf weniger als 50% der Norm (meistens 5-30%). Faktor VIII um 50% oder tiefer. Autosomal dominant, macht etwa 70% der Patienten aus. Cave: Menschen mit Blutgruppe O haben physiologisch tiefere vWF-Werte ohne klinische Blutungsneigung. Der Cut-Off wird hier auf 35% statt 50% gesetzt.
Typ 2A: Verminderung der vWF-Aktivität begleitet von normalem oder nur leicht vermindertem vWF-Antigen (Ratio vWF Aktivität/vWF Antigen <0.7). Die Analyse der Multimere ergibt eine Verminderung der funktionell besonders aktiven hochmolekularen Ketten. Faktor VIII normal oder grenzwertig reduziert. Autosomal dominante Vererbung (einige rezessiv).
Typ 2B: Wie Typ 2A, jedoch begleitet von einer paradoxen Verstärkung der Ristocetin-induzierten Aggregation des plättchenreichen Patientenplasmas, bedingt durch eine erhöhte Affinität abnormer vWF-Ketten für den GP Ib/V/IX-Rezeptor des Patienten (von Willebrand Faktor Rezeptor). Bei der vWF-Multimeren Analyse Verminderung der grossen und mittelgrossen VWF-Ketten. Stimulation der Freisetzung des abnormen vWF führt zur Thrombopenie. Autosomal dominante Vererbung.
Typ 2M: Schlechte Plättchenaffinität der vWF-Multimere. Im Unterschied zu den Typen 2A und 2B sind auch die grossen Multimere vorhanden, aber funktionsgestört, was sich in der erniedrigten vWF-Funktion bei noch normalem vWF-Antigen und normaler vWF-Multimerenelektrophorese äussert (jedoch abnorme Form der Banden-Tripletten der Multimere). Autosomal dominante Vererbung.
Typ 2N, vWF-Typ Normandy: vWF mit abnormer Bindungsstelle für den Faktor VIII, der wegen fehlender Bindung an den sonst quantitativ und qualitativ normalen vWF schnell eliminiert wird. Formal entsteht das Bild einer leichten Hämophilie A (auch bei Frauen) mit VIII um 5-30%. Autosomal rezessive Vererbung.
Typ 3: vWF funktionell und immunologisch nicht nachweisbar. Faktor VIII wegen fehlendem Transportprotein im Plasma auch auf wenige Aktivitätsprozente reduziert. Autosomal rezessive Vererbung. Blutungszeit stark verlängert.
Seltene Koagulopathien
Die sehr seltenen hereditären Verminderungen einzelner Faktoren äussern sich in abnormen Globaltests: II, V und X durch abnorme Quick/INR und abnorme APTT, VII durch abnorme Quick/INR bei normaler APTT und die Kontaktphasenproteine XII und Faktor XI durch isolierte Verlängerung der APTT. Bei Afibrinogenämie fehlt die Gerinnselbildung in allen Globaltests. Die Diagnose wird mit der spezifischen Faktorenbestimmung gestellt. Der Mangel an Faktor XII ist nicht mit einer Blutungsneigung verbunden, ebensowenig der Faktor VII-Mangel mit Restaktivitäten über 10% der Norm.
Hereditäre Thrombozytenfunktionsstörungen
Die schwersten Blutungen findet man bei den seltenen, klassischen Modellkrankheiten der Plättchen, der Thrombasthenie Glanzmann (Defekt des Fibrinogenrezeptors GP IIb/IIIa) und dem Bernard-Soulier-Syndrom (Defekt des vWF-Rezeptors GP Ib/V/IX). Die Blutungszeit ist in der Regel abnorm, spezifische Diagnostik mit Plättchenaggregationstests und Rezeptorenuntersuchung bestätigt den Defekt. Weitere, unterschiedlich charakterisierte Defekte betreffen andere Oberflächenrezeptoren, die Signalübertragung, die Alpha- und Dichte-Granula sowie den Arachidonsäuremetabolismus.
