Trastuzumab Deruxtecan hat sich bei Patientinnen mit zuvor behandeltem HER2-positivem fortgeschrittenem oder metastasiertem Brustkrebs als wirksam erwiesen. Die Wirksamkeit und Sicherheit von Trastuzumab Deruxtecan bei Patientinnen ohne vorherige Therapie für HER2-positiven fortgeschrittenen oder metastasierten Brustkrebs ist jedoch unklar.
Methoden
Studienleiterin Sara Tolaney und ihre Mitarbeiter führten eine Phase-3-Studie mit Patientinnen durch, die an HER2-positivem, fortgeschrittenem oder metastasiertem Brustkrebs litten und zuvor noch keine Chemotherapie oder HER2-gerichtete Therapie gegen metastasierte Erkrankungen erhalten hatten. Die Patientinnen wurden im Verhältnis 1:1:1 randomisiert und erhielten entweder Trastuzumab Deruxtecan plus Pertuzumab, Trastuzumab Deruxtecan plus Placebo oder die Kombination aus Taxan, Trastuzumab und Pertuzumab (THP). Der primäre Endpunkt war das progressionsfreie Überleben, das durch eine verblindete, unabhängige, zentrale Überprüfung bewertet wurde. Sekundäre Endpunkte waren das objektive Ansprechen, die Ansprechdauer und die Sicherheit.
Ergebnisse
Für diese vorab festgelegte Zwischenanalyse werden Daten zu Trastuzumab Deruxtecan plus Pertuzumab und zu THP berichtet. Die Daten zu Trastuzumab Deruxtecan plus Placebo bleiben bis zur endgültigen Analyse des progressionsfreien Überlebens verblindet. Zum Stichtag (26. Februar 2025) betrug das mediane progressionsfreie Überleben 40.7 Monate unter der Kombination aus Trastuzumab Deruxtecan und Pertuzumab (383 Patienten) sowie 26.9 Monate unter THP (387 Patienten) (Hazard Ratio für Progression oder Tod: 0.56; 95 %-Konfidenzintervall [KI]: 0.44 bis 0.71; p < 0.00001 [p-Wert-Grenze für Überlegenheit: 0.00043]). Die Inzidenz eines bestätigten Ansprechens betrug 85.1 % unter Trastuzumab Deruxtecan plus Pertuzumab und 78.6 % unter THP (vollständiges Ansprechen bei 15.1 % bzw. 8.5 %), mit einer medianen Ansprechdauer von 39.2 Monaten bzw. 26.4 Monaten. Die Sicherheit entsprach den bekannten Profilen der einzelnen Behandlungen. Die Inzidenz von unerwünschten Ereignissen des Grades 3 oder höher betrug 63.5 % unter Trastuzumab Deruxtecan plus Pertuzumab und 62.3 % unter THP. Am häufigsten traten unter Trastuzumab Deruxtecan plus Pertuzumab Neutropenie, Hypokaliämie und Anämie sowie unter THP Neutropenie, Leukopenie und Diarrhoe auf. Eine bestätigte, arzneimittelbedingte interstitielle Lungenerkrankung oder Pneumonitis trat bei 12.1 % der Patienten unter Trastuzumab Deruxtecan plus Pertuzumab (Grad 1 oder 2 bei 44 Patienten und Grad 5 [Tod] bei zwei Patienten) auf, während es unter THP nur 1.0 % waren (alle Grad 1 oder 2).
Schlussfolgerungen
Bei der Erstlinienbehandlung von HER2-positivem, fortgeschrittenem oder metastasiertem Brustkrebs führte Trastuzumab Deruxtecan plus Pertuzumab zu einem signifikant geringeren Risiko für Progression oder Tod als THP, ohne dass neue Sicherheitssignale auftraten. (Finanziert von AstraZeneca und Daiichi Sankyo, DESTINY-Breast09, ClinicalTrials.gov-Nummer NCT04784715).
Kommentar
Das sind sehr eindrucksvolle Zahlen zur Wirksamkeit von Trastuzumab-Deruxtecan. Die entscheidende Frage für die Praxis ist, wie wir dieses Medikament am besten einsetzen. Dazu gehören folgende Fragen: Welche Patientinnen sollen Trastuzumab-Deruxtecan als Erstlinienbehandlung erhalten? Wie lange soll das Medikament gegeben werden, wenn die Krankheit gut kontrolliert ist? Wohl nicht so, wie es in dieser Registrier-Firstline-Studie gemacht wurde, die ein Modell darstellt, um die Wirksamkeit bestmöglich und am sichersten zu beweisen. In der Studie wurde Trastuzumab-Deruxtecan + Pertuzumab offenbar über im median dreieinhalb Jahre kontinuierlich verabreicht. Das ist sicher keine gängige Praxis, zumindest nicht in unserer Umgebung. Zudem warten wir auf die Beantwortung der Frage, ob Pertuzumab bei dieser Behandlung essentiell und relevant ist. Es darf daran erinnert werden, dass Trastuzumab-Deruxtecan ohne Pertuzumab in der zweiten Therapielinie (Destiny-Breast03) ein medianes progressionsfreies Überleben von 29 Monaten erreichte. Diese Patientinnen erfreuten sich während der langen Zeit der Antikörpererhaltungstherapie ohne Chemotherapie einer sehr guten Lebensqualität. Und last but not least müssen auch die Kosten berücksichtigt werden.
Um den optimalen Einsatz dieses sehr wirksamen Medikaments bei fortgeschrittenem Brustkrebs zu bestimmen, müssen diese Fragen untersucht und beantwortet werden.
Quelle Tolaney et al.Trastuzumab Deruxtecan plus Pertuzumab for HER2-Positive Metastatic Breast Cancer. New Engl J Med. Published October 29, 2025. DOI: 10.1056/NEJMoa2508668.
Die Behandlung chronischer Wunden in der Praxis ist ein häufiges aber oft frustrierendes Unterfangen. Entscheidend ist es, die Wunde als Symptom einer Grunderkrankung zu sehen. Diese zu behandeln, ist eine absolute Bedingung für eine erfolgreiche Wundheilung. Die lokale Wundbehandlung erfolgt meist interdisziplinär und folgt dem «TIME»-Konzept, wobei das Débridement von Nekrosen und Fibrinbelägen und die Gewährleistung einer die Wundheilung unterstützenden Umgebung mit Hilfe von Wundauflagen im Vordergrund stehen. Bei insuffizientem Ansprechen auf die konservative Lokaltherapie kommen plastisch-chirurgische Massnahmen zum Einsatz, wobei auch eindrückliche komplexe rekonstruktive Massnahmen nur funktionieren, wenn die Grundvoraussetzungen für eine Wundheilung gewährleistet sind.
Treatment of chronic wounds in outpatient practice is a common but often frustrating undertaking. It is crucial to view the wound as a symptom of an underlying disease. Identifying and treating that disease is an absolute prerequisite for successful wound healing. Local wound care is usually interdisciplinary and follows the “TIME” concept, whereby debridement of necrosis and fibrin deposits and creating a supportive environment for wound healing using appropriate dressings are key. If there is insufficient response to conservative local therapy, plastic surgical interventions are often necessary. However even the most complex reconstructive procedures will only be successful, if the basic conditions for wound healing are ensured. Keywords: Chronische Wunden, Wundtherapie, Wundheilung, TIME-Konzept
Phasen der Wundheilung
Die physiologische Wundheilung verläuft über drei überlappende Phasen: Entzündungs-, Proliferations- und Remodellierungsphase.
Akute Wunden entstehen infolge eines zeitlich begrenzten Traumas, beispielsweise durch Schnitt-, Platz- oder Operationsverletzungen. Bei adäquater Versorgung heilen sie in der Regel innerhalb von zwei bis vier Wochen ohne weitere Intervention ab. Fehlen jedoch die Grundvoraussetzungen, dass dieser Prozess reibungslos abläuft, verbleibt die Wunde in der Entzündungsphase.
Von einer chronischen Wunde spricht man, wenn innerhalb von acht Wochen keine Heilungstendenz erkennbar ist. Sie muss vielmehr als Symptom einer Grundproblematik und nicht als eigene Erkrankung gesehen werden.
Störfaktoren der Wundheilung
Die häufigsten Störfaktoren einer physiologischen Wundheilung sind vordergründig eine unzureichende Gewebeoxygenierung, bakterielle Belastung, Biofilmbildung, sowie eine gestörte Zellmigration. Klinisch werden diese durch arterielle und venöse Durchblutungsstörungen, Druck- und Scherkräfte, Infektionen, Ödeme, Diabetes mellitus, Mangelernährung, Nikotinabusus, sowie die Einnahme immunsuppressiver oder zytotoxischer Medikamente verursacht. Ohne eine Behandlung oder zumindest Optimierung dieser Faktoren wird jede, noch so aufwändige (und teure) lokale Behandlung einer chronischen Wunde fruchtlos bleiben.
Differentialdiagnose chronischer Wunden
Im klinischen Alltag überwiegen das Ulcus cruris venosum, arterielle und gemischt-arterielle Ulzera, das diabetische Fusssyndrom, sowie der Dekubitus. Oft ist die Anamnese chronischer Wunden jedoch uneindeutig. So können einige Krankheitsbilder den Aspekt und eine suggestive Anamnese für eine chronische Wunde haben, jedoch eine komplett andere Behandlung verlangen. Zur Differentialdiagnose jeder chronischen Wunde, insbesondere bei fehlendem Therapieansprechen innerhalb von 2–4 Wochen, gehören:
• Autoimmunerkrankungen (Vaskulitiden, Lupus etc.),
• Pyoderma gangraenosum,
• maligne Entartungen (z. B. Marjolin-Ulkus),
• seltene Infektionen (z. B. Tropical ulcer),
• Drug-induced Skin Reactions (z. B. Marcoumar oder Steroide)
• faktitiöse Wunden
Diagnostik in/aus der Praxis
Die meisten Abklärungen für die Erstdiagnostik einer chronischen Wunde können gut in der Praxis durchgeführt oder veranlasst werden.
Für die klinische Wundbeurteilung ist die Dokumentation von Grösse, Tiefe, Exsudat, Wundrand, Wundumgebung und allfällig exponierte tiefere Strukturen (Knochen, Sehnen, Muskelfaszie) relevant. Hilfreich ist auch eine gute Fotodokumentation zur Verlaufsbeurteilung.
Zur weiteren Basisabklärung gehören:
– Gefässstatus (ABI, Duplex), ev. Angiographie
– Labor: Hämatogramm, Infektparameter, HbA1c,
Proteinhaushalt (Albumin/Präalbumin), Vitamine/Spurenelemente (Zink, Selen, B12, Folsäure)
– Wundabstrich (aus dem Wundgrund)
– Biopsie (immer aus dem Wundrand und nicht dem
Zentrum der Wunde)
– Röntgen/MRI (Frage nach Osteomyelitis)
– Vorliegen von Druck-/Scherkräften (schlechtes Schuhwerk, Lagerung/Sitzpolster bei Rollstuhlpatienten etc.)
Kausale Therapie als Voraussetzung der Heilung
Wurden die Grundursachen der Wunde identifiziert, ist deren Behandlung oder zumindest Optimierung vordergründig. Dazu zählen die Verbesserung der arteriellen Durchblutung (gegebenenfalls interventionell), die konsequente Druckentlastung (Spezialschuhe/Einlagen, Druckpolster), die Kompressionstherapie und die manuelle Lymphdrainage, die Optimierung der Stoffwechsellage (Einstellung des Blutzuckerspiegels, Ernährungsberatung, Substitution von Mängeln) sowie die Behandlung lokaler oder systemischer Infektionen.
Lokale Behandlung chronischer Wunden in der Praxis
Die lokale Behandlung chronischer Wunden ist oft pflegerisch sehr aufwändig und fordert sowohl vom Patienten als auch von den Fachpersonen viel Geduld. Entsprechend ist eine «Arbeitsteilung» mit einem interdisziplinären Team, bestehend aus Ärzten (Grundversorger, Chirurgen oder andere Fachspezialisten), Wundexperten/Pflegefachperson und Physiotherapeuten, unumgänglich.
Die moderne Wundtherapie orientiert sich am TIME-Konzept: Tissue (Débridement), Infection/Inflammation (Keimkontrolle), Moisture (Exsudatmanagement) und Edge (Förderung der Epithelisierung) (1).
Insbesondere beim Débridement wird oft zu zögerlich vorgegangen. Allgemein gilt: sämtliches infiziertes und/oder nekrotisches Gewebe muss entfernt werden. Dies auch wenn es sich hierbei um eine grundsätzlich relevante anatomische Struktur handelt (z. B. Haut, Sehne oder Muskel). Wundbeläge, beispielsweise Fibrin, sowie feuchtes, nekrotisches Gewebe sind ein ausserordentlich guter Nährboden für Bakterien, weshalb ein radikales, manchmal auch wiederholtes Débridement für die Heilung entscheidend ist. Kleinere Nekrosen oder Fibrinbeläge können gut ohne Anästhesie oder nur in Lokalanästhesie in der Praxis entfernt werden.
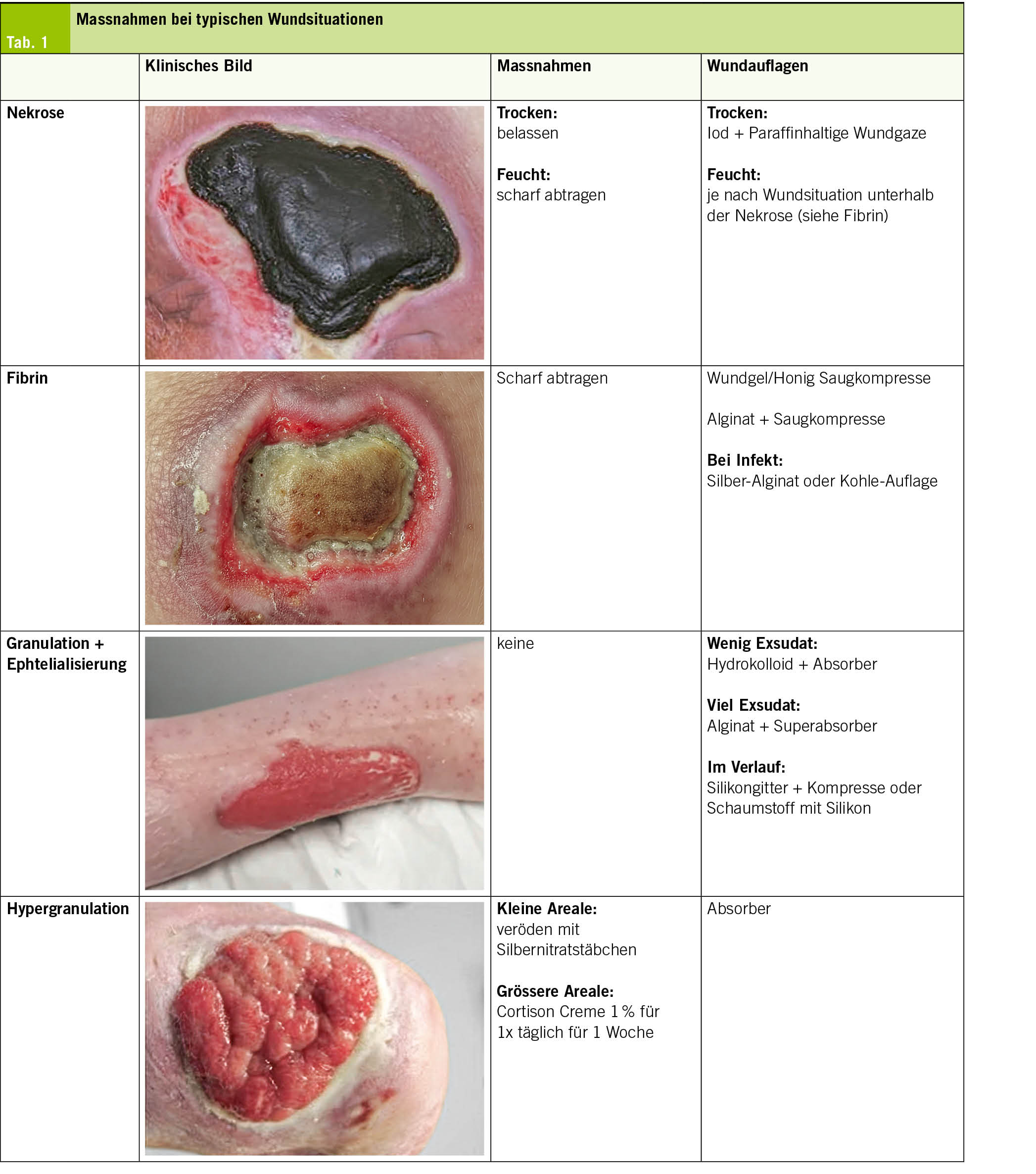
Ablauf einer typischen Wundbehandlung: (Tab. 1)
1. Beurteilung und Dokumentation
2. Spülung/Nassphase (keine Angst vor Leitungswasser!)
3. Débridement
4. Wundrandschutz
5. Auflage
6. Fixation
Wundauflagen
Die Infektionskontrolle, das Exsudatmanagement und die Förderung der Epithelisierung können durch die korrekte Wahl von Wundauflagen wesentlich gefördert werden. Die Plethora an Wundauflagen auf dem Markt ist überwältigend, was darauf hinweist, dass es keine «beste» Wundauflage gibt. Je nach Wundsituation kommen Schaumverbände, Alginate, Hydrogele, Hydrokolloide oder antimikrobielle Auflagen (z. B. Silber, Iod) zum Einsatz. Die Auswahl richtet sich nach der Wundphase und der Exsudatmenge (Tab. 2).
«Neuere» alternative Wundauflagen
Moderne dermale Matrizes bzw. Dermisäquivalente für chronische Wunden umfassen heute vor allem Kerecis, azelluläre dermale Matrizes und kollagenbasierte, proteasemodulierende Systeme. Kerecis ist eine azelluläre Fischhautmatrix (Kabeljau) mit natürlichem Kollagen und Omega-3-Fettsäuren. Sie dient als biologisches 3-D-Gerüst zur Zellmigration und Gefässneubildung. Ähnlich funktionieren auch azelluläre dermale Matrizes aus humaner Spenderhaut wie AlloDerm® oder DermaGraft® (dezellularisierte ECM), die bei diabetischen Fussulzera und komplexen chronischen Wunden genutzt werden. Xenogene Matrizes wie Oasis® (Schweinedarm-Submukosa), Integra® oder Matriderm® (Kollagen-/ECM-Gerüste) fördern insbesondere die Granulation und spätere Epithelisierung bei venösen und arteriellen Ulzera.
Ergänzend kommen Kollagen- bzw. proteasemodulierende Matrizes wie Promogran Prisma® oder Suprasorb® C zum Einsatz, die überschüssige Proteasen binden und so das gestörte Wundmilieu chronischer, vor allem diabetischer und venöser Ulzera stabilisieren. Alle diese «Advanced Methods» sind sehr teuer und in der Schweiz streng reguliert. Sie werden deshalb erst eingesetzt, wenn die Ursachenbehandlung erfolgt ist und unter optimaler Standardtherapie über mindestens 6–8 Wochen kein ausreichender Heilungsfortschritt erzielt wurde.
Wann soll die Überweisung zum Chirurgen erfolgen?
Der Verlauf chronischer Wunden ist auch unter besten Voraussetzungen, wie der Name impliziert, langwierig. Entsprechend kann es schwierig sein, den adäquaten Zeitpunkt für einen Strategiewechsel oder eine Strategieerweiterung zu identifizieren. Generell sollte – unter der Voraussetzung, dass allfällige Grunderkrankungen behandelt sind/werden – innerhalb von 3–4 Wochen nach Einleitung einer lokalen Behandlung eine sichtbare Verbesserung zu erkennen sein. Hierzu ist, wie erwähnt, die regelmässige Fotodokumentation sehr hilfreich. Ist dies nicht der Fall, ist eine konsiliarische Beurteilung durch einen (plastischen) Chirurgen empfehlenswert.
Zu den chirurgischen Massnahmen gehört an oberster Stelle das scharfe Débridement von sämtlichem nekrotischem Gewebe und die Drainage von allfälligen Abszessen, sowie gegebenenfalls die Entnahme von Gewebe-/Knochenbiopsien. Nicht selten wird zur sogenannten Wundkonditionierung vorübergehend eine NPWT-Therapie angewendet. Diese stimuliert die Perfusion und Neovaskularisation sowie die Bildung von Granulationsgewebe. Zudem wird Exsudat direkt abtransportiert, die Keimfreiheit gefördert und tägliche Verbandswechsel vermieden, was insbesondere bei ambulanten Behandlungen von Vorteil ist. Kleinere Wunden können mit NPWT oft ohne weitere Intervention zur Heilung gebracht werden. Bei grösseren Wunden kommen bei oberflächlichen Wunden Hauttransplantate und bei tieferen Wunden, insbesondere mit exponierten tieferen Strukturen wie Knochen oder Sehnen, gestielte oder freie Lappenplastiken zum Einsatz.
Copyright
Aerzteverlag medinfo AG
Dr. med. Natasha Forster
Swissparc – Klinik für Plastische Chirurgie
und Dermatologie
Steinentischstrasse 5
8002 Zürich
Die Autorin hat keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
Chronische Wunden sind als Symptom einer Grunderkrankung zu verstehen
Kausale Therapie steht vor der lokalen Wundbehandlung
Bei ungenügendem Therapieansprechen innerhalb 4 Wochen: Differentialdiagnosen ausschliessen, ggf. Chirurgen involvieren
Behandlung im Team (Ärzte, Wundexperten, Pflegefachpersonen, Physiotherapie)
1. AWMF S3-Leitlinie Lokaltherapie schwerheilender und/oder chronischer Wunden aufgrund von peripherer arterieller Verschlusskrankheit, Diabetes Mellitus oder chronischer venöser Insuffizienz (https://register.awmf.org/de/leitlinien/detail/091-001)
2. European Wound Management Association – The EWMA wound care resource library (https://ewma.org/resource-library)
Schützt Nachholungsschlaf am Wochenende vor Demenzerkrankung?
Schlafmangel wird mit einem höheren Demenzrisiko in Verbindung gebracht (1), aber die Rolle des Erholungsschlafs am Wochenende (weekend recovery sleep – WRS) bei der Minderung dieses Risikos ist noch unklar. Ziel einer prospektiven Kohortenstudie der UK-Biobank (2) ist es, dies zu untersuchen.
Methoden
Diese prospektive Kohortenstudie verfolgte 88 592 demenzfreie Erwachsene im Alter von 40 bis 79 Jahren aus der UK Biobank und verwendete Handgelenk-Beschleunigungsmesser, um die durchschnittliche Schlafdauer an Wochentagen und Wochenenden zu messen. Das Auftreten von Demenz (Demenz aller Ursachen, Alzheimer-Krankheit [AD], vaskuläre Demenz [VaD] und unspezifische Demenz) wurde anhand von Krankenakten ermittelt. Die Zusammenhänge wurden unter Verwendung von Cox-Proportional-Hazards-Modellen und eingeschränkten kubischen Splines (RCS) geschätzt.
Ergebnisse
Von den 88 592 Teilnehmern (Durchschnittsalter [SD]: 61.9 [7.9] Jahre) entwickelten 735 (0.83 %) eine Demenz, darunter 308 (0.35 %) Fälle von AD, 137 (0.15 %) Fälle von VaD und 319 (0.36 %) Fälle von unspezifischer Demenz. RCS-Analysen ergaben optimale Schlafdauern an Wochentagen, die mit dem geringsten Demenzrisiko verbunden waren: 8.38 Stunden (HR, 0.73; 95 % CI 0.64–0.84) für Demenz aller Ursachen, 8.33 Stunden (HR, 0,72; 95 % CI 0.58–0.89) für AD und 9.07 Stunden (HR, 0.59; 95 % CI 0.40–0.88) für VaD. In der Gruppe mit suboptimalem Schlaf (Schlafdauer an Wochentagen unter der optimalen Dauer) war eine längere WRS mit einem verringerten Risiko für Demenz aller Ursachen (HR, 0.801; 95 % KI 0.717–0.893) und VaD (HR, 0.747; 95 % KI 0.612–0.91) verbunden. In der Gruppe mit verlängertem Schlaf (Schlafdauer an Wochentagen über der optimalen Dauer) war eine längere WRS jedoch mit einem erhöhten Risiko für unspezifische Demenz verbunden (HR, 1.291; 95 % KI 1.087–1.533)
Schlussfolgerung
Eine angemessene WRS kann das Demenzrisiko, insbesondere für VaD, nach unzureichendem Schlaf an Wochentagen verringern, was die Bedeutung einer angemessenen WRS für die kognitive Gesundheit unterstreicht.
1. Müller, T. Langzeitstudie: Wenig Schlaf, erhöhtes Alzheimer-Risiko. InFo Neurologie 2021; 23: 58 https://doi.org/10.1007/s15005-021-2071-9
2. Zhao, B., Zhou, S., Chang, J. et al. Association between weekend recovery sleep and risk of incident dementia: a prospective cohort study in the UK Biobank. J Neurol 2025;272: 612. https://doi.org/10.1007/s00415-025-13363-y
Die Alzheimer-Krankheit steht exemplarisch für die grossen Herausforderungen – und zunehmend auch für die neuen Chancen – der modernen Altersmedizin.
Über Jahrzehnte war unsere therapeutische Rolle im Wesentlichen symptomlindernd, während Pathophysiologie und Krankheitsverlauf nur begrenzt beeinflussbar schienen. Mit den neuen Anti-Amyloid-Therapien zeichnet sich nun ein echter Paradigmenwechsel ab, der auch für die hausärztliche Versorgung von hoher Relevanz ist.
Der vorliegende Beitrag von Marc Aurel Busche und Kolleginnen und Kollegen ordnet diesen Wandel präzise, differenziert und praxisnah ein. Er zeigt, dass krankheitsmodifizierende Therapien wie Lecanemab und Donanemab weder Wundermittel noch ferne Zukunftsmusik sind, sondern einen klar definierten Platz im frühen Stadium der Alzheimer-Krankheit einnehmen. Entscheidend ist dabei weniger die einzelne Substanz als das dahinterliegende Prinzip: frühe Erkennung, präzise Diagnostik und strukturierte Zusammenarbeit.
Gerade Hausärztinnen und Hausärzte spielen in diesem neuen Versorgungspfad eine Schlüsselrolle. Sie sind meist die ersten Ansprechpersonen bei subtilen kognitiven Veränderungen und damit die entscheidenden «Gatekeeper», damit das therapeutische Zeitfenster nicht verpasst wird. Das Motto «time is brain» gilt heute nicht mehr nur für den Schlaganfall, sondern zunehmend auch für die Alzheimer-Diagnostik und -Therapie.
Der Artikel vermittelt nicht nur den aktuellen Stand der Evidenz, sondern auch die notwendige klinische Nüchternheit: Nutzen und Risiken müssen sorgfältig abgewogen, Patientinnen und Patienten gezielt ausgewählt und eng begleitet werden. Die Schweiz ist mit ihrem dichten Netzwerk an Memory Clinics und klaren nationalen Empfehlungen dafür hervorragend positioniert.
Als Herausgeber des Geriatrie Forums ist es mir ein besonderes Anliegen, solche Beiträge einzuordnen, die Brücken schlagen – zwischen Grundlagenforschung und klinischem Alltag, zwischen Spezialdisziplinen und Hausarztmedizin, zwischen internationaler Evidenz und nationaler Umsetzung.
Dieser Beitrag leistet genau das und bietet Ihnen eine fundierte, verlässliche Orientierung für Gespräche mit Patientinnen, Patienten und Angehörigen in einer Phase, in der sich die Demenzmedizin grundlegend weiterentwickelt.
Ich wünsche Ihnen eine anregende Lektüre.
Herausgeber Geriatrie Forum
PD Dr. med. Mathias Schlögl, MPH, EMBA HSG
Chefarzt Geriatrie
Stv. Leiter Department Innere Medizin
5017 Barmelweid
Demenzen werden in der Schweiz immer häufiger und zählen zu den führenden Todesursachen. Sie gehören zu den wichtigsten Erkrankungen hinsichtlich verlorener Lebensjahre, da sie reduzierte Lebenserwartung und krankheitsbedingten Verlust an Lebensqualität kombinieren. Die Alzheimer-Krankheit ist mit rund zwei Dritteln aller Fälle die häufigste Ursache einer Demenz. Mit Lecanemab und Donanemab stehen erstmals krankheitsmodifizierende Therapien zur Verfügung, die pathologisches Amyloid-β – ein Protein, das sich bei Alzheimer krankhaft im Gehirn ablagert – gezielt entfernen und den Krankheitsverlauf verlangsamen. Dieser Wandel von rein symptomatischer zu ursächlich wirksamer Behandlung rückt die Früherkennung und präzise Patientenselektion noch stärker in den Vordergrund. Hausärzt/-innen spielen dabei eine entscheidende Rolle, indem sie früheste kognitive Störungen erkennen und Betroffene bei Verdacht auf eine Alzheimer-Krankheit rasch an eine Memory Clinic überweisen. Ein dichtes Netz spezialisierter Memory Clinics sowie einheitliche nationale Empfehlungen sichern in der Schweiz eine koordinierte und leitliniengerechte Einführung dieser neuen Therapien, sobald eine Zulassung auch in der Schweiz erfolgt ist. Die neuen monoklonalen Antikörper stellen einen echten Fortschritt in der Alzheimer-Therapie dar. Sie eröffnen Patient/-innen im Frühstadium erstmals die Möglichkeit, den Krankheitsverlauf zu beeinflussen. Damit gewinnt das Prinzip «time is brain» auch in der Alzheimer-Diagnostik und -Therapie an Bedeutung.