Dr. med. Leda Leoncini1 Dr. med. Mario Uhr1 Dr. Yordanka Tirefort2 Prof. Dr. med. Dimitrios Tsakiris3 1 SYNLAB Suisse SA, Via Pianon 7, 6934 Bioggio (leda.leoncini@synlab.com, mario.uhr@synlab.com) 2 SYNLAB Suisse SA, Ch. d’Entre-Bois 21, 1018 Lausanne (yordanka.tirefort@synlab.com) 3 SYNLAB Suisse SA, Alpenquai 14, 6002 Luzern (dimitrios.tsakiris@synlab.com)
Copyright Aerzteverlag medinfo
Dr. med. Leda Leoncini
SYNLAB Suisse SA
Via Pianon 7
6934 Bioggio
leda.leoncini@synlab.com
Prof. Dr. med. Dimitrios Tsakiris
Klinik für Hämatologie
Hämatologische Diagnostik Labormedizin
Universitätsspital Basel und Blutspendezentrum beider Basel SRK
Petersgraben 4
4031 Basel
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel zu deklarieren.
◆ Eine gezielte Blutungsanamnese ist sehr hilfreich. Da die Patienten die vereinzelten früheren Blutungsepisoden oft vergessen oder verdrängen, sollten sie gezielt befragt werden.
◆ Junge Menschen mit weniger Expositionsrisiken fallen klinisch wegen Blutung nicht sofort auf im Vergleich zu den Älteren, deshalb ist eine gründliche, frühe Labordiagnostik sehr sinnvoll.
◆ Eine Basisdiagnostik ist bei Blutungsneigung der erste Schritt. Diese beinhaltet die Globaltests der Gerinnung Quick/INR, APTT, Fibrinogen, und der Plättchenfunktion (Thrombozytenokklusionstest PFA), wie auch den von Willebrand Faktor und Faktor XIII, weil diese durch die Globaltests nicht erkannt werden.
◆ Je nach Resultat der Basisdiagnostik wird die Abklärung mit Spezialanalysen erweitert, wie z.B. gezielte Bestimmung der einzelnen Gerinnungsfaktoren oder Analyse der Thrombozytenfunktion mittels Plättchenaggregation und/oder Immunphänotypisierung.
1. Hayward CPM. How I investigate for bleeding disorders. Int J Lab Hematol. 2018 May;40 Suppl 1:6-14. doi: 10.1111/ijlh.12822. PMID: 29741250.
2. Boender J, Kruip MJ, Leebeek FW. A diagnostic approach to mild bleeding disorders. J Thromb Haemost. 2016 Aug;14(8):1507-16. doi: 10.1111/jth.13368. Epub 2016 Jun 27. PMID: 27208505.
3. Hayward CPM, Moffat KA, Brunet J, Carlino SA, Plumhoff E, Meijer P, Zehnder JL. Update on diagnostic testing for platelet function disorders: What is practical and useful? Int J Lab Hematol. 2019 May;41 Suppl 1:26-32. doi: 10.1111/ijlh.12995. PMID: 31069975.
4. James PD. Women and bleeding disorders: diagnostic challenges. Hematology Am Soc Hematol Educ Program. 2020 Dec 4;2020(1):547-552. doi: 10.1182/hematology.2020000140. PMID: 33275722; PMCID: PMC7727580.
5. Rodeghiero F, Tosetto A, Abshire T, Arnold DM, Coller B, James P, Neunert C, Lillicrap D; ISTH/SSC joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010 Sep;8(9):2063-5. doi: 10.1111/j.1538-7836.2010.03975.x. PMID: 20626619
Als primären Hyperparathyreoidismus (pHPT) bezeichnet man den Zustand einer chronisch erhöhten, inadäquaten Sekretion von Parathormon (PTH) ohne erkennbaren physiologischen Stimulus. Der pHPT ist nach dem Diabetes mellitus und Schilddrüsenerkrankungen die dritthäufigste endokrine Erkrankung des Menschen und stellt die häufigste Ursache für eine ambulant entdeckte Hyperkalzämie dar.
Epidemiologie
Der primäre Hyperparathyreoidismus ist eine der häufigsten endokrinen Erkrankungen. Grundsätzlich kann der primäre Hyperparathyreoidismus in jeder Altersstufe auftreten, wobei Menschen über 50 Jahre und postmenopausale Frauen am häufigsten betroffen sind. Die Inzidenz wird in westlichen Industrienationen auf etwa 25-30/ 100 000 Einwohner/ Jahr geschätzt, was einer Prävalenz von 1-7/ 1000 Menschen in der Allgemeinbevölkerung entspricht (1-5).
Ätiologie
In 85% – 90% der Fälle handelt es sich um einen sporadisch auftretenden pHPT durch ein solitäres autonomes Nebenschilddrüsenadenom.
Hyperplasien mehrerer oder aller vier Nebenschilddrüsen kommen in bis zu 6% der Fälle vor (6) und können auch hinweisend auf eine genetische Ursache sein. Bei der genetischen Ursache muss insbesondere bei jungen Patienten mit einer multiglandulären Erkrankung an eine multiple endokrine Neoplasie (MEN) vom Typ 1 aber auch Typ 2a gedacht werden. Auch Lithium-haltige Medikamente und Thiaziddiuretika können die Entwicklung einer Mehrdrüsenerkrankung begünstigen. Sehr selten liegt ein Nebenschilddrüsenkarzinom vor (< 1% der Fälle).
Anatomie
Beim Menschen liegen in der Regel vier, selten auch fünf oder sechs, gelegentlich auch nur drei Nebenschilddrüsen (NSD, Epithelkörperchen) vor. Sie wiegen 20-50 mg bei einer Grösse von etwa 5 x 3 x 1 mm. Grundsätzlich sind NSD sehr variabel in Form, Grösse, Gewicht, Farbe und Konsistenz. Topographisch liegen die NSD in der Regel der Schilddrüsenkapsel auf, wobei die kranialen Nebenschilddrüsen meist dorsal des N. laryngeus recurrens und die kaudalen Nebenschilddrüsen ventral der Nerven liegen.
Pathophysiologie
Der Kalziumspiegel wird im Blut durch das Parathormon (PTH) und Vitamin D3 konstant zwischen 2.0 und 2.6 mmol/l gehalten, was für zahlreiche Körperfunktionen unentbehrlich ist (z.B. Muskelkontraktionen, Nervenleitung, Zellstoffwechsel). PTH wird von den Hauptzellen der NSD produziert und sezerniert. Die Regulation erfolgt dabei über sog. Kalzium-Sensing-Rezeptoren (CaSR), die auf der Oberfläche der NSD lokalisiert sind. Sie registrieren z.B. einen Abfall des ionisierten Kalziums im Blut und steigern die Produktion und Sekretion des PTH.
Zu den Wirkungen des PTH zählen:
1. eine verstärkte renale Kalziumrückresorption und Phosphatausscheidung
2. ein verstärkter Kalziumabbau am Knochen durch Aktivierung der Osteoklasten
3. eine verstärkte Absorption von Kalzium aus dem Darm durch gesteigerte Synthese von 1,25-Dihydroxy-Vitamin D aus 25-Hydroxyvitamin D3 durch Aktivierung der renalen 1α-Hydroxylase
Klinik
Bereits durch eine sorgfältige Anamnese können wertvolle Hinweise auf das Vorliegen wesentlicher Differentialdiagnosen gewonnen werden, wobei die Leitsymptome des primären Hyperparathyreoidismus meist recht unspezifisch sind. Die Symptome können zusammengefasst werden mit «Stein, Bein und Magenpein». Dies könnte ergänzt werden mit «Seelenkummer obendrein». Dabei steht das «Stein» für Nierensteine. Renal kann zudem eine Polyurie imponieren (7-12). «Bein» steht für Osteoporose. Klinisch imponieren dabei oft Knochen- bzw. Gelenkschmerzen, selten kommt es zu pathologischen Frakturen (11, 12). Zudem kann der Gastrointestinaltrakt betroffen sein mit peptischen Ulzera des Duodenums und Magens aber auch Pankreatitiden. Zuletzt können auch in bis zu 20% neuropsychologische Symptome vorliegen. Dies können depressive Verstimmungen, oder kognitive Veränderungen sowie auch vermehrte Müdigkeit, Konzentrationsstörungen und Stimmungsschwankungen sein (13, 14, 15). Nicht selten liegt eine asymptomatische Hyperkalzämie vor.