Dementia is becoming increasingly common in Switzerland and is one of the leading causes of death. It is one of the most significant diseases in terms of years of healthy life lost, as it combines reduced life expectancy with a disease-related loss of quality of life. Alzheimer’s disease is the most common cause of dementia, accounting for around two-thirds of all cases. Lecanemab and donanemab are the first disease-modifying therapies available that specifically remove pathological amyloid-ß – a protein that accumulates abnormally in the brain in Alzheimer’s disease – and slow the progression of the disease. This shift from purely symptomatic to causally effective treatment places even greater emphasis on early detection and precise patient selection. Family doctors play a crucial role in this by recognizing the earliest cognitive disorders and quickly referring those affected to a memory clinic if Alzheimer’s disease is suspected. A dense network of specialized memory clinics and uniform national recommendations ensure the coordinated and guideline-compliant introduction of these new therapies in Switzerland as soon as they are approved there. The new monoclonal antibodies represent a real advance in Alzheimer’s therapy. For the first time, they offer patients in the early stages the opportunity to influence the course of the disease. This means that the principle of «time is brain» is also gaining importance in Alzheimer’s diagnosis and therapy. Keywords: Alzheimer-Krankheit, Anti-Amyloid-Antikörper (Lecanemab/Donanemab), ARIA und MRT-Monitoring
Die Alzheimer-Krankheit ist die häufigste Ursache einer Demenz. Sie nimmt auch in der Schweiz weiter zu und verursacht bereits heute Kosten in Milliardenhöhe. Angesichts der schweren Belastung von Patient/-innen und Angehörigen durch den fortschreitenden Verlust von Selbstständigkeit, Pflegebedürftigkeit und verminderter Lebensqualität besteht ein enormer Bedarf an neuen Therapien, die den Krankheitsverlauf verlangsamen. Was bei Multipler Sklerose oder Krebserkrankungen längst selbstverständlich ist, wird nun auch in der Therapie der Alzheimer-Krankheit Realität: krankheitsmodifizierende Behandlungen. Erstmals stehen monoklonale Antikörper zur Verfügung, die pathologisches Amyloid-β (Aβ) gezielt entfernen und den Krankheitsverlauf nachweislich verlangsamen. Das markiert einen Paradigmenwechsel und rückt die Früherkennung sowie die präzise Patientenauswahl stärker in den Fokus.
Diagnostik und Therapie der Alzheimer-Krankheit in der Schweiz
Die Schweiz verfügt im internationalen Vergleich über ein hervorragendes Netzwerk an Memory Clinics, welche in Zusammenarbeit mit den Hausärzt/-innen sowie weiteren Fachspezialist/-innen die Versorgung der Menschen mit Demenz auf hohem Niveau sicherstellen. Im Verein Swiss Memory Clinics (SMC) sind alle grösseren Memory Clinics der Schweiz (aktuell: 54) organisiert. Alle Mitglieder folgen den gleichen Empfehlungen zur Diagnostik und Therapie, so dass schweizweit ein hoher einheitlicher Standard gewährleistet werden kann, wobei die regionalen Besonderheiten dennoch sehr gut berücksichtigt werden können. Mit der Verfügbarkeit krankheitsmodifizierender Therapien verschiebt sich die Schwelle für eine Überweisung. Ein abwartendes Vorgehen mit Kontrolle nach mehreren Monaten kann heute dazu führen, dass das therapeutische Zeitfenster verpasst wird. Bei neuen kognitiven Beschwerden im Frühstadium sollte daher die zeitnahe Abklärung in einer Memory Clinic niedrigschwellig erfolgen.
Unter der Leitung von SMC wurden in den vergangenen Jahren diverse Empfehlungen publiziert, welche in übersichtlicher Form im Internet frei zugänglich sind (https://www.swissmemoryclinics.ch/qualitaetsentwicklung/qualitaetsstandards/). Es existieren Empfehlungen für die Diagnostik von Demenzerkrankungen in der Schweiz (1, 2), ergänzt durch spezialisierte Empfehlungen für den Einsatz von Biomarkern (3) und PET-Diagnostik (4). 2024 wurden im Auftrag des Bundesamtes für Gesundheit (BAG) Empfehlungen für die Therapie der Demenz in der Schweiz publiziert (5), zudem unter der Führung der Schweizerischen Gesellschaft für Alterspsychiatrie (SGAP) spezifische Empfehlungen für die Therapie von Verhaltenssymptomen (BPSD) (6) und zuletzt auch Anwendungsempfehlungen für die neuen Anti-Amyloid-Antikörper (7).
Aβ als therapeutisches Ziel bei Alzheimer
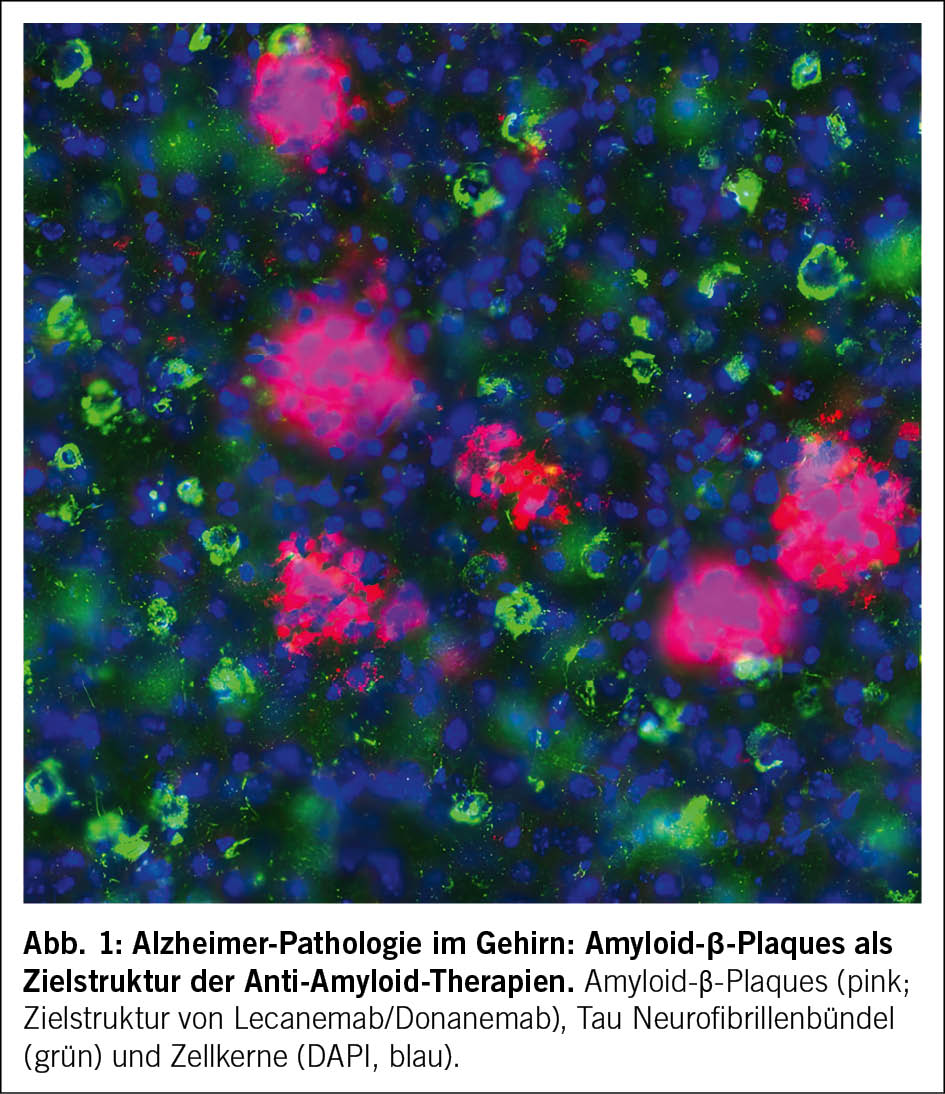
Amyloid (Aβ) und Tau sind die zentralen pathologischen Proteine der Alzheimer-Krankheit. Ihre Ablagerungen in Form von extrazellulären Aβ-Plaques und intrazellulären Tau-Neurofibrillenbündeln schädigen Synapsen und Nervenzellen und treiben die Neurodegeneration voran (8). Diese beiden typischen Proteinablagerungen (Aβ‑Plaques und Tau‑Neurofibrillenbündel) sind in Abb. 1 dargestellt. Die zentrale Rolle von Aβ wird durch genetische Befunde gestützt. Mutationen in den Genen, die an der Aβ-Produktion beteiligt sind, führen zu familiären, früh einsetzenden Formen der Erkrankung, während Varianten, die die Bildung von Aβ verringern, das Erkrankungsrisiko senken (9, 10).
Monoklonale Antikörper gegen Aβ – Wirkprinzip und klinische Bedeutung
Lecanemab ist ein humanisierter monoklonaler Antikörper, der gezielt lösliche protofibrilläre Aβ-Spezies bindet. Diese löslichen Aggregate gelten als besonders neurotoxisch, da sie früh synaptische Dysfunktion auslösen und zur Aktivierung von Mikroglia beitragen (11, 12). Neuere Daten zeigen zudem, dass Lecanemab auch fibrilläres (plaquegebundenes) Aβ bindet (13). Donanemab bindet bevorzugt N-terminal pyroglutamat-modifiziertes Aβ, eine besonders stabile Spezies, die in reifen Plaques angereichert ist. Durch die Bindung fördert Donanemab die Mikroglia-vermittelte Plaque Clearance, was zu einer Reduktion der Plaquelast führt (14).
Lecanemab und Donanemab sind inzwischen in zahlreichen Ländern zugelassen, teils beide, teils nur eine der Substanzen. Die Entscheidung der Swissmedic für die Schweiz steht aktuell (Stand Dezember 2025) noch aus. Diese neuen Therapien markieren einen wichtigen Fortschritt, ersetzen jedoch nicht die bestehenden symptomatischen Behandlungen (Cholinesterasehemmer und Memantin), die weiterhin konsequent angewendet werden sollten. Sie bilden nach wie vor die Grundlage einer modernen, ganzheitlichen Alzheimer-Therapie.
Lecanemab
In der Phase-3-Studie Clarity-AD wurde Lecanemab bei 1795 Personen mit früher Alzheimer-Krankheit, definiert als leichte kognitive Störung oder milde Demenz bei nachgewiesener Aβ-Positivität, untersucht (15). Die Teilnehmenden erhielten 10 mg/kg Lecanemab intravenös alle zwei Wochen oder Placebo über 18 Monate. Lecanemab reduzierte die Aβ-Plaques im Gehirn deutlich, im PET war bei vielen Patient/-innen eine nahezu vollständige Entfernung nachweisbar.
Der primäre klinische Endpunkt, die Veränderung der Clinical Dementia Rating – Sum of Boxes (CDR-SB), einer Skala zur Bewertung kognitiver und alltagspraktischer Fähigkeiten, zeigte eine 0.45-Punkte geringere Zunahme als unter Placebo (p < 0.001), entsprechend einer Verlangsamung der Krankheitsprogression um 27 %. Dies entspricht grob einer Verzögerung um fünf bis sechs Monate über 18 Monate, also einem späteren Übergang in ein schwereres klinisches Stadium. Die Number Needed to Treat (NNT, bezogen auf Verschlechterung im CDR‑Global über 18 Monate) lag bei rund 13 (17). Das heisst: Von 100 vergleichbaren Patient/-innen zeigen über 18 Monate etwa 8 weniger eine solche Verschlechterung als unter Placebo. Auch die sekundären Endpunkte, die die Gedächtnisleistung, die Denkgeschwindigkeit und die Alltagsfunktionen abbilden (z. B. ADAS-Cog14, ADCOMS, ADCS-MCI-ADL), fielen konsistent zugunsten von Lecanemab aus. In einigen Analysen zeigten sich zudem Vorteile in von Patient/-innen und Angehörigen berichteten Endpunkten zur Lebensqualität und Belastung (EQ 5D, QoL AD und das Zarit Burden Interview) (16). Biomarkeranalysen zeigten eine Abnahme von p-Tau181 (CSF und Plasma) und des glialen Aktivierungsmarkers GFAP, während NfL unverändert blieb.
Langzeitdaten über 48 Monate aus einer offenen Verlängerungsphase (18) deuten auf einen anhaltenden klinischen Vorteil hin. Im Vergleich zur unbehandelten ADNI- Kohorte war das Risiko, in ein fortgeschritteneres Krankheitsstadium überzugehen, um 34 %, und das Risiko, von einer leichten kognitiven Störung in eine Alzheimer-Demenz überzutreten, um 56 % reduziert. Etwa die Hälfte der Behandelten zeigte über vier Jahre keinen messbaren kognitiven Abfall.
Donanemab
In der Phase-3-Studie TRAILBLAZER-ALZ-2 wurde Donanemab bei 1736 Personen mit früher Alzheimer-Krankheit (leichte kognitive Störung oder milde Demenz) über 18 Monate untersucht (19). Die Teilnehmenden erhielten Donanemab intravenös einmal monatlich oder Placebo; in den ersten drei Zyklen wurden 700 mg verabreicht, danach 1400 mg bis Studienende. Die Teilnehmenden wurden anhand der Tau-Pathologie im Tau-PET in Gruppen mit niedriger/mittlerer und hoher Tau-Belastung eingeteilt. Donanemab reduzierte die Aβ-Plaques im Gehirn deutlich. Bereits nach 24 Wochen war bei über einem Drittel der Behandelten keine Plaques mehr nachweisbar, nach 18 Monaten bei mehr als 80 %.
In der Gruppe mit niedriger/mittlerer Tau-Belastung verlangsamte Donanemab die Krankheitsprogression hinsichtlich des primären Endpunktes (iADRS, kombiniert Kognition und Alltagsfunktionen) um 35 % gegenüber Placebo. Dies entspricht einer Verzögerung der Krankheitsprogression um rund fünf bis sechs Monate über 18 Monate sowie einer NNT (bezogen auf Verschlechterung im CDR‑Global über 18 Monate) von etwa 10 (17). Das heisst: Von 100 vergleichbaren Patient/-innen zeigen über 18 Monate etwa 10 weniger eine solche Verschlechterung als unter Placebo. Der Nutzen war am grössten bei niedriger oder mittlerer Tau-Belastung; bei fortgeschrittener Tau-Pathologie fiel er geringer aus. Auch die sekundären Endpunkte (CDR-SB, ADCS-iADL, ADAS-Cog13) zeigten eine konsistente Verlangsamung kognitiver und funktioneller Einbussen unter Donanemab. Begleitende Biomarkeranalysen belegten eine deutliche Abnahme von Plasma-p-Tau217 sowie eine stabile oder rückläufige Tau-PET-Signalintensität, was auf eine reduzierte Tau-Propagation hindeuten könnte.
Hervorzuheben ist der treat-to-target-Ansatz. Donanemab wird dabei so lange gegeben, bis im Amyloid-PET keine oder nur noch eine minimale Plaquelast nachweisbar ist, und kann dann pausiert oder beendet werden. In den bisherigen Verlaufsdaten blieb der klinische Vorteil nach Erreichen dieses Ziels über den beobachteten Zeitraum erhalten. Langzeitdaten über bis zu 36 Monate deuten darauf hin, dass sich der Abstand zu unbehandelten Beobachtungskohorten (z. B. ADNI) weiter vergrösserte (20); der Unterschied im CDR-SB nahm von 0.6 nach 18 Monaten auf 1.2 Punkte nach 36 Monaten zu. Eine frühe Behandlung reduzierte damit das Risiko einer klinischen Progression um 27 %.
Wirksamkeit in der Praxis
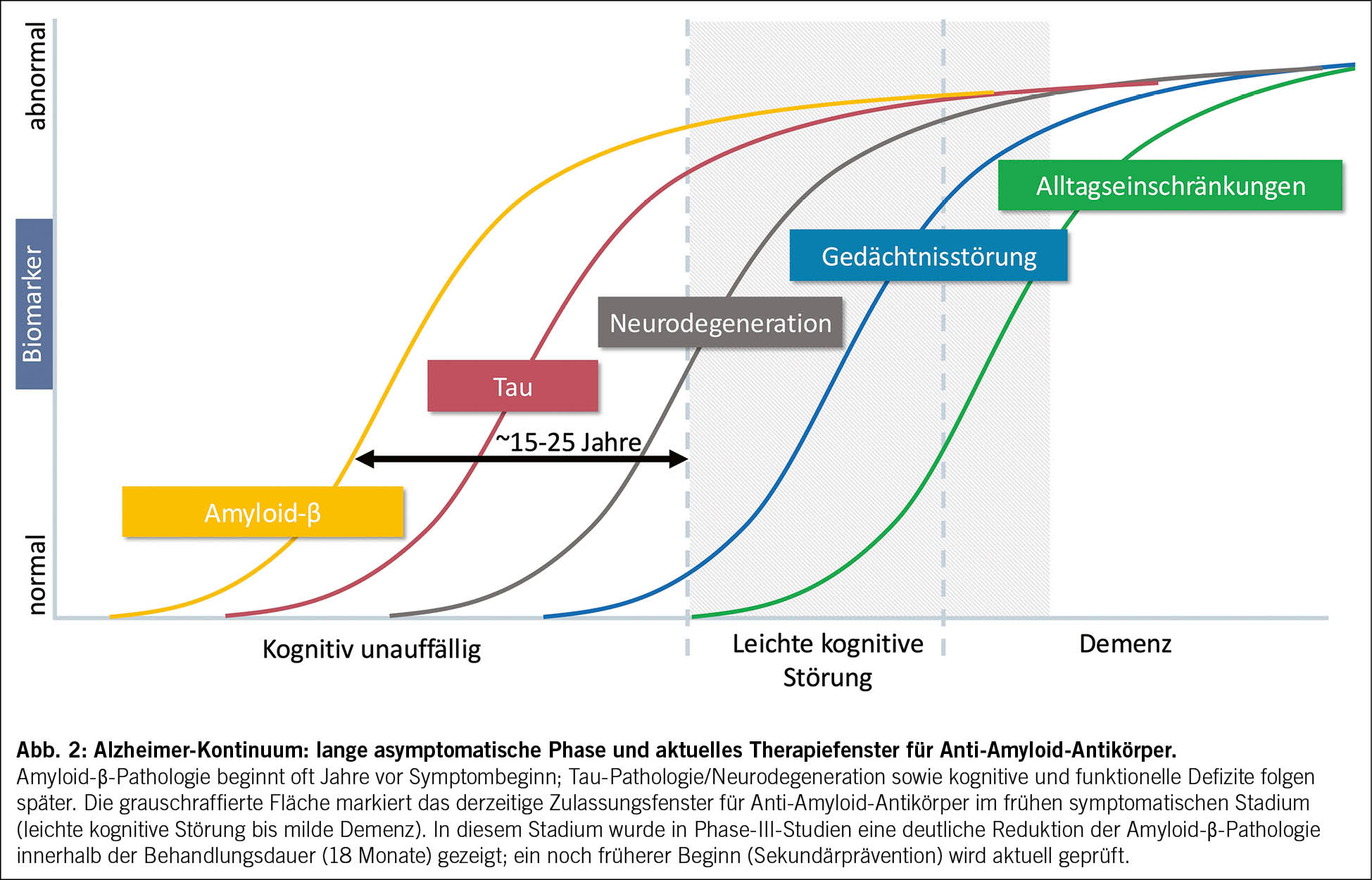
Lecanemab und Donanemab verlangsamen somit den klinischen Verlauf von Alzheimer um etwa ein Viertel bis ein Drittel, was einem Zeitgewinn von rund sechs Monaten über 18 Monate entspricht. Der grösste Nutzen wird bei Patient/-innen im Frühstadium einer Alzheimer-Krankheit erzielt, weshalb eine biomarkerbasierte Frühdiagnostik entscheidend ist. Mit einer NNT von etwa 10 bis 13 und einer Verzögerung des klinischen Verlaufs um 27–36 % liegen die Effektstärken dieser Antikörper in einer ähnlichen Grössenordnung wie jene etablierter Biologika bei anderen chronischen Erkrankungen, etwa bei Multipler Sklerose, Rheumatoider Arthritis oder onkologischen Erkrankungen (25). Angesichts der hohen gesellschaftlichen und pflegerischen Belastung durch Demenz dürfte eine Verzögerung der Krankheitsprogression auch ökonomisch relevant sein. Gleichzeitig wird nur ein kleiner Anteil der Patient/-innen die Kriterien für eine Antikörpertherapie erfüllen (22). Zu beachten ist, dass ein relevanter Teil der Patient/-innen, die aktiv eine Antikörpertherapie anstreben, bei Erstkontakt bereits zu weit fortgeschritten und damit nicht mehr geeignet ist. (26). Für die grosse Mehrheit bleibt eine strukturierte postdiagnostische Versorgung mit Beratung, Behandlung von Begleiterkrankungen und Angehörigenunterstützung zentral. Das Krankheitskontinuum und das derzeitige (internationale) Zulassungsfenster der Anti‑Amyloid‑Antikörper sind in Abb. 2 schematisch dargestellt.
Indikation, Durchführung und Monitoring
Lecanemab und Donanemab sind in der Schweiz derzeit noch nicht zugelassen (Stand Dezember 2025). Für die klinische Vorbereitung dienen die europäischen Zulassungen, internationale Appropriate Use Criteria (22, 23) und die Empfehlungen der Swiss Memory Clinics (7) als Orientierung. Beide Therapien sind für Menschen im Frühstadium der Alzheimer-Krankheit vorgesehen, das heisst bei leichter kognitiver Störung oder milder Demenz im Rahmen einer Alzheimer-Krankheit, wenn eine Aβ-Pathologie nachgewiesen ist und das zerebrale MRT keine relevanten Blutungen, keine superfizielle Siderose und keine ausgeprägte Mikroangiopathie zeigt. Der Nachweis einer Aβ-Pathologie kann über Liquorbiomarker (Aβ42/40-Ratio, p-Tau) oder ein Amyloid-PET erfolgen. Bluttests wie p-Tau217 gewinnen an Bedeutung, sind derzeit aber nur als ergänzende Nachweismethode geeignet. In der Praxis sind grenzwertige Befunde und Unterschiede zwischen Testplattformen zu berücksichtigen, weshalb Blutmarker derzeit vor allem zur gezielten Triage und zur Ergänzung, nicht als alleinige Bestätigung dienen sollten. Da das Risiko für Nebenwirkungen vom APOE-Genotyp abhängt, sollte dieser vor Therapiebeginn bestimmt und besprochen werden. In der EU und in Grossbritannien sind homozygote APOE-ε4-Trägerinnen und -Träger wegen des höheren ARIA-Risikos derzeit nicht zugelassen. Dabei ist zu beachten, dass die ApoE-Genotypisierung lebenslange Risikoinformationen berührt und auch für Angehörige relevant sein kann. Patient/-innen unterscheiden sich deutlich in ihrer Präferenz, genetische Risikoinformationen zu kennen, weshalb eine strukturierte Aufklärung und bei Bedarf genetische Beratung wichtig ist. Eine orale Antikoagulation sowie mehr als vier Mikroblutungen oder eine superfizielle Siderose im MRT schliessen die Therapie aus. Eine Monotherapie mit Thrombozyten-aggregationshemmern (z. B. ASS) ist in der Regel möglich; für eine duale Plättchenhemmung liegen bislang zu wenige Daten vor.
Lecanemab wird alle zwei Wochen als intravenöse Infusion verabreicht (10 mg/kg Körpergewicht, über etwa eine Stunde).
Donanemab wird einmal monatlich über etwa 30 Minuten intravenös infundiert. In der Phase-3-Studie (TRAILBLAZER-ALZ-2) erhielten die Teilnehmenden 700 mg Donanemab für die ersten drei Gaben und anschliessend 1400 mg alle vier Wochen bis Woche 72. In der nachfolgenden TRAILBLAZER-ALZ-6-Studie wurde ein langsames Titrationsschema getestet (350 → 700 → 1050 → 1400 mg), das die Häufigkeit von ARIA-E deutlich senkte bei unveränderter Aβ-Reduktion (24).
In den ersten Zyklen ist bei beiden Therapien eine etwas längere Überwachung vorgesehen, da zu Beginn bei 20–30 % milde Reaktionen auftreten können (z. B. Kopfschmerz, Schüttelfrost, Blutdruckanstieg). Die Behandlung wird beendet, wenn die Erkrankung in eine Demenz mittleren Schweregrades übergeht.
Für beide Anti‑Amyloid‑Therapien ist ein strukturiertes MRT‑Monitoring zur ARIA-Früherkennung obligat; die empfohlenen Zeitpunkte sind jedoch substanzspezifisch. Vor Therapiebeginn ist ein Baseline‑MRT (in der Regel innerhalb der letzten 6 Monate) erforderlich, um vorbestehende Mikroblutungen (insbesondere >4), eine superfizielle Siderose oder relevante Leukoenzephalopathie auszuschliessen. Unter Lecanemab sind gemäss EU‑Fachinformation Kontroll‑MRTs vor der 3., 5., 7. und 14. Gabe vorgesehen; bei Symptomen, die an ARIA denken lassen, sollen jederzeit zusätzliche MRTs erfolgen. Unter Donanemab erfolgen neben dem Baseline-MRT weitere Kontrollen vor der 2., 3., 4. und 7. Gabe; ein zusätzliches MRT nach einem Jahr Therapie (vor der 12. Infusion) ist bei erhöhtem ARIA‑Risiko (z. B. ApoE ε4‑Heterozygotie) und/oder nach vorausgegangenen ARIA‑Ereignissen empfohlen. Die Verlaufskontrollen sollten möglichst am gleichen Scanner und mit identischem Sequenzprotokoll (wenn verfügbar 3‑Tesla) erfolgen, um subtile Veränderungen zuverlässig zu erkennen. Donanemab kann im Rahmen des treat‑to‑target-Ansatzes nach dokumentierter Amyloid‑Clearance pausiert oder beendet werden; gemäss EU‑Zulassung soll die Behandlungsdauer 18 Monate nicht überschreiten.
Nebenwirkungen und Sicherheit
Die neuen krankheitsmodifizierenden Therapien gelten insgesamt als gut verträglich, erfordern jedoch ein strukturiertes Monitoring. Die Aufklärung und Einwilligung sind im Frühstadium nicht trivial, weil Nutzen und Risiken moderat bis relevant sind und die Behandlung für Patient/-innen und Angehörige durch regelmässige Infusionen und MRT-Monitoring belastend sein kann. Zusätzlich kann fehlende Krankheitseinsicht die gemeinsame Entscheidungsfindung erschweren, weshalb eine besonders personenzentrierte Kommunikation und die Einbindung von Angehörigen wichtig sind. Die klinisch wichtigsten Nebenwirkungen sind Infusionsreaktionen und Amyloid-bezogenen Bildgebungsanomalien (amyloid-related imaging abnormalities, ARIA).
Infusionsreaktionen treten vor allem zu Beginn der Behandlung auf, insbesondere in den ersten Infusionszyklen. In den Phase‑III‑Studien wurden akute Infusionsreaktionen unter Lecanemab bei etwa 1 von 4, unter Donanemab bei etwa 1 von 13 Behandelten beobachtet; schwere Reaktionen waren selten (~1.2 % unter Lecanemab, 0.3 % unter Donanemab). Real-World-Daten bestätigen diese Häufigkeit und zeigen, dass Infusionsreaktionen auch ausserhalb von Studien meist mild bis moderat verlaufen (Kopfschmerz, Schüttelfrost, Übelkeit oder Blutdruckanstieg) und gut beherrschbar sind (21, 26). Sie bessern meist nach kurzzeitiger Unterbrechung der Infusion oder symptomatischer Behandlung (z. B. Paracetamol, Antihistaminikum). Eine Standard-Prämedikation mit Antihistaminikum und Paracetamol kann das Risiko zusätzlich senken. Schwere Reaktionen oder Anaphylaxien sind selten (~ 1 %).
ARIA entstehen durch vorübergehende Flüssigkeitseinlagerungen oder Mikroblutungen im Gehirn. Diese sind im MRT gut zu erkennen, weshalb im Verlauf der Behandlung regelmässige MRT-Kontrollen erforderlich sind. Man unterscheidet ARIA-E («E» für edema), die auf vasogene Schwellungen hinweist, und ARIA-H («H» für haemorrhage), die Mikroblutungen oder Hämosiderinablagerungen im MRT beschreibt. ARIA können in Einzelfällen Symptome wie z. B. Kopfschmerzen, Schwindel, Sehstörungen oder Gangunsicherheit verursachen und sind in den meisten Fällen reversibel; seltene schwere Verläufe sind jedoch klinisch relevant und müssen konsequent behandelt werden. Die «80–80–80‑Regel» ist für die Aufklärung hilfreich: Rund 80 % der ARIA treten innerhalb der ersten vier Monate auf, etwa 80 % verlaufen ohne Symptome, und etwa 80 % bilden sich innerhalb von vier Monaten vollständig zurück (17). Das Risiko ist erhöht bei Träger/-innen des APOE-ε4-Gens, insbesondere bei homozygoten Personen, weshalb diese Gruppe in Europa und Grossbritannien derzeit nicht behandelt wird. In einer US-Kohorte lag die ARIA-Häufigkeit bei 44 % der homozygoten, 24 % der heterozygoten und 15 % der nicht-Träger (21). Daher sollte der APOE-Genotyp vor Therapiebeginn bestimmt und mit den Patient/-innen besprochen werden.
In ersten Real-World-Analysen zeigte sich ein vergleichbares Sicherheitsprofil. In einer israelischen Kohorte (n = 86) trat ARIA bei rund 19 % der Behandelten auf, überwiegend mild und asymptomatisch; nur eine Patientin benötigte eine kurzfristige Steroidtherapie (27). In einer Kohorte der Washington University (n = 234) lag die Häufigkeit bei etwa 22 %; die meisten Fälle waren leicht ausgeprägt, 5–6 % symptomatisch, 1 % schwer, ohne Todesfälle oder Makroblutungen (21). Schwere ARIA-Verläufe oder Hirnblutungen >1 cm erfordern den dauerhaften Therapieabbruch. Bei milden oder moderaten Veränderungen kann die Behandlung hingegen nach deren Abklingen in der Regel fortgesetzt werden.