Diagnostisches Vorgehen
Labordiagnostik
Ein pHPT wird durch den Nachweis eines erhöhten oder hochnormalen intakten Parathormons (PTH) und gleichzeitig bestehendem erhöhtem Albumin-korrigierten Gesamt-Kalzium oder ionisiertem Serumkalzium gestellt. Es ist also eine Diagnose, die primär laborchemisch gestellt wird. Andere Ursachen einer Hyperkalzämie bzw. ein erhöhter Wert des Parathormons sollten ausgeschlossen werden. Z.B. bei einer Paraneoplasie, einer Medikamentennebenwirkung, einer Niereninsuffizienz, einem Vitamin D-Mangel oder einer familiären hypokalziurischen Hyperkalzämie.
Die Bestimmung von Kreatinin und Harnstoff sowie der Kreatinin-Clearance dienen dem Ausschluss einer Niereninsuffizienz-bedingten Nebenschilddrüsenüberfunktion (sekundärer HPT), während die Bestimmung der Kalzium- und Kreatinin-Clearance im Spot-urin einen milden pHPT von einer familiären hypokalziurischen Hyperkalzämie (FHH) ohne Krankheitswert differenzieren kann. Dies ist deshalb so wichtig, weil eine FHH keine Therapie benötigt.
Die häufigste Ursache für einen reaktiv erhöhten PTH-Wert bei Normokalzämie ist ein Vitamin-D-Mangel, der durch Bestimmung des 25-Hydroxy-Vitamin D aufgedeckt werden kann.
Lokalisationsdiagnostik
Eine erfolgreiche Lokalisationsdiagnostik ermöglicht einen fokussierten und dadurch auch minimal-invasiven operativen Zugangsweg (16) und schliesst als Optionen 1. die zervikale Sonographie und 2. die Sestamibi-Szintigraphie bzw. Sestamibi-SPECT/ CT-Untersuchung mit ein.
Bei fehlender Lokalisation in der Sonographie und Szintigraphie oder auch bei diskordanten Befunden, steht neuerdings das Cholin-PET zu Verfügung, das eine erhöhte Sensitivität hat und entsprechend eine tiefere falsch-negative Rate (17).
Therapeutisches Vorgehen
Bei laborchemisch diagnostiziertem pHPT und typischer Symptomatik, einem sogenannten symptomatischen pHPT, ist eine chirurgische Therapie in Form einer Parathyreoidektomie klar indiziert. Aber auch bei asymptomatischen Patienten ist die Operationsindikation oft gegeben, weil auch sogenannt asymptomatische Patienten postoperativ häufig über eine Verbesserung ihrer Lebensqualität berichten, und der pHPT auch bei asymptomatischen Patienten einen nachteiligen Effekt z.B. auf den Knochen hat und bei einem gewissen Prozentsatz dieser Patienten binnen zehn Jahren auch zu Symptomen führt. Empfohlen wird eine Operation grundsätzlich bei asymptomatischen Patienten, wenn die Hyperkalzämie sehr ausgeprägt ist, wenn eine Niereninsuffizienz vorliegt, oder auch wenn die Patienten jünger als 50 Jahre alt sind. Auch eine Osteoporose kann eine Operationsindikation sein. Hier geht es immer um eine Nutzen Risiko-Abwägung, die idealerweise von einem Endokrinologen mitabgewogen werden sollte.
Alternativ zur chirurgischen Therapie, insbesondere bei asymptomatischen Verlaufsformen des pHPT, sollten regelmässig Kontrollen durchgeführt werden. Zudem müssen die Patienten dazu angehalten werden ausreichend zu trinken und eine Immobilisation zu vermeiden.