Für die Praxis entscheidend ist die Unterscheidung zwischen ARIA und einem Schlaganfall. Wenn ein Patient unter Antikörpertherapie plötzlich fokal-neurologische Symptome zeigt, wie etwa eine Sprachstörung, Lähmung oder Sehstörung, sollte umgehend ein zerebrales MRT veranlasst werden, nicht nur ein CT, da frühe ARIA-Veränderungen sonst übersehen werden können. Eine intravenöse Thrombolyse ist während einer Antikörpertherapie mit hohen Risiken verbunden, da das Blutungsrisiko deutlich erhöht ist; eine mechanische Thrombektomie ist die Methode der Wahl, sofern eine Gefässokklusion gesichert ist.
Eine Antikoagulation sollte vermieden werden, da sie das Risiko schwerer Hirnblutungen erhöht. Eine einfache Thrombozytenhemmung (z. B. ASS) ist in der Regel möglich, eine duale sollte jedoch nach Möglichkeit unterbleiben. Hypertonie ist ein relevanter modifizierbarer ARIA‑Risikofaktor (mittlerer arterieller Druck >107 mmHg), daher trägt eine konsequente Blutdruckkontrolle wesentlich zur Reduktion des ARIA-Risikos bei (17, 23). Patient/-innen sollten stets eine patient alert card mitführen, die in Notfallsituationen auf die laufende Therapie hinweist.
Zuletzt wird in der Bildgebung gelegentlich eine scheinbare Hirnvolumenabnahme beobachtet (amyloid-related pseudoatrophy). Diese gilt als Folge des Amyloid-Abtransports und der Rückbildung lokaler Entzündungsreaktionen und wird derzeit nicht notwendigerweise als Ausdruck zusätzlicher Schädigung interpretiert (28).
Fazit für die Praxis
Für die ärztliche Praxis ist entscheidend, mögliche Warnzeichen einer ARIA früh zu erkennen und bei Verdacht sofort Kontakt mit der behandelnden Memory Clinic oder dem neurologischen Notfalldienst aufzunehmen. Neu auftretende Symptome wie Kopfschmerzen, Schwindel, Gangunsicherheit, Sehstörungen, Sprachstörungen oder Krampfanfälle erfordern umgehend ein MRT, da ein CT frühe Veränderungen häufig nicht erfasst. Patient/-innen und Angehörige müssen darüber aufgeklärt sein, bei solchen Symptomen unverzüglich die Memory Clinic oder die Notfallambulanz zu kontaktieren. In den bislang publizierten Real‑World‑Erfahrungen zeigten sich keine neuen Sicherheitssignale; die ARIA‑Raten lagen nicht über den in den Zulassungsstudien beobachteten.
Antikoagulation sollte vermieden werden, und eine konsequente Blutdruckkontrolle senkt das Risiko für ARIA deutlich. Bei akuten Schlaganfallsymptomen sollte keine i.v.-Thrombolyse erfolgen, während eine mechanische Thrombektomie bei gesicherter Gefässokklusion in Betracht gezogen werden kann. Patient/-innen sollten stets eine patient alert card mitführen, und die laufende Therapie sollte in der Praxisdokumentation vermerkt sein, damit im Notfall rasch reagiert werden kann. ARIA treten unter Anti‑Amyloid‑Therapien in der Grössenordnung von ~20 % auf; in Zulassungsstudien lagen die Raten je nach Antikörper und Dosierung höher, während frühe Real‑World‑Kohorten bislang um ~20 % berichten. Meist sind ARIA asymptomatisch/mild und reversibel, selten schwer. Infusionsreaktionen sind in der Regel ebenfalls mild und lassen sich durch eine Prämedikation mit Antihistaminikum und Paracetamol gut vermeiden. Real-World-Daten sprechen dafür, dass die Therapien bei konsequentem Monitoring insgesamt sicher und gut handhabbar sind.
Zusammenfassung
Neue krankheitsmodifizierende Therapien markieren einen Wendepunkt in der Alzheimer-Behandlung. Der Weg dahin war lang und von Rückschlägen geprägt. Abb. 3 ordnet die heutigen Anti-Amyloid-Therapien in die wichtigsten Meilensteine ein, von der Erstbeschreibung der Alzheimer‑Krankheit über die Identifikation von Aβ bis zum präklinischen Proof‑of‑concept einer anti-Aβ‑Immunisierung (29) und den zuletzt erfolgreichen Phase‑III‑Studien mit ersten internationalen Zulassungen. Zwischen dem ersten Plaque‑Clearing im Tiermodell und der klinischen Anwendung beim Menschen lagen demnach über zwei Jahrzehnte. Erste Erfahrungen aus Ländern mit bereits zugelassenen Antikörpern zeigen, dass die Anwendung in spezialisierten Memory Clinics sicher und effektiv möglich ist, bei gleichzeitig ähnlichen oder sogar geringeren Nebenwirkungsraten als in den Zulassungsstudien (21). Entscheidend bleiben die sorgfältige Patientenselektion, die enge interdisziplinäre Zusammenarbeit und ein strukturiertes Monitoring. Direkte Head‑to‑Head‑Vergleichsstudien fehlen; die derzeitigen Sicherheits‑ und Wirksamkeitsvergleiche zwischen Lecanemab und Donanemab beruhen auf indirekten Vergleichen und sind daher mit methodischer Unsicherheit behaftet. Für die Umsetzung sind neben den Medikamentenkosten vor allem Infrastruktur und Personal relevant, etwa für regelmässige Infusionen, häufige MRT-Kontrollen, Biomarkerbestätigung, ApoE-Testung und Beratung. Diese Investitionen müssen so geplant werden, dass andere zentrale Bereiche der Demenzversorgung nicht verdrängt werden. Je früher eine geeignete Therapie bei passenden Patient/-innen beginnt, desto grösser ist der potenzielle Nutzen – «time is brain» gewinnt damit auch bei der Alzheimer‑Versorgung zusätzlich an Relevanz. Diese Therapien eröffnen reale Chancen für Betroffene im Frühstadium, vergleichbar mit dem Beginn der modernen MS-Therapie vor 25 Jahren, die heute mehr als 20 hochwirksame Optionen umfasst. Jetzt gilt es, Erfahrungen zu sammeln, Strukturen zu schaffen und offene Fragen zu klären, insbesondere zu Langzeitnutzen, optimaler Patientenselektion und Sicherheit im Versorgungsalltag.
Copyright
Aerzteverlag medinfo AG
Prof. Dr. Dr. med. Marc Aurel Busche
– Departement für Demenzielle Erkrankungen, Universitäre Altersmedizin Felix Platter, Universität Basel
– Departement Biomedizin, Universität Basel
– Departement Klinische Forschung, Universität Basel
– UK Dementia Research Institute at University College London
Dr. med. Ansgar Felbecker
– Universitätsklinik für Neurologie, Inselspital Bern
– Neuro im Zentrum, St. Gallen
Dr. med. Hannah Schneider-Häner
Departement für Demenzielle Erkrankungen
Universitäre Altersmedizin Felix Platter
Universität Basel
Die Autorenschaft hat keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
Die monoklonalen Antikörper Lecanemab und Donanemab entfernen pathologisches Amyloid-β aus dem Gehirn und verlangsamen den Krankheitsverlauf von Alzheimer im Frühstadium nachweislich; sie bewirken jedoch keine Heilung oder einen Stopp der Erkrankung.
Geeignet sind Patient/-innen mit leichter kognitiver Störung oder milder Demenz, nachgewiesener Amyloid-Pathologie und ohne relevante vaskuläre oder hämorrhagische Läsionen im Gehirn.
Therapien entfalten den grössten Nutzen in frühen Krankheitsstadien. Offene Verlängerungsdaten und Beobachtungsdaten deuten darauf hin, dass ein klinischer Vorteil mit längerer Behandlungsdauer erhalten bleiben kann. Daher sind das frühzeitige Erkennen von Gedächtnis- und anderen kognitiven Störungen und die enge Zusammenarbeit zwischen Hausärzt/-innen und Memory Clinics entscheidend, um Betroffene rasch einer spezialisierten biomarkergestützten Abklärung zuzuführen.
Bei klarer Indikation und regelmässigem MRT-Monitoring sind die Therapien sicher anwendbar; erste Real-World-Daten legen eine gute Sicherheit und Handhabbarkeit im klinischen Alltag nahe.
Memory Clinics entwickeln sich von rein diagnostischen und supportiven Angeboten zu einem aktiven therapeutischen Setting. Das ist ein Paradigmenwechsel in der klinischen Versorgung von Alzheimer, der auch Impulse für Diagnostik und Therapie bei anderen Demenzursachen geben dürfte.
1. Bürge M, Bieri G, Brühlmeier M, Colombo F, Demonet JF, Felbecker A, Georgescu D, Gietl A, Brioschi Guevara A, Jüngling F, Kirsch E, Kressig RW, Kulic L, Monsch AU, Ott M, Pihan H, Popp J, Rampa L, Rüegger-Frey B, Schneitter M, Unschuld PG, von Gunten A, Weinheimer B, Wiest R, Savaskan E. Recommendations of Swiss Memory Clinics for the Diagnosis of Dementia. Praxis (Bern 1994). 2018 Apr;107(8):1-17. German. doi: 10.1024/1661-8157/a003374. PMID: 31589108.
2. Popp J, Meyer-Heim T, Bürge M, Ehrensperger MM, Felbecker A, Pihan H, et al. Die Empfehlungen der Swiss Memory Clinics für die Diagnostik der Demenzerkrankungen – ein Update. Praxis (Bern 1994). 2025;114(4):127–136. doi:10.23785/PRAXIS.2025.04.002. PMID: 40336391.
3. Popp J, Georgescu D, Bürge M, Mundwiler-Pachlatko E, Bernasconi L, Felbecker A. Biomarker in der Diagnostik kognitiver Störungen – Empfehlungen der Swiss Memory Clinics. Praxis (Bern 1994). 2022;111(13):738–744. doi:10.1024/1661-8157/a003913. PMID: 36221969.
4. Juengling FD, Allenbach G, Bruehlmeier M, Klaeser B, Wissmeyer MP, Garibotto V, et al. Appropriate use criteria for dementia amyloid imaging in Switzerland-mini-review and statement on behalf of the Swiss Society of Nuclear Medicine and the Swiss Memory Clinics. Nuklearmedizin. 2021;60(1):7–9. doi:10.1055/a-1277-6014. PMID: 33080626.
5. Klöppel S, Meyer-Heim T, Ehrensperger M, Rüttimann A, Weibel I, Schnelli A, et al. Die Empfehlungen der Swiss Memory Clinics für die Therapie der Demenzerkrankungen. Praxis (Bern 1994). 2024;113(8):187–194. doi:10.23785/PRAXIS.2024.08.003. PMID: 39508540.
6. Savaskan E, Georgescu D, Becker S, Benkert B, Blessing A, Bürge M, et al. Recommendations for the diagnostic and therapy of behavioural and psychological symptoms of dementia (BPSD). Praxis (Bern 1994). 2024;113(2):34–43. PMID: 38536191.
7. Felbecker A, Rouaud O, Lathuiliere A, Allali G, Sollberger M, Meyer-Heim T, Monsch AU, Lövblad KO, Becker S, Barro-Belaygues N, Popp J, Bürge M, Lindheimer K, Gietl A, Jung HH, Georgescu D, Meyer R, Frisoni GB. Anti-Amyloid Monoclonal Antibodies for the Treatment of Alzheimer Disease: Intersocietal Recommendations for Their Appropriate Use in Switzerland. Neurodegener Dis. 2025;25(3):114-125. doi: 10.1159/000545799. Epub 2025 Apr 14. PMID: 40222353.
8. Busche MA, Hyman BT. Synergy between amyloid-ß and tau in Alzheimer‘s disease. Nat Neurosci. 2020 Oct;23(10):1183-1193. doi: 10.1038/s41593-020-0687-6. Epub 2020 Aug 10. PMID: 32778792; PMCID: PMC11831977.
9. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer‘s disease at 25 years. EMBO Mol Med. 2016 Jun 1;8(6):595-608. doi: 10.15252/emmm.201606210. PMID: 27025652; PMCID: PMC4888851.
10. Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jönsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K. A mutation in APP protects against Alzheimer‘s disease and age-related cognitive decline. Nature. 2012 Aug 2;488(7409):96-9. doi: 10.1038/nature11283. PMID: 22801501.
11. Rajani RM, Ellingford R, Hellmuth M, Harris SS, Taso OS, Graykowski D, et al. Selective suppression of oligodendrocyte-derived amyloid beta rescues neuronal dysfunction in Alzheimer’s disease. PLoS Biol. 2024;22(7):e3002727. doi:10.1371/journal.pbio.3002727.
12. Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer‘s disease. Proc Natl Acad Sci U S A. 2012 May 29;109(22):8740-5. doi: 10.1073/pnas.1206171109. Epub 2012 May 16. PMID: 22592800; PMCID: PMC3365221.
13. Butler PM, Francis A, Meunier AL, Anderson AK, Hennessey EL, Miller MB, et al. Anti-amyloid antibody equilibrium binding to Aß aggregates from human Alzheimer disease brain. bioRxiv. 2025:2025.05.20.654902. doi:10.1101/2025.05.20.654902.
14. Kang C. Donanemab: first approval. Drugs. 2024;84:1313–1318. doi:10.1007/s40265-024-02087-4.
15. Van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388(1):9–21. doi:10.1056/NEJMoa2212948.
16. Cohen S, van Dyck CH, Gee M, Doherty T, Kanekiyo M, Dhadda S, Li D, Hersch S, Irizarry M, Kramer LD. Lecanemab Clarity AD: Quality-of-Life Results from a Randomized, Double-Blind Phase 3 Trial in Early Alzheimer‘s Disease. J Prev Alzheimers Dis. 2023;10(4):771-777. doi: 10.14283/jpad.2023.123. PMID: 37874099.
17. Fox NC, Belder C, Ballard C, Kales HC, Mummery C, Caramelli P, et al. Treatment for Alzheimer’s disease. Lancet. 2025;406:1408–1423.
18. Van Dyck CH, Sperling R, Li D, Kanekiyo M, Dhadda S, Hersch S, et al. The Lecanemab Clarity AD open-label extension in early Alzheimer’s disease: initial findings from the 48-month analysis. Presented at: Alzheimer’s Association International Conference (AAIC); 2025 Jul 27–31; Toronto, Canada.
19. Sims JR, Zimmer JA, Evans CD, Lu M, Ardayfio P, Sparks J, Wessels AM, Shcherbinin S, Wang H, Monkul Nery ES, Collins EC, Solomon P, Salloway S, Apostolova LG, Hansson O, Ritchie C, Brooks DA, Mintun M, Skovronsky DM; TRAILBLAZER-ALZ 2 Investigators. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA. 2023 Aug 8;330(6):512-527. doi: 10.1001/jama.2023.13239. PMID: 37459141; PMCID: PMC10352931.
20. Zimmer JA, Sims JR, Evans CD, Nery ESM, Wang H, Wessels AM, Tronchin G, Sato S, Raket LL, Andersen SW, Sapin C, Paget MA, Gueorguieva I, Ardayfio P, Khanna R, Brooks DA, Matthews BR, Mintun MA; Alzheimer’s Disease Neuroimaging Initiative. Donanemab in early symptomatic Alzheimer‘s disease: results from the TRAILBLAZER-ALZ 2 long-term extension. J Prev Alzheimers Dis. 2025 Dec 1:100446. doi: 10.1016/j.tjpad.2025.100446. Epub ahead of print. PMID: 41330788.
21. Paczynski M, Hofmann A, Posey Z, Gregersen M, Rudman M, Ellington D, Aldinger M, Musiek ES, Holtzman DM, Bateman RJ, Long JM, Ghoshal N, Carr DB, Dow A, Namazie-Kummer S, Jana N, Xiong C, Morris JC, Benzinger TLS, Schindler SE, Snider BJ. Lecanemab Treatment in a Specialty Memory Clinic. JAMA Neurol. 2025 Jul 1;82(7):655-665. doi: 10.1001/jamaneurol.2025.1232. Erratum in: JAMA Neurol. 2025 Jul 1;82(7):754. doi: 10.1001/jamaneurol.2025.2446. PMID: 40354064; PMCID: PMC12070285.
22. Cummings J, Apostolova L, Rabinovici GD, Atri A, Aisen P, Greenberg S, Hendrix S, Selkoe D, Weiner M, Petersen RC, Salloway S. Lecanemab: Appropriate Use Recommendations. J Prev Alzheimers Dis. 2023;10(3):362-377. doi: 10.14283/jpad.2023.30. PMID: 37357276; PMCID: PMC10313141.
23. Rabinovici GD, Selkoe DJ, Schindler SE, Aisen P, Apostolova LG, Atri A, Greenberg SM, Hendrix SB, Petersen RC, Weiner M, Salloway S, Cummings J. Donanemab: Appropriate use recommendations. J Prev Alzheimers Dis. 2025 May;12(5):100150. doi: 10.1016/j.tjpad.2025.100150. Epub 2025 Mar 27. PMID: 40155270; PMCID: PMC12180672.
24. Wang H, Serap Monkul Nery E, Ardayfio P, Khanna R, Otero Svaldi D, Gueorguieva I, Shcherbinin S, Andersen SW, Hauck PM, Engle SE, Brooks DA, Collins EC, Fox NC, Greenberg SM, Salloway S, Mintun MA, Sims JR. Modified titration of donanemab reduces ARIA risk and maintains amyloid reduction. Alzheimers Dement. 2025 Apr;21(4):e70062. doi: 10.1002/alz.70062. Erratum in: Alzheimers Dement. 2025 Aug;21(8):e70576. doi: 10.1002/alz.70576. PMID: 40172303; PMCID: PMC11963282.
25. Frisoni GB, Ribaldi F, Aho E, Brayne C, Walsh S, Ciccarelli O, et al. The new clinical landscape in Alzheimer’s disease: controversies and future directions. Part 3. Lancet. 2025;406:1424–1442. doi:10.1016/S0140-6736(25)01389-3.
26. Rubin R. Treating Alzheimer Disease With Antiamyloid Therapies-The Real-World Experience Grows. JAMA. 2025 Sep 23;334(12):1041-1044. doi: 10.1001/jama.2025.14180. PMID: 40844810
27. Bregman N, Nathan T, Shir D, Omer N, Levy MH, Bar David A, et al. Lecanemab in clinical practice: real-world outcomes in early Alzheimer’s disease. Alzheimers Res Ther. 2025;17:119. doi:10.1186/s13195-025-01763-1.
28. Belder CRS, Boche D, Nicoll JAR, Jaunmuktane Z, Zetterberg H, Schott JM, Barkhof F, Fox NC. Amyloid-related iatrogenic atrophy of the brain: data transparency is an urgent safety priority – Authors‘ reply. Lancet Neurol. 2025 Mar;24(3):190. doi: 10.1016/S1474-4422(25)00030-4. PMID: 39986300.
29. Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999 Jul 8;400(6740):173-7. doi: 10.1038/22124. PMID: 10408445.
Die korrekte Einordnung von Kopfschmerzen im klinischen Alltag gestaltet sich aufgrund der Vielzahl an Differenzialdiagnosen oft als herausfordernd. Besonders wichtig ist dabei die Unterscheidung zwischen den häufigeren primären Kopfschmerzen, bei denen der Kopfschmerz die eigentliche Erkrankung darstellt, und den sekundären Kopfschmerzen, die teilweise lebensbedrohliche Ursachen haben können. Die diagnostischen Entscheidungswege beginnen daher mit der Identifizierung von sekundären Kopfschmerzen.
In clinical practice, correctly diagnosing headaches can be challenging due to the many differential diagnoses. It is particularly important to differentiate between the more common primary headache types, where the headache itself is the main condition, and secondary headaches, which may have potentially lifethreatening causes. The diagnostic approach initially focuses on identifying secondary headaches. Keywords: Secondary headaches, primary headaches, red flags, green flags
Überblick
Viele Erkrankungen können sich durch Kopfschmerzen äussern. Kopfschmerzen zählen zu den häufigsten Beschwerden, die zur Vorstellung beim Arzt führen. Epidemiologisch gesehen, liegt in der Europäischen Union die Ein-Jahres-Prävalenz von Kopfschmerzen bei 79 % (1). Neben der erheblichen psychischen Belastung und der Beeinträchtigung der Lebensqualität können Kopfschmerzen auch die Arbeitsleistung deutlich mindern (2). Die Internationale Kopfschmerzgesellschaft (IHS) unterscheidet in der aktuellen Kopfschmerzklassifikation 276 verschiedene Kopfschmerzerkrankungen (3). Migräne ist eine der häufigsten Erkrankungen. Etwa 1.1 Milliarden Menschen weltweit sind von Migräne betroffen (4). Unter den neurologischen Erkrankungen löst sie die höchste Erkrankungslast aus (5). Viele Patienten mit Migräne stellen sich notfallmässig vor, v. a. bei erstmaligem Auftreten einer Aura.
Primär versus sekundär
Bei der Vorstellung von Kopfschmerzpatienten steht oft die Frage nach der Ursache der Beschwerden im Mittelpunkt. Viele Betroffene erhoffen sich durch die Behandlung nicht nur eine Linderung der Symptome, sondern auch die vollständige Beseitigung der Ursache und damit eine Heilung ihrer Erkrankung. Tatsächlich stellt die Unterscheidung zwischen primären und sekundären Kopfschmerzen eine grosse Herausforderung dar, da Kopfschmerzerkrankungen heterogen sind. Das Ziel ist zunächst, einen potenziell bedrohlichen sekundären oder symptomatischen Kopfschmerz mit einer hohen Sensitivität zu identifizieren (6). Während primäre Kopfschmerzen wie Migräne eine eigenständige Erkrankung darstellen, ist bei sekundären oder symptomatischen Kopfschmerzen eine andere Erkrankung zugrunde liegend. Die WHO schätzte im Jahr 2021, dass bis zu 18 % der Patienten, die sich mit Kopfschmerzen vorstellen, an einem sekundären Kopfschmerz leiden (6). In einer norwegischen populationsbasierten Studie betrug die Ein-Jahres-Prävalenz von sekundären Kopfschmerzen in der Altersgruppe zwischen 30 und 44 Jahren hingegen nur 2.14 %. Der überwiegende Anteil dieser Patienten hatte einen Medikamentenübergebrauchskopfschmerz, welcher keinen Notfall darstellt und rein durch die Anamnese diagnostiziert werden kann (7). Dementsprechend ist davon auszugehen, dass nur wenige Menschen, die sich in dieser Altersspanne mit Kopfschmerzen vorstellen, tatsächlich an einem potenziell gefährlichen sekundären Kopfschmerz leiden. Da die Ursache von sekundären Kopfschmerzen eine hohe Morbidität oder gar Mortalität haben kann, müssen sie zuverlässig und ggf. schnell erkannt werden. Besteht aufgrund der Anamnese und klinischen Untersuchung bereits der Verdacht auf einen sekundären Kopfschmerz, erhöht sich auch die Wahrscheinlichkeit, eine relevante Pathologie zu finden (sog. Vortest-Wahrscheinlichkeit) (8). Die Identifizierung der zugrunde liegenden Pathologie ist zudem entscheidend, da auch die erfolgreiche Behandlung der Kopfschmerzen oft erst durch die gezielte Therapie der verursachenden Erkrankung möglich wird.
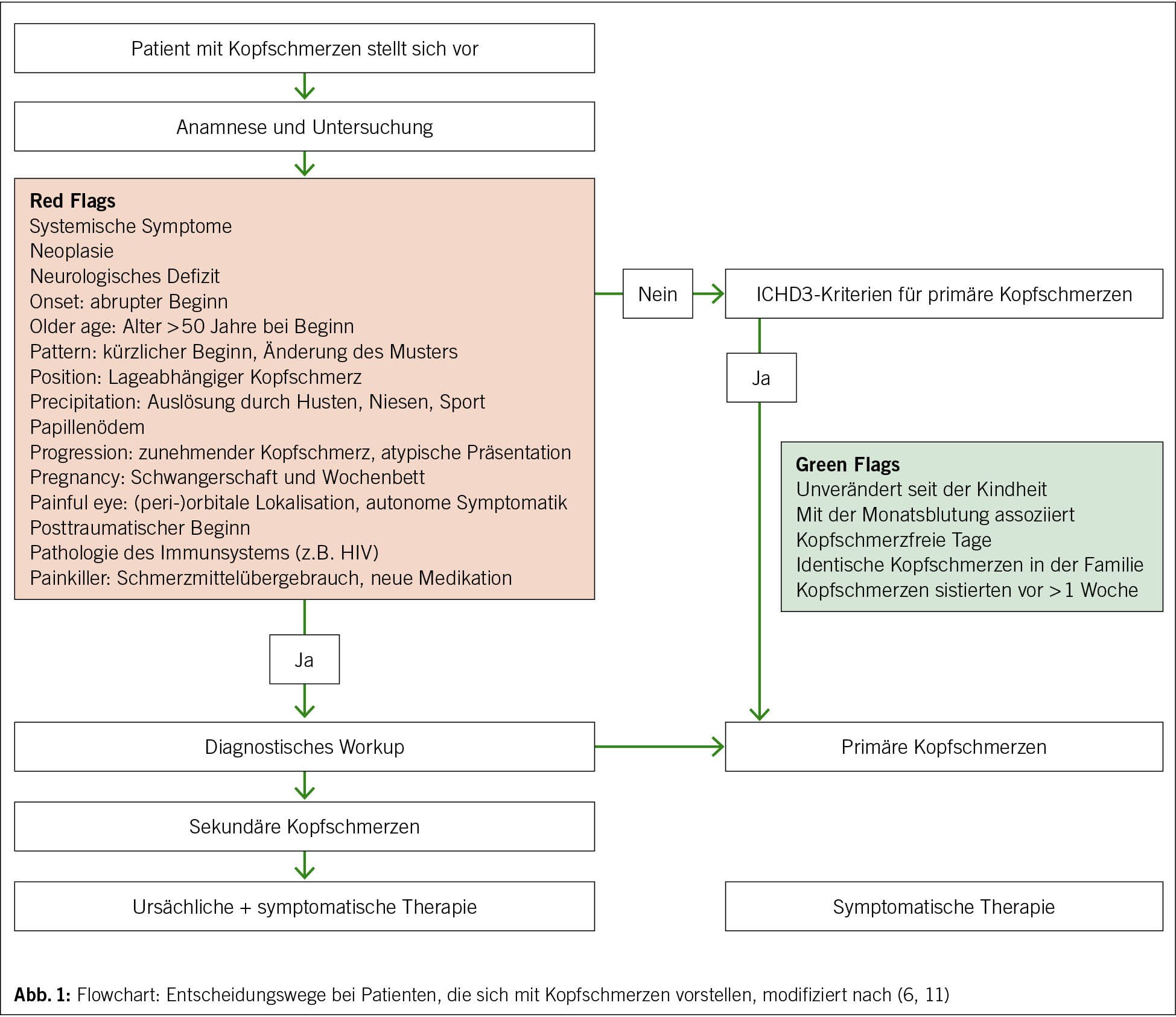
«Red Flags»
In der Konsultation mit Kopfschmerzpatienten ist es empfehlenswert, von Beginn an auf Warnzeichen, die auf sekundäre Kopfschmerzen hinweisen, sogenannte Red Flags, zu achten. Das Akronym SNNOOP10 aus dem Englischen umfasst 15 wichtige Punkte, die als Gedankenstütze bei der Identifikation von «Red Flags» für sekundäre Kopfschmerzen hilfreich sind (9). In der strukturierten Anamnese gibt die Kopfschmerzdynamik bereits wichtige Hinweise, wie akut der Kopfschmerz auftritt, und hilft, einen vorbestehenden von den aktuellen, neuartigen Kopfschmerzen zu unterscheiden. Begleitphänomene, Medikation und Vorerkrankungen können zudem wichtige Informationen für die Genese der Kopfschmerzen liefern. Im nächsten Schritt kann der Kopfschmerzphänotyp dann genauer analysiert werden.
Kurzkasuistik
Eine 45-jährige Patientin mit einer bekannten Migräne ohne Aura stellt sich notfallmässig mit stärksten Kopfschmerzen vor, welche plötzlich begonnen hätten. Sie kommt gerade aus dem Skiurlaub zurück. Während des Urlaubes sei es ihr sehr gut gegangen. Ein Trauma wird verneint. Der Kopfschmerz habe sich nicht, wie sonst üblich, nach Einnahme eines Triptans gebessert. Da sich die Kopfschmerzen wie die bekannte Migräne präsentieren, wird die Patientin nach unauffälligem CT-Schädel und regredienten Schmerzen unter Analgesie wieder nach Hause entlassen. Wenige Tage später stellt sie sich erneut mit einem Donnerschlagkopfschmerz vor. Nun zeigen sich bildgebend nicht nur eine Subarachnoidalblutung in der Konvexität, sondern auch Gefässspasmen. Klinisch finden sich eine leichte linksseitige Armschwäche, Wortfindungsstörungen und eine Abduzensparese. Es wird die Diagnose eines reversiblen zerebralen Vasokonstriktionssyndroms (RCVS), eines sekundären Kopfschmerzes, gestellt. Während der intensivmedizinischen Behandlung kommt es rezidivierend zu Kopfschmerzexazerbationen. Die fokal neurologischen Defizite sind im Verlauf vollständig regredient.