Präoperatives Management
Vitamin D3
Patienten mit einem pHPT haben häufig einen Vitamin-D-
Mangel. Vitamin-D3-Mangel ist v.a. im Winter stark verbreitet. Da ein Vitamin-D3-Mangel die Parathormonsekretion
stimuliert, sollte vor einer Operation bei Vorliegen eines Vitamin-D3-Mangels eine Supplementation erfolgen (18, 19). Dabei sind kurzfristige Kontrollen des Serumkalziums erforderlich. Aktuell wird eine Vitamin D Gabe von 800 bis 1000 IU/Tag bei Erwachsenen empfohlen (20).
Laryngoskopie
Eine prä- und postoperative Laryngoskopie mit Dokumentation der Stimmlippenbeweglichkeit soll bei allen Patienten mit einem pHPT aus Qualitätszwecken erfolgen. Dies, weil v.a. die kranialen Nebenschilddrüsen sehr nah am N. laryngeus recurrens liegen und dieser bei einer Operation geschädigt werden kann. Dies hätte eine Stimmbandfunktionseinschränkung mit Heiserkeit beim Patienten zur Folge.
Operation
Das chirurgische Vorgehen unterscheidet sich wesentlich zwischen Ersteingriffen und Rezidiveingriffen und ist von der zugrundeliegenden Ätiologie abhängig. Im Folgenden wird aus Gründen der Übersicht das Vorgehen eines Ersteingriffes beschrieben. Voraussetzung für alle operativen Verfahren ist die Notwendigkeit einer exakten Kenntnis der Anatomie der Nebenschilddrüsen und ihrer potentiell ektopischen Lagevariationen.
Besteht aufgrund der Lokalisationsdiagnostik präoperativ der Verdacht auf ein singuläres Nebenschilddrüsenadenom, kann eine fokussierte Parathyreoidektomie und damit potentiell auch eine minimalinvasive Operationstechnik avisiert werden. Bei Verdacht auf einen multiglandulären Befall wird je nach Lokalisation eine uni- oder bilaterale Halsexploration durchgeführt.
Die Operation erfolgt in der Regel in Vollnarkose. Die konventionelle, offene Entfernung eines Nebenschilddrüsenadenoms erfolgt über einen vorderen oder lateralen Zugang. Der vordere Zugang entspricht dem üblichen Zugang für die Thyreoidektomie, kann aber durch eine kleinere Inzision durchgeführt werden (20).
Da das Parathormon eine sehr kurze Halbwertszeit von weniger als 5 Minuten aufweist, ermöglicht die intraoperative PTH-
Bestimmung nach Entfernung des betroffenen Nebenschilddrüsenadenoms eine unmittelbare Erfolgskontrolle noch während des Eingriffes (19). Dabei wird zu verschiedenen Zeiten perioperativ Parathormon bestimmt, meist vor der Operation, zum Zeitpunkt der Adenomentfernung sowie 10 bis 20 Minuten nach Adenom-entfernung. Bei nicht adäquatem Abfall des Parathormonwertes kann dann die operative Strategie entsprechend angepasst werden, d.h., wenn das Parathormon nicht mindestens 50% abfällt binnen
10 Minuten nach Entfernung des Nebenschilddrüsenadenoms, muss nach einer weiteren erkrankten Nebenschilddrüse gesucht werden. Der Eingriff muss also erweitert werden. Auch ein intraoperativer Schnellschnitt kann hilfreich sein, insbesondere, wenn die Identifizierung des Nebenschilddrüsenadenoms aspektmässig nicht einwandfrei möglich ist.
Ein intraoperatives Neuromonitoring kann helfen die Stimmbandnerven zu identifizieren und wird regelhaft eingesetzt (21). Besteht eine Indikation zur Schilddrüsenoperation wird empfohlen, diese zusammen mit der Nebenschilddrüsenoperation durchzuführen.
Grundsätzlich sind Komplikationen nach Nebenschilddrüsenoperationen selten. Durch exakte anatomische Kenntnisse und vorsichtige Präparationstechnik können die wesentlichen Komplikationen auf ein Minimum reduziert werden. Zu den möglichen Komplikationen zählen:
1. Stimmbandnervlähmung (sog. Rekurrensparese)
2. Nachblutung
3. Persistenz und Rezidiv
Zur Beurteilung der postoperativen Nebenschilddrüsenfunktion sollte eine Bestimmung des Albumin- korrigierten Serumkalziums und des intakten Parathormons am 1. postoperativen Tag erfolgen, um das Risiko einer therapierelevanten postoperativen Hypokalz-ämie zu erfassen. Nach erfolgreicher Parathyreoidektomie werden in 1,8% – 42% der Fälle erniedrigte Serumcalciumwerte gemessen (24, 25). Dies entspricht durchaus einem regelhaften postoperativen Verlauf und ist zunächst nicht als postoperative Komplikation zu werten.
Eine Bestimmung insbesondere des Albumin-korrigierten Serumkalziums, fakultativ auch des PTHs sollte, auch bei unkompliziertem Verlauf, nach 6 Monaten durchgeführt werden, um eine Persistenz bzw. ein Rezidiv des pHPT zu erfassen.
Nachblutungen sind eine seltene, aber potenziell lebensbedrohliche Komplikation nach Nebenschilddrüsenoperation. Sie treten meist innerhalb der ersten 24 h postoperativ auf und erfordern eine unverzügliche operative Revision mit vordringlicher Sicherung der Atemwege. Aus diesem Grunde sind Nebenschilddrüsenoperationen auch weiterhin keine ambulanten Eingriffe.
Eine Persistenz und oder ein Rezidiv eines Hyperparathyreoidismus wird definiert als anhaltende Hyperkalzämie binnen bzw. nach 6 Monaten und findet sich in 2-5% (22, 23).
Die Autorinnen haben im Zusammenhang mit diesem Artikel keine Interessenkonflikte deklariert.
◆ Die Diagnose eines primären Hyperparathyreoidismus wird laborchemisch gestellt.
◆ Zur Diagnose des pHPT sollte eine wiederholte Bestimmung des Albumin-korrigierten Serumkalziums und des intakten Parathormons erfolgen und eine sekundäre Komponente durch Bestimmung des Kreatinins und des 25(OH)- Vitamin D ausgeschlossen werden. Gegebenenfalls soll Vitamin D substituiert werden.
◆ Die Bestimmung der Calcium Clearance im Spoturin dient dem Ausschluss einer familiären hypokalziurischen Hyperkalzämie, welche nicht therapiebedürftig ist.
◆ Die Operation ist die bisher einzige ursächliche Therapie.
1. Wilhelm SM, Wang TS, Ruan DT, Lee JA, Asa SL, Duh QY, et al. The American Association of Endocrine Surgeons Guidelines for Definitive Management of Primary Hyperparathyroidism. JAMA Surg. 2016; 151(10):959-68
2. Abood A, Vestergaard P. Increasing incidence of primary hyperparathyroidism in Denmark. Dan Med J 2013; 60(2): A4567
3. Griebeler ML, Kearns AE, Ryu E, Hathcock MA, Melton LJ 3rd Wermers RA. Secular trends in the incidence of primary hyperparathyroidism over five decades (1965-2010). Bone 2015; 73:1-7
4. Nilsson IL. Primary hyperparathyroidism: should surgery be performed on all patients? Current evidence and residual uncertainties. J Intern Med 2019; 285(2):149-164
5. Siilin H, Lundgren E, Mallmin H, Mellström, Ohlsson C, Karlsson M, Ljunggren O. Prevalence of primary hyperparathyroidism and impact on bone mineral density in elderly men: MrOs Sweden. World J Surg 2011; 35(6):1266-1272
6. Ruda JM, Hollenbeak CS, Stack BC Jr, A systematic review of the diagnosis and treatment of primary hyperparathyroidism from 1995 to 2003, Otolaryngol Head Neck Surg. 2005;132(3):359.