Diese Kasuistik zeigt, dass es schwierig sein kann, anhand des Phänotyps einen primären und vorbestehenden Kopfschmerz von einem sekundären, potenziell bedrohlichen Kopfschmerz zu unterscheiden (10). Bei dieser Patientin äusserte sich der sekundäre Kopfschmerz im Rahmen eines reversiblen zerebralen Vasokonstriktionssyndroms (RCVS) ähnlich wie eine schwere Migräneattacke. Auffallend waren der schnelle Beginn – bei der Migräne gibt es in der Regel eine Vorphase –, die hohe Intensität (Donnerschlagkopfschmerz) und das fehlende Ansprechen auf die gewohnte Akutmedikation. Die Kopfschmerzdynamik, Intensität und Medikation lieferten hier genauere Informationen als die Analyse des Phänotyps. Es ist nicht ungewöhnlich, dass sekundäre Kopfschmerzen Merkmale eines Migräne- oder Spannungskopfschmerzes aufweisen. Dieser Fall zeigt auch, dass die meist auf dem Notfall durchgeführte Bildgebung eines CT die Pathologie nicht immer aufzeigt.
Ausschlaggebend für die Differenzierung von primären und sekundären Kopfschmerzen ist das Gesamtbild, das sich aus gezielter Anamnese und klinischer Untersuchung ergibt.
Anamnese
Im Folgenden sind die wichtigsten Punkte für eine strukturierte Anamnese von Kopfschmerzpatienten aufgelistet (Abb. 1) (11,12). Die Punkte sind aktiv zu erfragen (Holprinzip). Jeder Punkt, der nicht vorbestehend ist, sollte zunächst als «Red Flag» gewertet werden.
Kopfschmerzspezifische Anamnese
• Dynamik
– Seit wann besteht der aktuelle Kopfschmerz?
– Wie hat er beim ersten Mal begonnen?
– Wie häufig trat der Kopfschmerz zu Beginn auf?
– Wie hat sich die Kopfschmerzfrequenz im Verlauf
verändert?
– Wie häufig tritt der Kopfschmerz aktuell auf?
• Phänotyp
– Lokalisation, Intensität, Qualität und Dauer der einzelnen Attacken
– Begleitbeschwerden wie Photophobie, Phonophobie, Osmophobie, Übelkeit und Erbrechen
– Zunahme bei körperlicher Anstrengung, Ruhebedürfnis, psychomotorische Unruhe
– Kranioautonome Symptome, z. B. Augentränen,
Nasenlaufen, verstopfte Nase, gerötetes Auge, Lidödem, verstopftes Ohr, Schwitzen auf der Stirn, einseitige Ptose
– Aura (visuell, sensibel, motorisch), Dauer und Zeitpunkt des Auftretens
– Nicht-Kopfschmerz-Symptome, z. B. Konzentrationsstörungen, Reizbarkeit, Müdigkeit oder Hungergefühl
– Vorboten, z. B. Müdigkeit, häufiges Gähnen, Gereiztheit, Euphorie
– Triggerfaktoren, z. B. Lageabhängigkeit, Husten, Niesen, Pressen, Trauma
Allgemeine Anamnese
• Aktuelle Medikation
– Akut- und Basistherapie (Dosis, Dauer, Effekt, Nebenwirkungen)
– Weitere Medikation
• Frühere Kopfschmerzmedikation (getrennt nach Akut- und Basistherapie)
– Dosis, Dauer, Effekt, Nebenwirkungen
• Vorerkrankungen, z. B. Tumor, Immundefizite
• Systemanamnese inkl. Schlaf, Stimmung, B-Symptomatik etc.
• Familienanamnese
• Sozialanamnese inkl. Auswirkungen der Kopfschmerzen
• Bisher erfolgte Untersuchungen
Untersuchung
Bei jeder Erstvorstellung sowie bei neuen anamnestischen Aspekten sollte eine umfassende körperliche Untersuchung mit besonderem Fokus auf neurologische Defizite, Meningismus, Schmerzlokalisation und systemische Zeichen, die auf einen sekundären Kopfschmerz hinweisen können, erfolgen.
Die körperliche Untersuchung sollte folgende Aspekte umfassen:
• Erhebung der Vitalparameter
• Vollständige neurologische Untersuchung mit Beurteilung der Hirnnerven, fokal neurologischer Defizite, Meningismus
• Untersuchung der Austrittspunkte des Nervus trigeminus
• Prüfung auf bulbären Druck- oder Bewegungsschmerz
• Inspektion der Schleimhäute und des Zahnstatus
• Beurteilung der Beweglichkeit der Halswirbelsäule
• Erfassung einer möglichen Druckschmerzhaftigkeit der perikranialen Muskulatur
• Untersuchung auf Schmerzen bei Kieferöffnung oder -okklusion
• Palpation der Arteria temporalis superficialis
• Systemische Auffälligkeiten wie Hautveränderungen
Diagnostisches Work-up
Auf Basis der erhobenen Anamnese und klinischen Untersuchung kann eine erste Arbeitshypothese zur Kopfschmerzursache erstellt werden. Wenn «Red Flags» vorliegen, sollte abhängig von der Verdachtsdiagnose ein gezieltes diagnostisches Work-up durchgeführt werden. Dabei unterscheidet sich die Abklärung eines akuten, neu aufgetretenen Kopfschmerzes wesentlich von einem seit Monaten bestehenden chronischen Kopfschmerz. Während bei Ersterem eine sofortige Bildgebung und ggf. Lumbalpunktion erforderlich sein könnte, ist zur weiteren Abklärung systemischer Ursachen beispielsweise eine Blutuntersuchung zunächst ausreichend. So kann Kopfschmerz z. B. auch als Symptom von Elektrolytstörungen, beispielsweise im Rahmen einer Dialyse, auftreten. Darüber hinaus können Kopfschmerzen bei rheumatologischen Erkrankungen, endokrinologischen Störungen oder autoimmunen Enzephalitiden auftreten (13). Je nach klinischer Symptomatik sind daher spezifische Laboruntersuchungen indiziert, um mögliche systemische Ursachen zu identifizieren. Die Diagnostik und Therapie komplexer Kopfschmerzerkrankungen erfolgen häufig in interdisziplinärer Zusammenarbeit mit verschiedenen Fachrichtungen, u.a. der Radiologie, Kardiologie, Ophthalmologie, HNO, Schlafmedizin, Psychosomatik, Infektiologie, Endokrinologie und Zahnmedizin.
«Smarter Medicine»
Im klinischen Alltag ist es teilweise schwierig zu entscheiden, bei welchen Patienten eine ausführliche Diagnostik erfolgen sollte und bei welchen beispielsweise auf eine wiederholte Bildgebung verzichtet werden sollte. Im Rahmen des Expertenkonsens «Smarter Medicine» wurden Situationen definiert, in denen auf weiterführende Diagnostik verzichtet werden kann (14).
Diese 5 Empfehlungen sind im Folgenden aufgelistet:
• Keine Wiederholung der zerebralen Bildgebung bei unverändertem Kopfschmerzphänotyp
• Keine Computertomographie des Schädels zur Diagnostik nicht akuter Kopfschmerzen
• Keine Zahnextraktion zur Behandlung eines anhaltenden idiopathischen Gesichtsschmerzes
• Keine Migränechirurgie
• Keine Entfernung von Amalgamfüllungen zur Kopfschmerzbehandlung
«Green Flags»
Um mit hoher Sicherheit einen primären Kopfschmerz diagnostizieren zu können, ist das Konzept «Green Flags» entwickelt worden. «Green Flags» sind Symptome und Befunde, die nach Ausschluss von «Red Flags» mit hoher Wahrscheinlichkeit auf einen primären Kopfschmerz hindeuten (15). Dieses Konzept beruht auf einem Expertenkonsens. Eine Validierung der «Red Flags» nach der SNNOOP10-Liste und der «Green Flags» steht noch aus (6). Zusätzlich wird in der Forschung weiter nach Biomarkern für primäre Kopfschmerzerkrankungen, wie z. B. der Migräne, die im klinischen Alltag eingesetzt werden können, gesucht.
Entscheidungswege bei Kopfschmerzpatienten
Das Vorgehen im klinischen Alltag zeigt Abb. 1. Stellen sich Patienten mit Kopfschmerzen vor, wird in Anamnese und klinischer Untersuchung auf «Red Flags» und «Green Flags» geachtet. Bei fehlenden «Red Flags» sollte geprüft werden, ob der berichtete Kopfschmerz mit den ICHD3-Diagnosekriterien für primäre Kopfschmerzen übereinstimmt. Ist dies nicht der Fall, sind die «Green Flags» Hinweise auf das wahrscheinliche Vorliegen eines primären Kopfschmerzes. Beim Vorliegen von «Red Flags» wird weitere Diagnostik veranlasst. Zeigen sich hier unauffällige Befunde, ist ebenfalls mit einer hohen Wahrscheinlichkeit von dem Vorliegen eines primären Kopfschmerzes auszugehen.
Therapie sekundärer Kopfschmerzen
Bei sekundären Kopfschmerzen sollte primär die zugrunde liegende Ursache therapiert werden. Die Schmerzbehandlung erfolgt dabei durchaus nach WHO-Stufenschema, wobei Opiate in der Kopfschmerztherapie aufgrund des Gewöhnungseffektes und des Risikos der Entwicklung eines Medikamentenübergebrauchskopfschmerzes zurückhaltend eingesetzt werden sollten. Aus praktischer Sicht empfiehlt sich vielmehr die Wahl der Akut- und Basistherapie anhand des Kopfschmerzphänotyps. Für die primären Kopfschmerzerkrankungen gibt es Therapieempfehlungen der Schweizerischen Kopfwehgesellschaft (www.headache.ch).
Differenzialdiagnosen sekundärer Kopfschmerzen
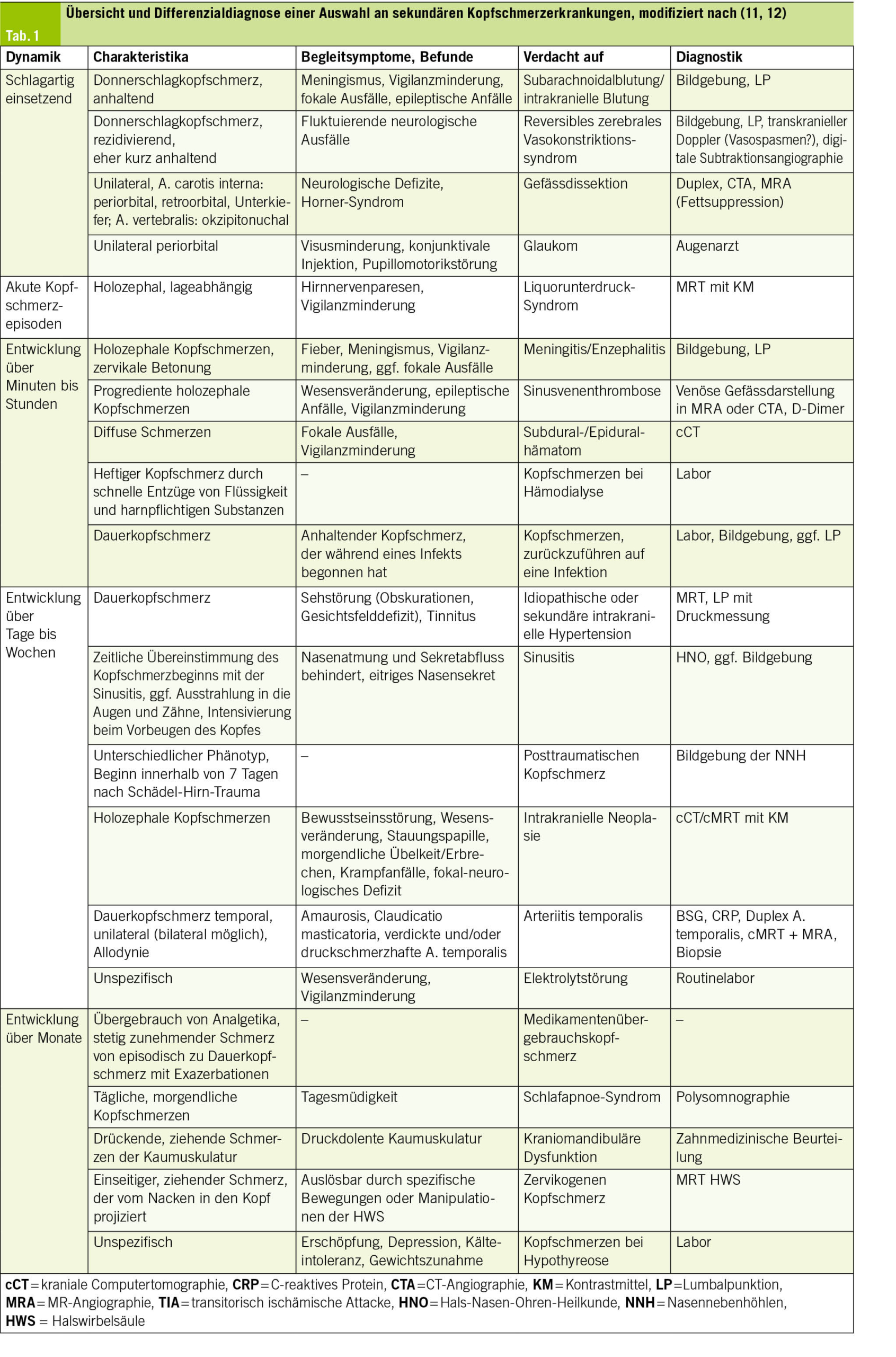
Mögliche Ursachen sekundärer Kopfschmerzen sind in der Tab. 1 aufgeführt.
Chronische sekundäre Kopfschmerzen
Zu den chronischen sekundären Kopfschmerzformen, die im klinischen Alltag zwar selten sind, aber leicht mit primären Kopfschmerzen verwechselt werden können und eine spezifische Diagnostik benötigen, zählen die idiopathische intrakranielle Hypertension (IIH) und die spontane intrakranielle Hypotension (SIH). An beide sollte bei Patienten mit unklaren chronischen Kopfschmerzen gedacht werden, da eine effektive Therapie zwingend die Ursache behandelt.