7. Suh JM, Cronan JJ, Monchik JM. Primary hyperparathyroidism: is there an increased prevalence of renal stone disease? AJR Am J Roentgenol. 2008; 191(3):908-911
8. Rejnmark L, Vestergaard P, Mosekilde L. Nephrolithiasis and renal calcifications in primary hyperparathyroidism. J Clin Endocrinol Metab. 2011;96(8):2377-2385
9. Ejlsmark-Svensson H, Bislev LS, Rolighed L, Sikjaer T, Rejnmark L. Predictors of renal function and calcifications in primary hyperparathyroidism: A nested case-control study. J Clin Endocrinol Metab 2018; 103(9):3574-3583
10. Tay YD, Liu M, Bandeira L, Bucovsky M, Lee JA, Silverberg SJ, Walker MD. Occult urolithiasis in asymptomatic primary hyperparathyroidism. Endocr Res 2018; 43(2):106-115
11. Reid LJ, Muthukrishnan B, Patel D, Seckl JR, Gibb FW. Predictors of nephrolithiasis, osteoporosis, and mortality in primary hyperparathyroidism. J Clin Endocrinol Metab 2019; 104(9):3692-3700
12. Castellano E, Attanasio R, Boriano A, Pellegrino M, Garino F, Gianotti L, Borretta G. Sex difference in the clinical presentation of primary hyperparathyroidism: Influence of menopausal status. J Clin Endocrinol Metab 2017; 102(11):4148-4152
13. Blanchard C, Mathonnet M, Sebag F, Caillard C, Hamy A, Volteau C et al. Surgery for ‚asymptomatic‘ mild primary hyperparathyroidism improves some clinical symptoms postoperatively. Eur J Endocrinol 2013; 169(5):665-672
14. Murray SE, Pathak PR, Schaefer SC, Chen H, Sippel RS. Improvement of sleep disturbance and insomnia following parathyroidectomy for primary hyperparathyroidism. World J Surg 2014; 38(3):542- 548
15. Weber T, Eberle J, Messelhauser U, Schiffmann L, Nies C, Schabram J et al. Parathyroidectomy, elevated depression scores, and suicidal ideation in patients with primary hyperparathyroidism: results of a prospective multicenter study. JAMA Surg 2013; 148(2):109-115
16. Bergenfelz AO, Hellman P, Harrison B, Sitges-Serra A, Dralle H. Positional statement of the European Society of Endocrine Surgeons (ESES) on modern techniques in pHPT surgery. Langenbecks Arch Surg 2009; 394(9):761-764
17. Lezaic L, Rep S, Jensterle SM, Hocevar M, Fettich J. 18F-fluorocholine PET/CT for localization of hyper functioning parathyroid tissue in primary hyper-parathyroidism: a pilot study. Eur J Nucl Med Mol Imaging. 2014; 41:2083–9
18. Marcocci C, Bollerslev J, Khan AA, Shobach DM. Medical management of primary hyperparathyroidism: Proceedings of the Fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism. J Clin Endocrinol Metab 2014; 99(10):3607-3618
19. Eastell R, Brandi ML, Costa AG, D’Amour P, Shoback DM, Thakker RV. Diagnosis of asymptomatic primary hyperparathyroidism: Proceedings of the Fourth International Workshop. J Clin Endocrinol Metab 2014; 99(10):3570-3579
20. Udelsman R, Lin Z, Donovan P. The superiority of minimally invasive parathyroidectomy based on 1650 consecutive patients with primary hyperparathyroidism. Ann Surg2011; 253(3):585-591
21. Lin HS, Terris DJ. An update on the status of nerve monitoring for thyroid/parathyroid surgery. Curr Opin Oncol2017; 29(1):14-19
22. Guerin C, Paladino NC, Lowery A, Castinetti F, Taieb D, Sebag F. Persistent and recurrent hyperparathyroidism. Updates Surg 2017; 69:161-169
23. Bagul A, Patel HP, Chadwick D, Harrison BJ, Balasubramanian SP. Primary hyperparathyroidism: ananalysis of failure of parathyroidectomy. World J Surg 2014; 38(3):534-541
24. Allendorf J, DiGorgi M, Spanknebel K, Inabnet W, Chabot J, Logerfo P. 1112 consecutive bilateral neck explorations for primary hyperparathyroidism. World J Surg 2007; 31 (11): 2075-2080
25. Mittendorf EA, Merlino Jl, McHenry CR. Post-parathyroidectomy hypocalcemia: incidence, risk factors, and management. Am Surg. 2004; 70(2):114-119