Die idiopathische intrakranielle Hypertension weist eine erhöhte Komorbidität mit Migräne auf, stellt aber gleichzeitig auch eine Differenzialdiagnose zur chronischen Migräne dar. Zu den «Red Flags», die für IIH sprechen, gehören u. a. lageabhängige Kopfschmerzen, die im Liegen zunehmen. Das Auftreten von Sehstörungen, welche sich in Form von kurz anhaltendem, wolkenartigem Verschwommensehen (visuellen Obskurationen), Photopsien oder Diplopie äussern und für ein Papillenödem sprechen, werden von etwa zwei Dritteln der Patienten berichtet. Seltener treten auch Gesichtsfelddefekte oder Abduzensparesen auf. Etwa die Hälfte der Patienten leidet an einem pulsatilen Tinnitus, der sich typischerweise im Liegen verstärkt. Auch Schwindel ist ein häufiges Symptom. IIH betrifft überwiegend Frauen im gebärfähigen Alter und ist mit Adipositas assoziiert. Eine Gewichtsreduktion kann zu einer Besserung der Symptomatik führen. Bei Verdacht auf IIH sollte eine Bildgebung vor Lumbalpunktion mit Liquordruckmessung durchgeführt werden. Die MRT spielt eine entscheidende Rolle bei der Diagnosestellung der IIH. Typische bildgebende Indikatoren für IIH sind eine Empty Sella, gewundene Sehnerven, eine Erweiterung der Sehnervenscheide, eine Abflachung des hinteren Augapfels, eine Vorwölbung des Sehnervenkopfes sowie eine Stenose des transversalen Sinus. Zur medikamentösen Behandlung gibt es nur Off-label-Optionen. Es werden Acetazolamid und Topiramat eingesetzt, insbesondere bei milden Verlaufsformen. Auch für die Therapie mit GLP-1-Agonisten zeigten sich in Bezug auf Kopfschmerzen und Gewichtsabnahme gute Ergebnisse (16). Bei schweren Verlaufsformen, die auf die medikamentöse Therapie nicht ausreichend ansprechen, oder bei Patienten mit Papillenödem wird neben der Optikusscheidenfenestrierung und der Anlage eines ventrikuloperitonealen Shunts auch eine Stentimplantation evaluiert, wenn eine Stenose des Sinus transversus vorliegt (17, 18). Wenn die Kopfschmerzen persistieren, nachdem sich der Liquordruck normalisiert hat, kommen CGRP-Antikörper ins Spiel (19).
Das Leitsymptom der spontanen intrakraniellen Hypotension (SIH) ist ein orthostatischer Kopfschmerz, der im Stehen stärker ausgeprägt ist und im Tagesverlauf zunimmt. Charakteristisch ist ein plötzlicher Beginn, wobei viele Betroffene den genauen Tag des Auftretens der Beschwerden benennen können. Die Diagnose wird meist anhand der Anamnese und der bildgebenden Befunde gestellt. Dabei können unspezifische Beschwerden auftreten. Typisch für SIH sind neben lageabhängigen Kopfschmerzen Nackenschmerzen, Tinnitus, Übelkeit und Sehstörungen. Der neurologische Status ist in der Regel unauffällig. Gelegentlich treten jedoch Doppelbilder infolge von Hirnnervenparesen auf.
Da der Liquordruck bei etwa zwei Drittel der Patienten mit SIH normal ist und eine Lumbalpunktion das Krankheitsbild durch ein zusätzliches postpunktionelles Syndrom verschlechtern kann, ist die Lumbalpunktion primär nicht indiziert. Zur Sicherung der Diagnose kommt die zerebrale und spinale Magnetresonanztomographie zum Einsatz. Mithilfe eines Scores kann die Wahrscheinlichkeit für das Vorliegen eines Liquorlecks errechnet werden. Folgende typische MRT-Befunde werden hierbei berücksichtigt: subdurale Flüssigkeitsansammlung, pachymeningeales Enhancement, Vergrösserung der Sinus durae matris, Distanz der suprasellären Zisterne, pontomamilläre Distanz und präpontine Zisterne (20). Die häufigste Ursache für SIH sind ventrale Duralecks, die vorwiegend im thorakalen Bereich auftreten und durch Mikrosporne oder Verkalkungen verursacht werden. Weniger häufig sind meningeale Divertikel, Ektasien, Schwachstellen der Dura an den spinalen Nervenwurzeln oder direkte Fisteln zwischen dem Liquorraum und den epiduralen Venen als Auslöser anzutreffen (21). Symptomatische Therapien mit Koffein, Theophyllin, Gabapentin und Hydrokortison können versucht werden. Bei fehlendem Effekt sollte rasch eskaliert werden. Neben dem epiduralen Blutpatch, der ggf. auch wiederholt werden muss, muss das Liquorleck oftmals mikrochirurgisch verschlossen oder endovaskulär, transvenös embolisiert werden. Zu beachten ist, dass der SIH-Kopfschmerz sich oft über Jahre bessern kann und seine Lageabhängigkeit verliert. Da das Leck aber oft doch nicht verschlossen ist, kann es zu schwerwiegenden Komplikationen wie zerebraler Siderose und bibrachialer Atrophie kommen.
Wann überweise ich Patienten zu Kopfschmerzspezialisten?
Eine Überweisung zu Neurologen oder Kopfschmerzspezialisten ist zu jedem Zeitpunkt richtig. Insbesondere in folgenden Situationen ist eine Mitbeurteilung indiziert:
• Neuartige Kopfschmerzen und «Red Flags». Ausnahme ist der Verdacht auf einen dringend zu behandelnden sekundären Kopfschmerz, bei dem eine unmittelbare Zuweisung auf einen Notfall notwendig ist.
• Unklare Kopfschmerzen
• Beginnende Chronifizierung mit Zunahme der Kopfschmerzfrequenz oder -intensität
• Chronische Kopfschmerzen
• Fehlendes Ansprechen auf Basistherapien wie Betablocker oder Amitriptylin
• Komorbiditäten
Fazit
Bei der Vorstellung von Patienten mit Kopfschmerzen gelingt die Unterscheidung zwischen primären und sekundären Kopfschmerzen durch eine strukturierte und kopfschmerzspezifische Anamnese und neurologische Untersuchung. Hilfreiche Instrumente in der Differenzialdiagnostik sind «Red Flags» und «Green Flags». Je nach Verdachtsdiagnose kann dann entschieden werden, welche Diagnostik und Therapie für den individuellen Kopfschmerzpatienten gewählt werden sollten.
Copyright
Aerzteverlag medinfo AG
Zweitabdruck aus Therapeutische Umschau 03-25
Dr. med. Laura Weichsel
Universitätsklinik für Neurologie
Inselspital
Rosenbühlgasse 25
3010 Bern
Prof. Dr. med. Christoph Schankin
– Universitätsklinik für Neurologie
Inselspital
Rosenbühlgasse 25
3010 Bern
– Zentrum für Migräne und Kopfschmerzen
Bellevue Medical Group
Theaterstrasse 22
8001 Zürich
christoph.schankin@bmg-swiss.ch
Die Autorenschaft hat keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Hoglinger G, Trenkwalder C. Parkinson-Krankheit, S2k-Leitlinie, 2023. Deutsche Gesellschaft für Neurologie (Hrsg), Leitlinien für Diagnostik und Therapie in der Neurologie. 2023.
2. Schapira AHV, Chaudhuri KR, Jenner P. Non-motor features of Parkinson disease. Nat Rev Neurosci. 2017 Aug;18(8):509.
3. Fereshtehnejad SM, Zeighami Y, Dagher A, Postuma RB. Clinical criteria for subtyping Parkinson’ s disease: biomarkers and longitudinal progression. Brain. 2017 Jul 1;140(7):1959-76.
4. Mylius V, Perez Lloret S, Cury RG, Teixeira MJ, Barbosa VR, Barbosa ER, et al. The Parkinson disease pain classification system: results from an international mechanism-based classification approach. Pain. 2021 Apr 1;162(4):1201-10.
5. Zaja-Milatovic S, Milatovic D, Schantz AM, Zhang J, Montine KS, Samii A, et al. Dendritic degeneration in neostriatal medium spiny neurons in Parkinson disease. Neurology. 2005 Feb 8;64(3):545-7.
6. Storch A, Schneider CB, Wolz M, Sturwald Y, Nebe A, Odin P, et al. Nonmotor fluctuations in Parkinson disease: severity and correlation with motor complications. Neurology. 2013 Feb 26;80(9):800-9.
7. Lee MA, Walker RW, Hildreth TJ, Prentice WM. A survey of pain in idiopathic Parkinson’ s disease. J Pain Symptom Manage. 2006 Nov;32(5):462-9.
8. Mylius V, Moller JC, Bohlhalter S, Ciampi de Andrade D, Perez Lloret S. Diagnosis and Management of Pain in Parkinson’ s Disease: A New Approach. Drugs Aging. 2021 Jul;38(7):559-77.
9. Mylius V, Perez Lloret S, Brook CS, Kruger MT, Hagele-Link S, Gonzenbach R, et al. The new Parkinson’ s disease pain classification system (PD-PCS). Nervenarzt. 2022 Oct;93(10):1019-27.
10. Mylius V, Moisset X, Rukavina K, Rosner J, Korwisi B, Marques A, et al. New ICD-11 diagnostic criteria for chronic secondary musculoskeletal pain associated with Parkinson disease. Pain. 2024 Jan 11.
11. Cohen SP, Vase L, Hooten WM. Chronic pain: an update on burden, best practices, and new advances. Lancet. 2021 May 29;397(10289):2082-97.
12. Finnerup NB, Haroutounian S, Kamerman P, Baron R, Bennett DL, Bouhassira D, et al. Neuropathic pain: an updated grading system for research and clinical practice. Pain. 2016 Aug;157(8):1599-606.
13. Bouhassira D, Attal N, Alchaar H, Boureau F, Brochet B, Bruxelle J, et al. Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). Pain. 2005 Mar;114(1-2):29-36.
14. Chaudhuri KR, Rizos A, Trenkwalder C, Rascol O, Pal S, Martino D, et al. King’ s Parkinson’ s disease pain scale, the first scale for pain in PD: An international validation. Mov Disord. 2015 Oct;30(12):1623-31.
15. Brefel-Courbon C, Payoux P, Thalamas C, Ory F, Quelven I, Chollet F, et al. Effect of levodopa on pain threshold in Parkinson’ s disease: a clinical and positron emission tomography study. Mov Disord. 2005 Dec;20(12):1557-63.
16. Boura E, Stamelou M, Vadasz D, Ries V, Unger MM, Kagi G, et al. Is increased spinal nociception another hallmark for Parkinson’ s disease? J Neurol. 2017 Mar;264(3):570-5.
17. Mylius V, Brebbermann J, Dohmann H, Engau I, Oertel WH, Moller JC. Pain sensitivity and clinical progression in Parkinson’ s disease. Mov Disord. 2011 Oct;26(12):2220-5.
18. Mylius V, Engau I, Teepker M, Stiasny-Kolster K, Schepelmann K, Oertel WH, et al. Pain sensitivity and descending inhibition of pain in Parkinson’ s disease. J Neurol Neurosurg Psychiatry. 2009 Jan;80(1):24-8.
19. Gerdelat-Mas A, Simonetta-Moreau M, Thalamas C, Ory-Magne F, Slaoui T, Rascol O, et al. Levodopa raises objective pain threshold in Parkinson’ s disease: a RIII reflex study. J Neurol Neurosurg Psychiatry. 2007 Oct;78(10):1140-2.
20. Mylius V, Baars JH, Witt K, Benninger D, de Andrade DC, Kagi G, et al. Deep Brain Stimulation Improves Parkinson’ s Disease-Associated Pain by Decreasing Spinal Nociception. Mov Disord. 2024 Feb;39(2):447-9.
21. Stocchi F, Rascol O, Kieburtz K, Poewe W, Jankovic J, Tolosa E, et al. Initiating levodopa/carbidopa therapy with and without entacapone in early Parkinson disease: the STRIDE-PD study. Ann Neurol. 2010 Jul;68(1):18-27.
22. Kassubek J, Chaudhuri KR, Zesiewicz T, Surmann E, Boroojerdi B, Moran K, et al. Rotigotine transdermal system and evaluation of pain in patients with Parkinson’ s disease: a post hoc analysis of the RECOVER study. BMC Neurol. 2014 Mar 6;14:42.
23. Rascol O, Zesiewicz T, Chaudhuri KR, Asgharnejad M, Surmann E, Dohin E, et al. A Randomized Controlled Exploratory Pilot Study to Evaluate the Effect of Rotigotine Transdermal Patch on Parkinson’ s Disease-Associated Chronic Pain. J Clin Pharmacol. 2016 Jul;56(7):852-61.
24. Borgohain R, Szasz J, Stanzione P, Meshram C, Bhatt MH, Chirilineau D, et al. Two-year, randomized, controlled study of safinamide as add-on to levodopa in mid to late Parkinson’ s disease. Mov Disord. 2014 Sep;29(10):1273-80.
25. Nebe A, Ebersbach G. Pain intensity on and off levodopa in patients with Parkinson’ s disease. Mov Disord. 2009 Jun 15;24(8):1233-7.
26. Li J, Zhu BF, Gu ZQ, Zhang H, Mei SS, Ji SZ, et al. Musculoskeletal Pain in Parkinson’ s Disease. Front Neurol. 2021;12:756538.
27. Buhmann C, Wrobel N, Grashorn W, Fruendt O, Wesemann K, Diedrich S, et al. Pain in Parkinson disease: a cross-sectional survey of its prevalence, specifics, and therapy. J Neurol. 2017 Apr;264(4):758-69.
28. Trenkwalder C, Chaudhuri KR, Martinez-Martin P, Rascol O, Ehret R, Valis M, et al. Prolonged-release oxycodone-naloxone for treatment of severe pain in patients with Parkinson’ s disease (PANDA): a double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2015 Dec;14(12):1161-70.
29. Chagas MH, Zuardi AW, Tumas V, Pena-Pereira MA, Sobreira ET, Bergamaschi MM, et al. Effects of cannabidiol in the treatment of patients with Parkinson’ s disease: an exploratory double-blind trial. J Psychopharmacol. 2014 Nov;28(11):1088-98.
30. Freo U, Furnari M, Ori C. Effects of tapentadol on pain, motor symptoms and cognitive functions in Parkinson’ s disease. J Pain Res. 2018;11:1849-56.
31. Rieu I, Degos B, Castelnovo G, Vial C, Durand E, Pereira B, et al. Incobotulinum toxin A in Parkinson’ s disease with foot dystonia: A double blind randomized trial. Parkinsonism Relat Disord. 2018 Jan;46:9-15.
32. Djaldetti R, Yust-Katz S, Kolianov V, Melamed E, Dabby R. The effect of duloxetine on primary pain symptoms in Parkinson disease. Clin Neuropharmacol. 2007 Jul-Aug;30(4):201-5.
33. Iwaki H, Ando R, Tada S, Nishikawa N, Tsujii T, Yamanishi Y, et al. A double-blind, randomized controlled trial of duloxetine for pain in Parkinson’ s disease. J Neurol Sci. 2020 Jul 15;414:116833.
34. Brefel-Courbon C, Harroch E, Marques A, Devos D, Thalamas C, Rousseau V, et al. Oxycodone or Higher Dose of Levodopa for the Treatment of Parkinsonian Central Pain: OXYDOPA Trial. Mov Disord. 2024 Jun 8.
35. Reuter I, Mehnert S, Leone P, Kaps M, Oechsner M, Engelhardt M. Effects of a flexibility and relaxation programme, walking, and nordic walking on Parkinson’ s disease. J Aging Res. 2011;2011:232473.
36. Huissoud M, Boussac M, Joineau K, Harroch E, Brefel-Courbon C, Descamps E. The effectiveness and safety of non-pharmacological intervention for pain management in Parkinson’ s disease: A systematic review. Rev Neurol (Paris). 2023 Oct 11.
37. Cury RG, Galhardoni R, Fonoff ET, Dos Santos Ghilardi MG, Fonoff F, Arnaut D, et al. Effects of deep brain stimulation on pain and other nonmotor symptoms in Parkinson disease. Neurology. 2014 Oct 14;83(16):1403-9.